
Significance of Specific Oxidoreductases in the Design of Hypoxia-Activated Prodrugs and Fluorescent Turn off–on Probes for Hypoxia Imaging


Abstract
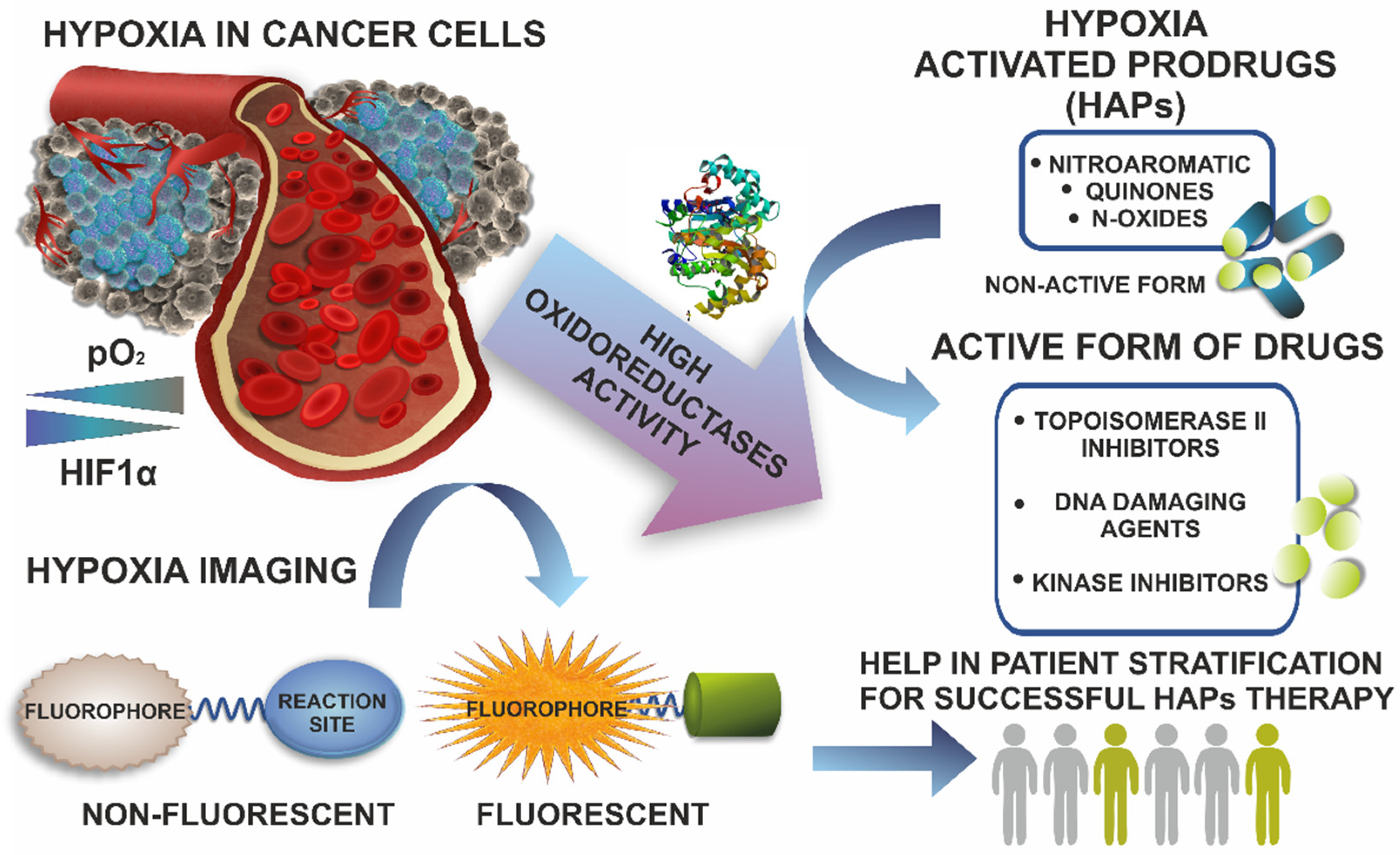
:Simple Summary
Abstract

1. Introduction
2. Hypoxia-Inducible Factor 1—Adaptive Response to Hypoxia
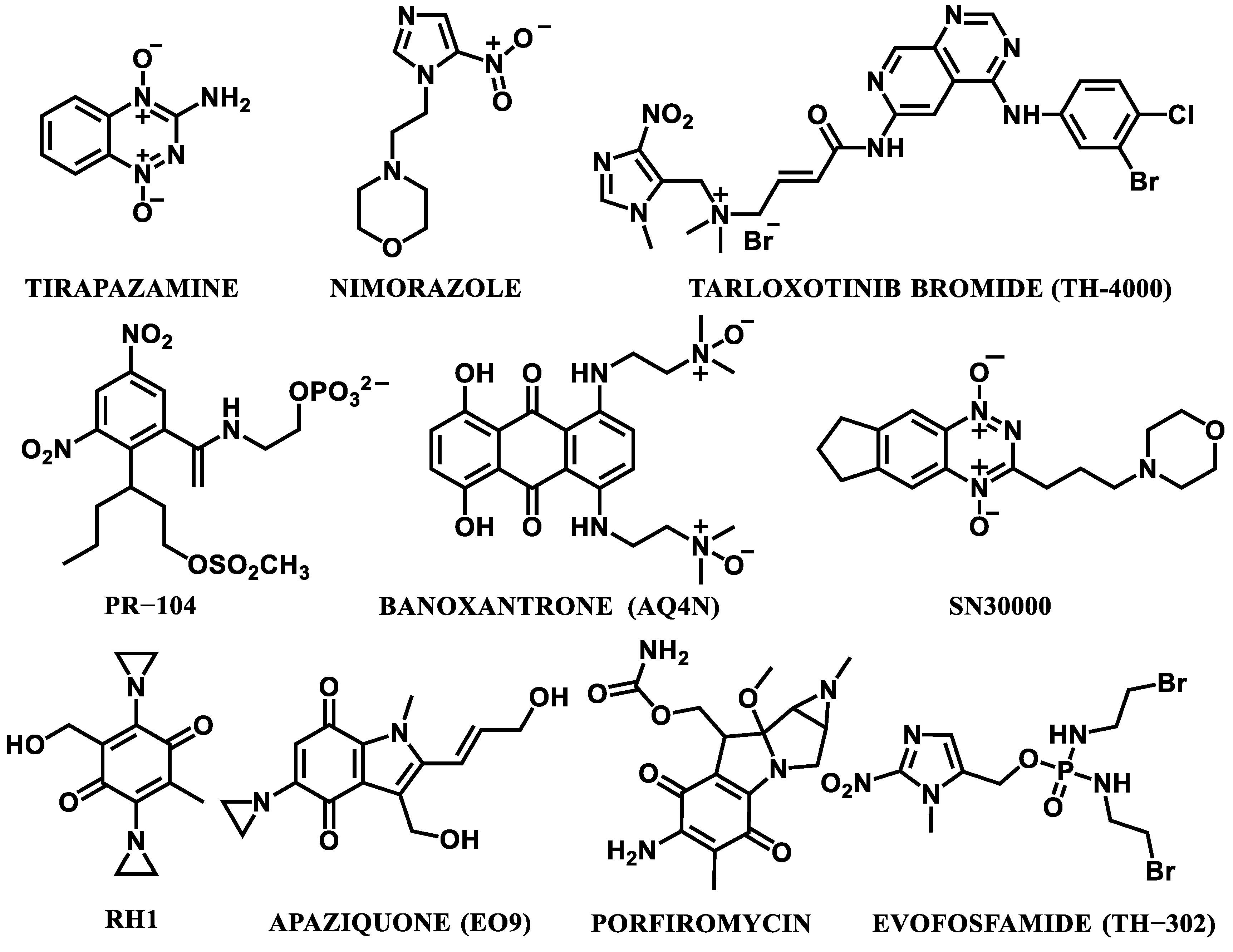
3. Hypoxia-Activated Prodrugs (HAPs)
- The development of screening methods (e.g., PET/CT imaging) for the selection of the best potential prodrugs [1].
- The development of HAPs that are preferably activated to release molecularly targeted protein ligands, rather than DNA-damaging cytotoxins, in order to limit the toxicity effects [38].
- Further development of gene-directed enzyme-prodrug therapy (GDEPT). This kind of therapy engages an enzyme–prodrug combination in order to generate high levels of bystander cell killing [71]. A genetically encoded therapeutic enzyme is indirectly delivered to the tumor milieu, and this process is mediated by a tumor-tropic bacterial or viral vector. Subsequently, the enzyme transforms the delivered non-toxic prodrug into a potent cytotoxin and the therapeutic effect is much stronger, compared to non-targeted traditional agents [72]. Enzymes, particularly bacterial nitroreductases, known to activate anticancer nitroaromatic prodrugs, are very promising for their use in GDEPT [73,74].
4. Oxidoreductases
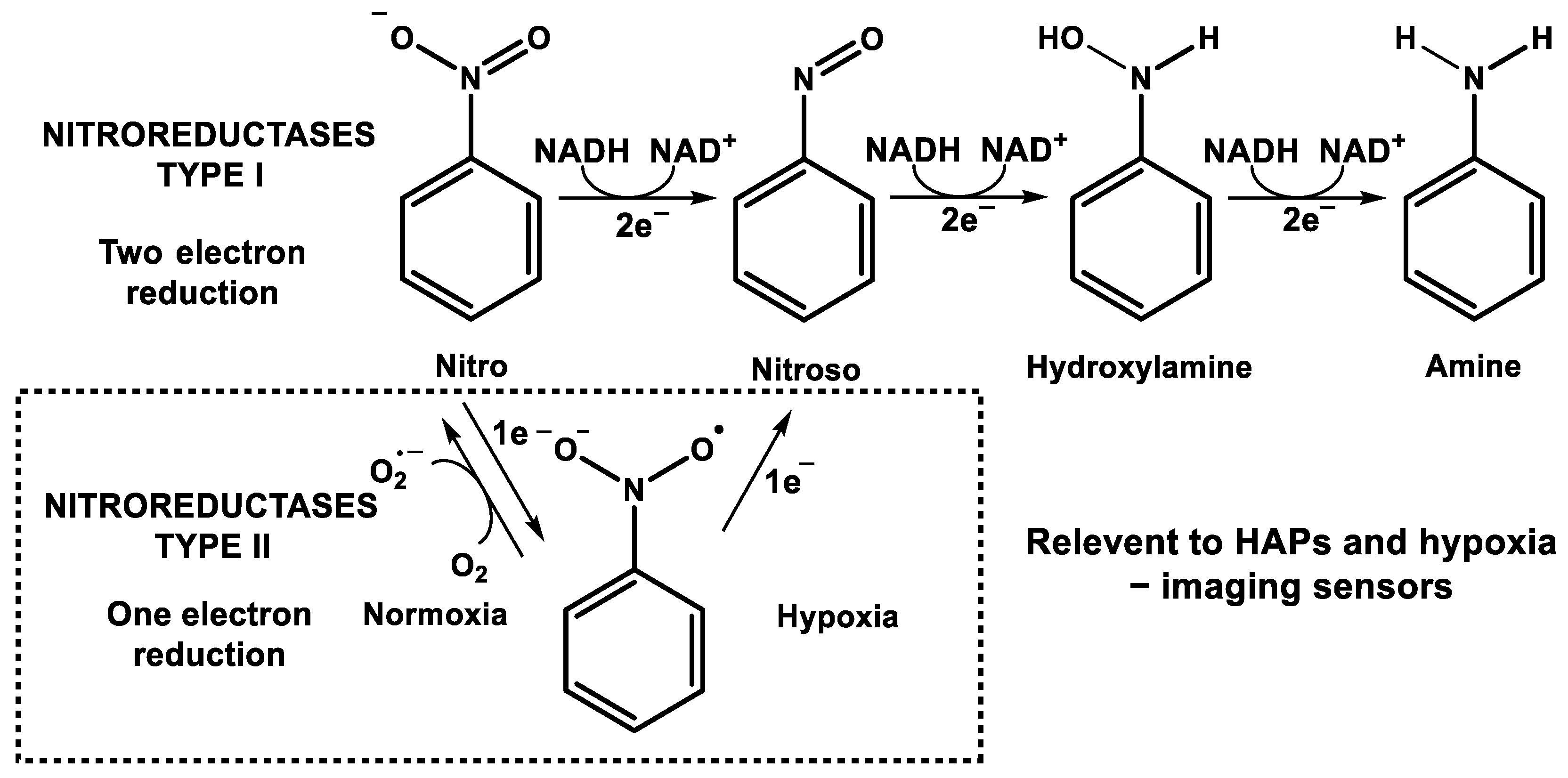
4.1. Nitroreductases (NTRs)
4.2. Azoreductases
4.3. Cytochrome p450 Reductase (POR)
4.4. Xanthine Oxidase (XO)/Xanthine Oxidoreductase (XOR)
4.5. Cytochrome b5 Reductase (CYB5R)
4.6. Critical Implications
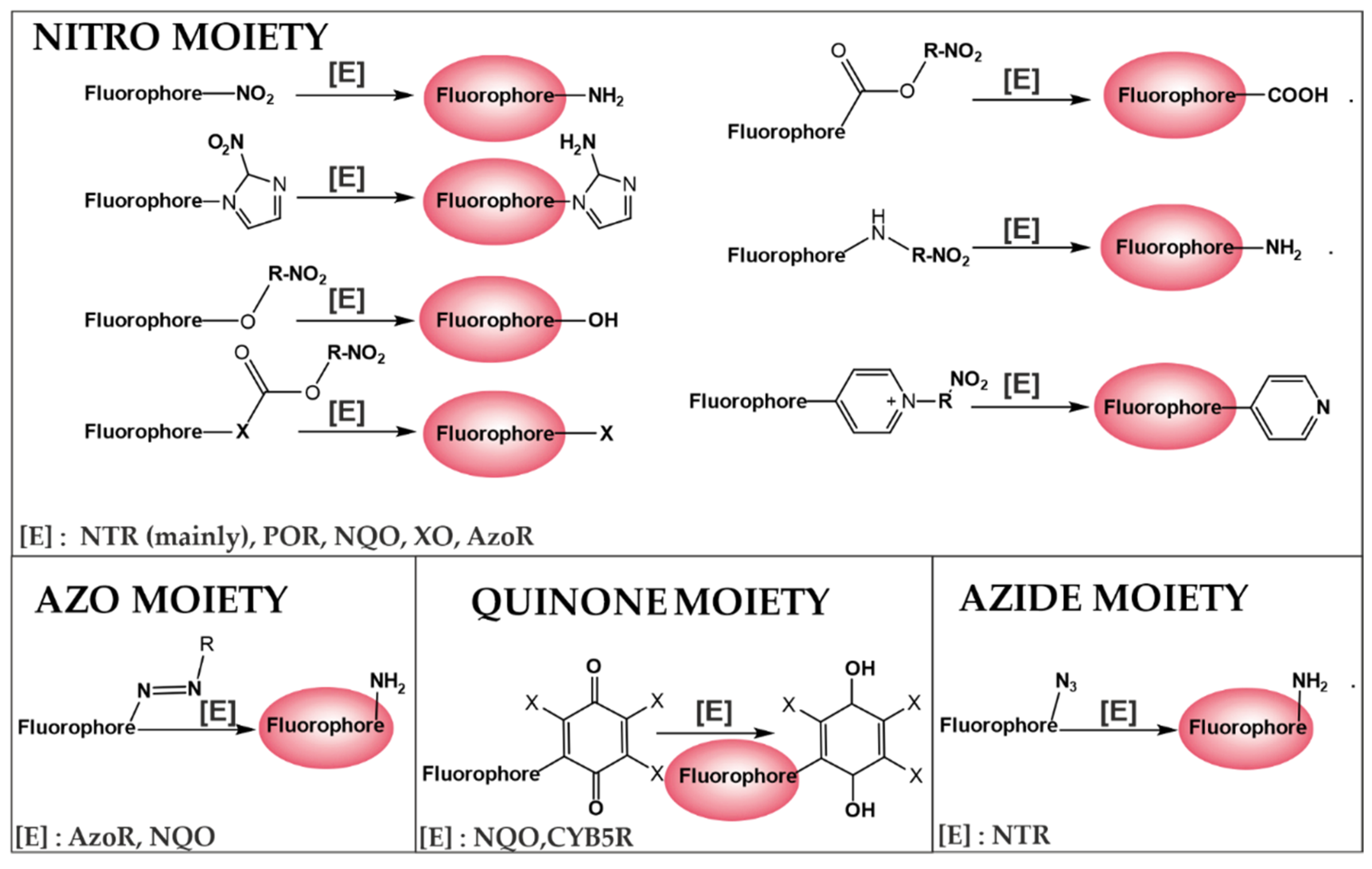
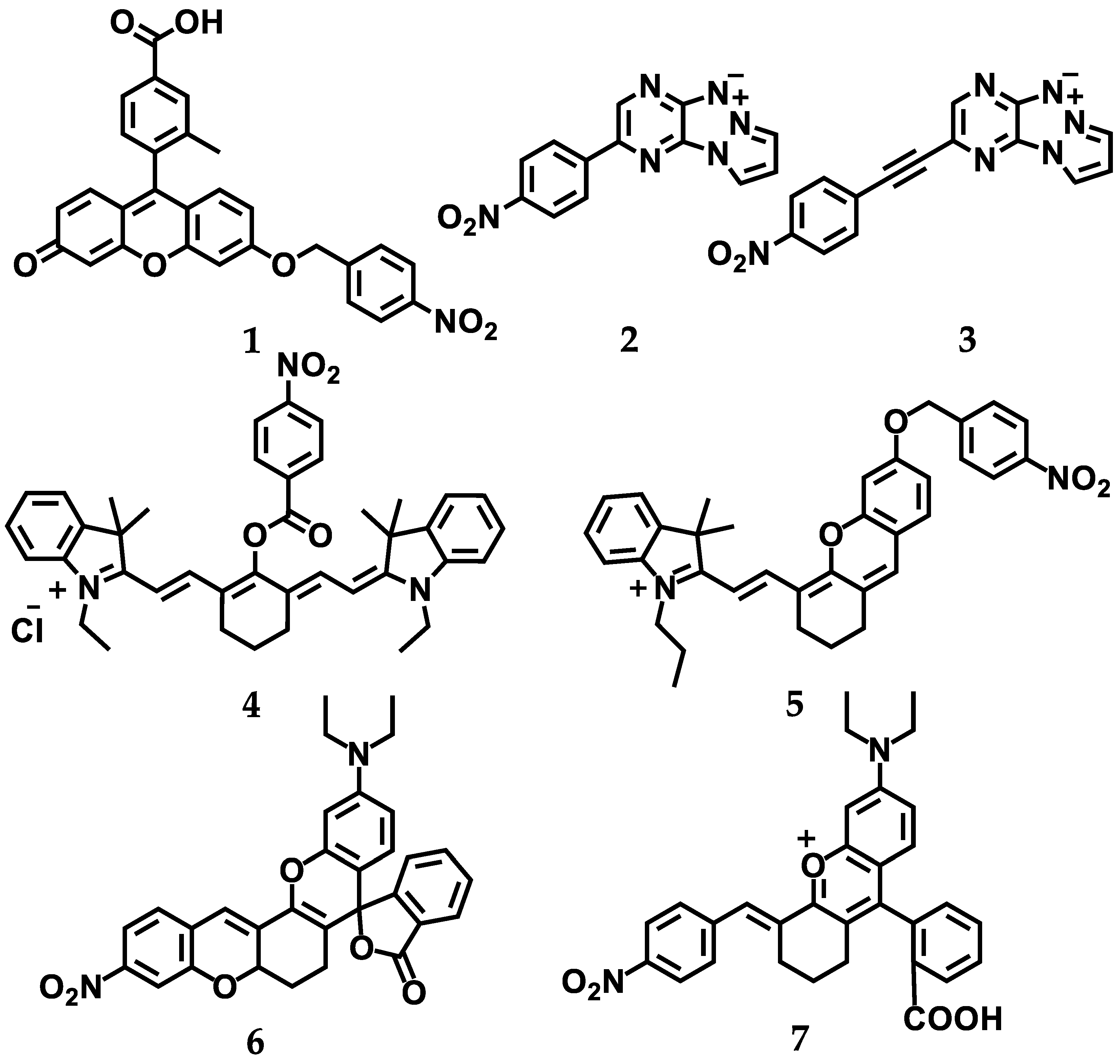
5. Visualization of the Level of Hypoxia
5.1. Design Strategies for Fluorescent Turn off–on Probes for Hypoxia Imaging
5.2. Evaluation of NTR Expression under Hypoxic Conditions—Correlation between Fluoresent Imaging Probes and Biochemial Evaluation
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Li, Y.; Zhao, L.; Li, X.F. Targeting Hypoxia: Hypoxia-Activated Prodrugs in Cancer Therapy. Front. Oncol. 2021, 11, 2920. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.C.; Lebedev, A.; Aten, E.; Madsen, K.; Marciano, L.; Kolb, H.C. The Clinical Importance of Assessing Tumor Hypoxia: Relationship of Tumor Hypoxia to Prognosis and Therapeutic Opportunities. Antioxid. Redox Signal. 2014, 21, 1516–1554. [Google Scholar] [CrossRef] [PubMed]
- Bayer, C.; Shi, K.; Astner, S.T.; Maftei, C.A.; Vaupel, P. Acute versus chronic hypoxia: Why a simplified classifi-cation is simply not enough. Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Challapalli, A.; Carroll, L.; Aboagye, E.O. Molecular mechanisms of hypoxia in cancer. Clin. Transl. Imaging 2017, 5, 225–253. [Google Scholar] [CrossRef] [Green Version]
- Wouters, A.; Pauwels, B.; Lardon, F.; Vermorken, J.B. Review: Implications of in vitro research on the effect of radiotherapy and chemotherapy under hypoxic conditions. Oncologist 2007, 12, 690–712. [Google Scholar] [CrossRef] [Green Version]
- Vaupel, P.; Harrison, L. Tumor hypoxia: Causative factors, compensatory mechanisms, and cellular response. Oncologist 2004, 9 (Suppl. 5), 4–9. [Google Scholar] [CrossRef] [Green Version]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metas-tasis, and resistance to therapy. Hypoxia 2015, 3, 83. [Google Scholar] [CrossRef] [Green Version]
- Li, X.F.; O’Donoghue, J.A. Hypoxia in microscopic tumors. Cancer Lett. 2008, 264, 172–180. [Google Scholar] [CrossRef] [Green Version]
- Vaupel, P. Physiological Mechanisms of Treatment Resistance; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar] [CrossRef]
- Moeller, B.J.; Richardson, R.A.; Dewhirst, M.W. Hypoxia and radiotherapy: Opportunities for improved out-comes in cancer treatment. Cancer Metastasis Rev. 2007, 26, 241–248. [Google Scholar] [CrossRef]
- Rofstad, E.K.; Sundfør, K.; Lyng, H.; Tropé, C.G. Hypoxia-induced treatment failure in advanced squamous cell carcinoma of the uterine cervix is primarily due to hypoxia-induced radiation resistance rather than hypox-ia-induced metastasis. Br. J. Cancer 2000, 83, 354–359. [Google Scholar] [CrossRef]
- Singleton, D.C.; Macann, A.; Wilson, W.R. Therapeutic targeting of the hypoxic tumour microenvironment. Nat. Rev. Clin. Oncol. 2021, 18, 751–772. [Google Scholar] [CrossRef] [PubMed]
- Jubb, A.M.; Buffa, F.M.; Harris, A.L. Assessment of tumour hypoxia for prediction of response to therapy and cancer prognosis. J. Cell. Mol. Med. 2010, 14, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.J.; Evans, S.M. Optimizing Hypoxia Detection and Treatment Strategies. Semin. Nucl. Med. 2015, 45, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.Y.; Lee, J.Y. Targeting Tumor Adaption to Chronic Hypoxia: Implications for Drug Resistance, and How It Can Be Overcome. Int. J. Mol. Sci. 2017, 18, 1854. [Google Scholar] [CrossRef] [Green Version]
- Evans, S.M.; Koch, C.J. Prognostic significance of tumor oxygenation in humans. Cancer Lett. 2003, 195, 1–16. [Google Scholar] [CrossRef]
- Dhani, N.; Fyles, A.; Hedley, D.; Milosevic, M. The clinical significance of hypoxia in human cancers. Semin. Nucl. Med. 2015, 45, 110–121. [Google Scholar] [CrossRef]
- Koch, C.J. Measurement of absolute oxygen levels in cells and tissues using oxygen sensors and 2-nitroimidazole EF5. Methods Enzym. 2002, 352, 3–31. [Google Scholar] [CrossRef]
- Godet, I.; Doctorman, S.; Wu, F.; Gilkes, D.M. Detection of Hypoxia in Cancer Models: Significance, Challenges, and Advances. Cells 2022, 11, 686. [Google Scholar] [CrossRef]
- Xue, F.; Chen, J.; Chen, H. Design strategy of optical probes for tumor hypoxia imaging. Sci. China Life Sci. 2020, 63, 1786–1797. [Google Scholar] [CrossRef]
- Qi, Y.L.; Guo, L.; Chen, L.L.; Li, H.; Yang, Y.S.; Jiang, A.Q.; Zhu, H.L. Recent progress in the design principles, sensing mechanisms, and applications of small-molecule probes for nitroreductases. Coord. Chem. Rev. 2020, 421, 213460. [Google Scholar] [CrossRef]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, P.H. The HIF pathway in cancer. Semin. Cell Dev. Biol. 2005, 16, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Ohh, M. Ubiquitin Pathway in VHL Cancer Syndrome. Neoplasia 2006, 8, 623–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayan, F.; Mazure, N.M.; Brahimi-Horn, M.C.; Pouysségur, J. A Dialogue between the Hypox-ia-Inducible Factor and the Tumor Microenvironment. Cancer Microenviron. 2008, 1, 53–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Oxygen Sensing, Homeostasis, and Disease. Mech. Dis. 2011, 365, 537–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378. [Google Scholar] [CrossRef] [Green Version]
- Brahimi-Horn, M.C.; Chiche, J.; Pouysségur, J. Hypoxia and cancer. J. Mol. Med. 2007, 85, 1301–1307. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Infantino, V.; Santarsiero, A.; Convertini, P.; Todisco, S.; Iacobazzi, V. Cancer Cell Metabolism in Hypoxia: Role of HIF-1 as Key Regulator and Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 5703. [Google Scholar] [CrossRef]
- Dengler, V.L.; Galbraith, M.D.; Espinosa, J.M. Transcriptional Regulation by Hypoxia Inducible Factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, Q.T.; Courter, D. Clinical Biomarkers for Hypoxia Targeting. Cancer Metastasis Rev. 2008, 27, 351. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HIF-1 and HIF-2 transcription factors--similar but not identical. Mol. Cells 2010, 29, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.Y.; Powis, G. Passing the baton: The HIF switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, F.W.; Wouters, B.G.; Wilson, W.R. Hypoxia-activated prodrugs: Paths forward in the era of personal-ised medicine. Br. J. Cancer 2016, 114, 1071–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegelberg, L.; Houben, R.; Niemans, R.; de Ruysscher, D.; Yaromina, A.; Theys, J.; Guise, C.P.; Smaill, J.B.; Patterson, A.V.; Lambin, P.; et al. Hypoxia-activated prodrugs and (lack of) clinical progress: The need for hypoxia-based biomarker patient selection in phase III clinical trials. Clin. Transl. Radiat. Oncol. 2019, 15, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Mistry, I.N.; Thomas, M.; Calder, E.D.D.; Conway, S.J.; Hammond, E.M. Clinical Advances of Hypox-ia-Activated Prodrugs in Combination With Radiation Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2017, 98, 1183–1196. [Google Scholar] [CrossRef] [Green Version]
- Rischin, D.; Peters, L.; Fisher, R.; Macann, A.; Denham, J.; Poulsen, M.; Jackson, M.; Kenny, L.; Penniment, M.; Carry, J.; et al. Tirapazamine, Cisplatin, and Radiation versus Fluorouracil, Cisplatin, and Ra-diation in patients with locally advanced head and neck cancer: A randomized phase II trial of the Trans-Tasman Radiation Oncology Group (TROG 98.02). J. Clin. Oncol. 2005, 23, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Le, Q.T.; Taira, A.; Budenz, S.; Dorie, M.J.; Goffinet, D.R.; Fee, W.E.; Goode, R.; Bloch, D.; Koong, A.; Brown, J.M.; et al. Mature results from a randomized phase II trial of cisplatin plus 5-fluorouracil and radiotherapy with or without tirapazamine in patients with resectable stage IV head and neck squamous cell carcinomas. Cancer 2006, 106, 1940–1949. [Google Scholar] [CrossRef]
- Rischin, D.; Peters, L.J.; O’Sullivan, B.; Giralt, J.; Fisher, R.; Yuen, K.; Trotti, A.; Bernier, J.; Bourhis, J.; Ringash, J.; et al. Tirapazamine, cisplatin, and radiation versus cisplatin and radiation for advanced squa-mous cell carcinoma of the head and neck (TROG 02.02, HeadSTART): A phase III trial of the Trans-Tasman Radiation Oncology Group. J. Clin. Oncol. 2010, 28, 2989–2995. [Google Scholar] [CrossRef]
- van Cutsem, E.; Lenz, H.-J.; Furuse, J.; Tabernero, J.; Heinemann, V.; Ioka, T.; Bazin, I.; Ueno, M.; Csõszi, T.; Wasan, H.; et al. Evofosfamide (TH-302) in combination with gemcitabine in previously untreated patients with metastatic or locally advanced unresectable pancreatic ductal adenocarcinoma: Primary analysis of the randomized, double-blind phase III MAESTRO study. J. Clin. Oncol. 2016, 34, 193. [Google Scholar] [CrossRef]
- Larue, R.T.H.M.; van de Voorde, L.; Berbée, M.; van Elmpt, W.J.C.; Dubois, L.J.; Panth, K.M.; Peeters, S.G.J.A.; Claessens, A.; Schreurs, W.M.J.; Nap, M.; et al. A phase 1 “window-of-opportunity” trial testing evofosfamide (TH-302), a tumour-selective hypoxia-activated cy-totoxic prodrug, with preoperative chemoradiotherapy in oesophageal adenocarcinoma patients. BMC Cancer 2016, 16, 644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overgaard, J.; Hansen, H.S.; Overgaard, M.; Bastholt, L.; Berthelsen, A.; Specht, L.; Lindeløv, B.; Jørgensen, K. A randomized double-blind phase III study of nimorazole as a hypoxic radiosensitizer of primary radiotherapy in supraglottic larynx and pharynx carcinoma. Results of the Danish Head and Neck Cancer Study (DA-HANCA) Protocol 5-85. Radiother. Oncol. 1998, 46, 135–146. [Google Scholar] [CrossRef]
- Overgaard, J.; Eriksen, J.G.; Nordsmark, M.; Alsner, J.; Horsman, M.R. Plasma osteopontin, hypoxia, and re-sponse to the hypoxia sensitiser nimorazole in radiotherapy of head and neck cancer: Results from the DA-HANCA 5 randomised double-blind placebo-controlled trial. Lancet Oncol. 2005, 6, 757–764. [Google Scholar] [CrossRef]
- Metwally, M.A.H.; Ali, R.; Kuddu, M.; Shouman, T.; Strojan, P.; Iqbal, K.; Prasad, R.; Grau, C.; Overgaard, J. IAEA-HypoX. A randomized multicenter study of the hypoxic radiosensitizer nimorazole concomitant with accelerated radiotherapy in head and neck squamous cell carcinoma. Radiother. Oncol. 2015, 116, 15–20. [Google Scholar] [CrossRef] [PubMed]
- McLean, L.S.; Morris, T.A.; Gramza, A.; Liu, S.; Khan, S.A.; Colevas, A.D.; Pearce, T.; Rischin, D. A phase II study of tarloxotinib (a hypoxia activated prodrug of a pan-erb tyrosine kinase inhibitor) in patients with recurrent or metastatic squamous cell carcinoma of the head and neck or skin. Investig. New Drugs 2022, 1–7. [Google Scholar] [CrossRef] [PubMed]
- McKeage, M.J.; Jameson, M.B.; Ramanathan, R.K.; Rajendran, J.; Gu, Y.; Wilson, W.R.; Melink, T.J.; Tche-kmedyian, N.S. PR-104 a bioreductive pre-prodrug combined with gemcitabine or docetaxel in a phase Ib study of patients with advanced solid tumours. BMC Cancer 2012, 12, 496. [Google Scholar] [CrossRef] [Green Version]
- Steward, W.P.; Middleton, M.; Benghiat, A.; Loadman, P.M.; Hayward, C.; Waller, S.; Ford, S.; Halbert, G.; Patterson, L.H.; Talbot, D. The use of pharmacokinetic and pharmacodynamic end points to determine the dose of AQ4N, a novel hypoxic cell cytotoxin, given with fractionated radiotherapy in a phase I study. Ann. Oncol. 2007, 18, 1098–1103. [Google Scholar] [CrossRef]
- Haffty, B.G.; Wilson, L.D.; Son, Y.H.; Cho, E.I.; Papac, R.J.; Fischer, D.B.; Rockwell, S.; Sartorelli, A.C.; Ross, D.A.; Sasaki, C.T.; et al. Concurrent chemo-radiotherapy with mitomycin C compared with porfiromycin in squamous cell cancer of the head and neck: Final results of a randomized clinical trial. Int. J. Radiat. Oncol. Biol. Phys. 2005, 61, 119–128. [Google Scholar] [CrossRef]
- Zeman, E.M.; Brown, J.M.; Lemmon, M.J.; Hirst, V.K.; Lee, W.W. SR-4233: A new bioreductive agent with high selective toxicity for hypoxic mammalian cells. Int. J. Radiat. Oncol. Biol. Phys. 1986, 12, 1239–1242. [Google Scholar] [CrossRef]
- Doherty, N.; Hancock, S.L.; Kaye, S.; Coleman, C.N.; Shulman, L.; Marquez, C.; Mariscal, C.; Rampling, R.; Senan, S.; Roemeling, R.V. Muscle cramping in phase I clinical trials of tirapazamine (SR 4233) with and without radiation. Int. J. Radiat. Oncol. Biol. Phys. 1994, 29, 379–382. [Google Scholar] [CrossRef]
- Saunders, M.P.; Patterson, A.V.; Chinje, E.C.; Harris, A.L.; Stratford, I.J. NADPH:cytochrome c (P450) reductase activates tirapazamine (SR4233) to restore hypoxic and oxic cytotoxicity in an aerobic resistant derivative of the A549 lung cancer cell line. Br. J. Cancer 2000, 82, 651–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hicks, K.O.; Siim, B.G.; Jaiswal, J.K.; Pruijn, F.B.; Fraser, A.M.; Patel, R.; Hogg, A.; Liyanage, H.D.S.; Dorie, M.J.; Brown, J.M.; et al. Pharmacokinetic/pharmacodynamic modeling identifies SN30000 and SN29751 as tirapazamine analogues with improved tissue penetration and hypoxic cell killing in tumors. Clin. Cancer Res. 2010, 16, 4946–4957. [Google Scholar] [CrossRef] [Green Version]
- Nytko, K.J.; Grgic, I.; Bender, S.; Ott, J.; Guckenberger, M.; Riesterer, O.; Pruschy, M. The hypoxia-activated pro-drug evofosfamide in combination with multiple regimens of radiotherapy. Oncotarget 2017, 8, 23702–23712. [Google Scholar] [CrossRef] [Green Version]
- Peeters, S.G.J.A.; Zegers, C.M.L.; Biemans, R.; Lieuwes, N.G.; van Stiphout, R.G.P.M.; Yaromina, A.; Sun, J.D.; Hart, C.P.; Windhorst, A.D.; van Elmpt, W.; et al. TH-302 in Combination with Radiotherapy En-hances the Therapeutic Outcome and Is Associated with Pretreatment [18F]HX4 Hypoxia PET Imaging. Clin. Cancer Res. 2015, 21, 2984–2992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borad, M.J.; Reddy, S.G.; Bahary, N.; Uronis, H.E.; Sigal, D.; Cohn, A.L.; Schelman, W.R.; Stephenson, J.; Chi-orean, E.G.; Rosen, P.J.; et al. Randomized Phase II Trial of Gemcitabine Plus TH-302 Versus Gemcitabine in Patients With Advanced Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 1475–1481. [Google Scholar] [CrossRef] [Green Version]
- Chawla, S.P.; Cranmer, L.D.; van Tine, B.A.; Reed, D.R.; Okuno, S.H.; Butrynski, J.E.; Adkins, D.R.; Hendifar, A.E.; Kroll, S.; Ganjoo, K.N. Phase II study of the safety and antitumor activity of the hypoxia-activated prodrug TH-302 in combination with doxorubicin in patients with advanced soft tissue sarcoma. J. Clin. Oncol. 2014, 32, 3299–3306. [Google Scholar] [CrossRef]
- Ganjoo, K.N.; Cranmer, L.D.; Butrynski, J.E.; Rushing, D.; Adkins, D.; Okuno, S.H.; Lorente, G.; Kroll, S.; Langmuir, V.K.; Chawla, S.P. A phase I study of the safety and pharmacokinetics of the hypoxia-activated pro-drug TH-302 in combination with doxorubicin in patients with advanced soft tissue sarcoma. Oncology 2011, 80, 50–56. [Google Scholar] [CrossRef]
- Weiss, G.J.; Infante, J.R.; Chiorean, E.G.; Borad, M.J.; Bendell, J.C.; Molina, J.R.; Tibes, R.; Ramanathan, R.K.; Lewandowski, K.; Jones, S.F.; et al. Phase 1 study of the safety, tolerability, and pharmacokinetics of TH-302, a hypoxia-activated prodrug, in patients with ad-vanced solid malignancies. Clin. Cancer Res. 2011, 17, 2997–3004. [Google Scholar] [CrossRef] [Green Version]
- Smaill, J.B.; Lu, G.; van Leeuwen, W.; Abbattista, M.; Anderson, R.F.; Denny, W.A.; Doñate, F.; Jaswail, J.; Ma-roz, A.; Puryer, M.; et al. Abstract C46: Design and identification of the nov-el hypoxia-activated irreversible pan-HER inhibitor SN29966. Mol. Cancer Ther. 2009, 8, C46. [Google Scholar] [CrossRef]
- Phillips, R.M. Targeting the hypoxic fraction of tumours using hypoxia-activated prodrugs. Cancer Chemother. Pharmacol. 2016, 77, 441–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, D.; Yang, H.; Baines, H.; Miles, E.; Bolton, S.; West, C.; Slevin, N. NIMRAD-a phase III trial to inves-tigate the use of nimorazole hypoxia modification with intensity-modulated radiotherapy in head and neck cancer. Clin. Oncol. (R Coll. Radiol.) 2014, 26, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Tharmalingham, H.; Hoskin, P. Clinical trials targeting hypoxia. Br. J. Radiol. 2019, 92, 20170966. [Google Scholar] [CrossRef] [PubMed]
- Vilaplana-Lopera, N.; Besh, M.; Moon, E.J. Targeting hypoxia: Revival of old remedies. Biomolecules 2021, 11, 1604. [Google Scholar] [CrossRef]
- Bonnet, M.; Hong, C.R.; Wong, W.W.; Liew, L.P.; Shome, A.; Wang, J.; Gu, Y.; Stevenson, R.J.; Qi, W.; Anderson, R.F.; et al. Next-Generation Hypoxic Cell Radiosensi-tizers: Nitroimidazole Alkylsulfonamides. J. Med. Chem. 2018, 61, 1241–1254. [Google Scholar] [CrossRef]
- Mortensen, L.S.; Buus, S.; Nordsmark, M.; Bentzen, L.; Munk, O.L.; Keiding, S.; Overgaard, J. Identifying hypoxia in human tumors: A correlation study between 18F-FMISO PET and the Eppendorf oxygen-sensitive elec-trode. Acta Oncol. 2010, 49, 934–940. [Google Scholar] [CrossRef]
- Rasey, J.S.; Koh, W.J.; Evans, M.L.; Peterson, L.M.; Lewellen, T.K.; Graham, M.M.; Krohn, K.A. Quantifying re-gional hypoxia in human tumors with positron emission tomography of [18F]fluoromisonidazole: A pretherapy study of 37 patients. Int. J. Radiat. Oncol. Biol. Phys. 1996, 36, 417–428. [Google Scholar] [CrossRef]
- Walton, W.I.; Sugget, N.; Workman, P. The role of human and rodent DT-diaphorase in the reductive metabo-lism of hypoxic cell cytotoxins. Int. J. Radiat. Oncol. Biol. Phys. 1992, 22, 643–647. [Google Scholar] [CrossRef]
- Zhang, R.; Feng, L.; Dong, Z.; Wang, L.; Liang, C.; Chen, J.; Ma, Q.; Zhang, R.; Chen, Q.; Wang, Y.; et al. Glucose & oxygen exhausting liposomes for combined cancer starvation and hypoxia-activated therapy. Biomaterials 2018, 162, 123–131. [Google Scholar] [CrossRef]
- Sharrock, A.v.; McManaway, S.P.; Rich, M.H.; Mumm, J.S.; Hermans, I.F.; Tercel, M.; Pruijn, F.B.; Ackerley, D.F. Engineering the Escherichia coli Nitroreductase NfsA to Create a Flexible Enzyme-Prodrug Activation Sys-tem. Front. Pharmacol. 2021, 12, 701456. [Google Scholar] [CrossRef]
- Williams, E.M.; Little, R.F.; Mowday, A.M.; Rich, M.H.; Chan-Hyams, J.V.E.; Copp, J.N.; Smaill, J.B.; Patter-son, A.V.; Ackerley, D.F. Nitroreductase gene-directed enzyme prodrug therapy: Insights and advances toward clinical utility. Biochem. J. 2015, 471, 131–153. [Google Scholar] [CrossRef] [PubMed]
- Copp, J.N.; Mowday, A.M.; Williams, E.M.; Guise, C.P.; Ashoorzadeh, A.; Sharrock, A.V.; Flanagan, J.U.; Smaill, J.B.; Patterson, A.V.; Ackerley, D.F. Engineering a Multifunctional Nitroreductase for Improved Activa-tion of Prodrugs and PET Probes for Cancer Gene Therapy. Cell Chem. Biol. 2017, 24, 391–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan-Hyams, J. Characterisation and Optimisation of Nitroreductase-Prodrug Combinations for Bacterial-Directed Enzyme-Prodrug Therapy; Victoria University of Wellington, Nowa Zelandia: Wellington, New Zealand, 2020. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Chandel, N.S. Hypoxia. 2. Hypoxia regulates cellular metabolism. Am. J. Physiol. Cell Physiol. 2011, 300, C385–C393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimmons, S.A.; Workman, P.; Grever, M.; Paull, K.; Camalier, R.; Lewis, A.D. Reductase Enzyme Expression Across the National Cancer Institute Tumor Cell Line Panel: Correlation With Sensitivity to Mitomycin C and EO9. J. Natl. Cancer Inst. 1996, 88, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Kelley, E.E.; Hock, T.; Khoo, N.K.H.; Richardson, G.R.; Johnson, K.K.; Powell, P.C.; Giles, G.I.; Agarwal, A.; Lancaster, J.R.; Tarpey, M.M. Moderate hypoxia induces xanthine oxidoreductase activity in arterial endothelial cells. Free. Radic. Biol. Med. 2006, 40, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Janczy-Cempa, E.; Mazuryk, O.; Sirbu, D.; Chopin, N.; Zarnik, M.; Zastawna, M.; Colas, C.; Hiebel, M.-A.; Su-zenet, F.; Brindell, M. Nitro-Pyrazinotriazapentalene scaffolds-nitroreductase quantification and in vitro fluo-rescence imaging of hypoxia. Sens. Actuators B Chem. 2021, 346, 925–4005. [Google Scholar] [CrossRef]
- Luo, S.; Zou, R.; Wu, J.; Landry, M.P. A Probe for the Detection of Hypoxic Cancer Cells. ACS Sens. 2017, 2, 1139–1145. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, X.F.; Chen, Q.; Cao, X.Q.; Shen, S.L. A novel near-infrared fluorescence off-on probe for im-aging hypoxia and nitroreductase in cells and in vivo. Sens. Actuators B Chem. 2022, 353, 131–145. [Google Scholar] [CrossRef]
- Li, Y.; Sun, Y.; Li, J.; Su, Q.; Yuan, W.; Dai, Y.; Han, C.; Wang, Q.; Feng, W.; Li, F. Ultrasensitive near-infrared fluo-rescence-enhanced probe for in vivo nitroreductase imaging. J. Am. Chem. Soc. 2015, 137, 6407–6416. [Google Scholar] [CrossRef]
- Hettie, K.S.; Klockow, J.L.; Moon, E.J.; Giaccia, A.J.; Chin, F.T. A NIR fluorescent smart probe for imaging tumor hypoxia. Cancer Rep. 2021, 4, e1384. [Google Scholar] [CrossRef] [PubMed]
- Punganuru, S.R.; Madala, H.R.; Arutla, V.; Zhang, R.; Srivenugopal, K.S. Characterization of a highly specific NQO1-activated near-infrared fluorescent probe and its application for in vivo tumor imaging. Sci. Rep. 2019, 9, 8577. [Google Scholar] [CrossRef] [PubMed]
- Manley, E.; Waxman, D.J. Impact of tumor blood flow modulation on tumor sensitivity to the bioreductive drug banoxantrone. J. Pharmacol. Exp. Ther. 2013, 344, 368–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Dwyer, P.J.; Yao, K.-S.; Ford, P.; Godwin, A.K.; Clayton, M. Effects of Hypoxia on Detoxicating Enzyme Ac-tivity and Expression in HT29 Colon Cells. Cancer Res. 1994, 54, 3082–3087. [Google Scholar] [PubMed]
- Linder, N.; Martelin, E.; Lapatto, R.; Raivio, K.O. Posttranslational inactivation of human xanthine oxidore-ductase by oxygen under standard cell culture conditions. Am. J. Physiol. Physiol. 2003, 285, 48–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassoun, P.M.; Yu, F.S.; Cote, C.G.; Zulueta, J.J.; Sawhney, R.; Skinner, K.A.; Skinner, H.B.; Parks, D.A.; Lanzillo, J.J. Upregulation of xanthine oxidase by lipopolysaccharide, interleukin-1, and hypoxia. Role in acute lung inju-ry. Am. J. Respir. Crit. Care Med. 1998, 158, 299–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayyali, U.S.; Donaldson, C.; Huang, H.; Abdelnour, R.; Hassoun, P.M. Phosphorylation of xanthine dehydro-genase/oxidase in hypoxia. J. Biol. Chem. 2001, 276, 14359–14365. [Google Scholar] [CrossRef] [Green Version]
- Poss, W.B.; Huecksteadt, T.R.; Panus, P.C.; Freeman, B.A.; Hoidal, J.R. Regulation of xanthine dehydrogenase and xanthine oxidase activity by hypoxia. Am. J. Physiol. 1996, 270. [Google Scholar] [CrossRef]
- Oliveira, I.M.; Bonatto, D.; Antonio, J.; Henriques, P.; Vargas, R.F. Nitroreductases: Enzymes with Environ-mental, Biotechnological and Clinical Importance. In Current Research, Technology and Education in Applied Microbiology and Microbial Biotechnology; Mendez-Vilas, A., Ed.; Formatex Research Center: Badajoz, Spain, 2010; pp. 1008–1019. [Google Scholar]
- Haynes, C.A.; Koder, R.L.; Miller, A.F.; Rodgers, D.W. Structures of Nitroreductase in Three States: Effects of inhibitor binding and reduction. J. Biol. Chem. 2002, 277, 11513–11520. [Google Scholar] [CrossRef] [Green Version]
- Qiao, J.; Wang, M.; Cui, M.; Fang, Y.; Li, H.; Zheng, C.; Li, Z.; Xu, Y.; Hua, H.; Li, D. Small-molecule probes for fluo-rescent detection of cellular hypoxia-related nitroreductase. J. Pharm. Biomed. Anal. 2021, 203. [Google Scholar] [CrossRef]
- Guillén, H.; Curiel, J.A.; Landete, J.M.; Muñoz, R.; Herraiz, T. Characterization of a Nitroreductase with Selec-tive Nitroreduction Properties in the Food and Intestinal Lactic Acid Bacterium Lactobacillus plantarum WCFS1. J. Agric. Food Chem. 2009, 57, 10457–10465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastorekova, S.; Gillies, R.J. The role of carbonic anhydrase IX in cancer development: Links to hypoxia, acido-sis, and beyond. Cancer Metastasis Rev. 2019, 38, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Huizing, F.J.; Hoeben, B.A.W.; Lok, J.; Boerman, O.C.; Heskamp, S.; Bussink, J. Imaging carbonic anhydrase IX as a method for monitoring hypoxia-related radioresistance in preclinical head and neck cancer models. Phys. Imaging Radiat. Oncol. 2021, 19, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Misal, S.A.; Gawai, K.R. Azoreductase: A key player of xenobiotic metabolism. Bioresour. Bioprocess. 2018, 5, 17. [Google Scholar] [CrossRef]
- Sharma, R.; Rawal, R.K.; Gaba, T.; Singla, N.; Malhotra, M.; Matharoo, S.; Bhardwaj, T.R. Design, synthesis and ex vivo evaluation of colon-specific azo based prodrugs of anticancer agents. Bioorg. Med. Chem. Lett. 2013, 23, 5332–5338. [Google Scholar] [CrossRef]
- Kennedy, D.A.; Vembu, N.; Fronczek, F.R.; Devocelle, M. Synthesis of mutual azo prodrugs of an-ti-inflammatory agents and peptides facilitated by α-aminoisobutyric acid. J. Org. Chem. 2011, 76, 9641–9647. [Google Scholar] [CrossRef]
- Ryan, A.; Kaplan, E.; Laurieri, N.; Lowe, E.; Sim, E. Activation of nitrofurazone by azoreductases: Multiple ac-tivities in one enzyme. Sci. Rep. 2011, 1, 63. [Google Scholar] [CrossRef]
- Wu, K.; Knox, R.; Sun, X.Z.; Joseph, P.; Jaiswal, A.K.; Zhang, D.; Deng, P.S.K.; Chen, S. Catalytic properties of NAD(P)H:quinone oxidoreductase-2 (NQO2), a dihydronicotinamide riboside dependent oxidoreductase. Arch Biochem. Biophys. 1997, 347, 221–228. [Google Scholar] [CrossRef]
- Mikami, K.; Shirakusa, T.; Tsuruo, T. DT-diaphorase: Redox potential, steady-state, and rapid reaction studies. J. Biol. Chem. 1995, 270, 1198–1204. [Google Scholar] [CrossRef] [Green Version]
- Vasiliou, V.; Ross, D.; Nebert, D.W. Update of the NAD(P)H:quinone oxidoreductase (NQO) gene family. Hum. Genom. 2006, 2, 329. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.-S.; Ham, W.; Kim, J. Roles of NAD(P)H:quinone Oxidoreductase 1 in Diverse Diseases. Life 2021, 11, 1301. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Chen, D.; Ma, K.; Wu, X.; Hao, H.; Jiang, S. NAD(P)H:Quinone Oxidoreductase 1 (NQO1) as a Thera-peutic and Diagnostic Target in Cancer. J. Med. Chem. 2018, 61, 6983–7003. [Google Scholar] [CrossRef] [PubMed]
- Koyasu, S.; Kobayashi, M.; Goto, Y.; Hiraoka, M.; Harada, H. Regulatory mechanisms of hypoxia-inducible fac-tor 1 activity: Two decades of knowledge. Cancer Sci. 2018, 109, 560–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiyose, K.; Hanaoka, K.; Oushiki, D.; Nakamura, T.; Kajimura, M.; Suematsu, M.; Nishimatsu, H.; Yamane, T.; Terai, T.; Hirata, Y.; et al. Hypoxia-sensitive fluorescent probes for in vivo real-time fluorescence imaging of acute ischemia. J. Am. Chem. Soc. 2010, 132, 15846–15848. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yuan, Y.; Zeng, G.; Li, X.; Yang, Z.; Li, X.; Jiang, R.; Hu, W.; Sun, P.; Wang, Q.; et al. A wa-ter-soluble conjugated polymer with azobenzol side chains based on “turn-on” effect for hypoxic cell imag-ing. Polym. Chem. 2016, 7, 6890–6894. [Google Scholar] [CrossRef]
- Ma, D.; Huang, C.; Zheng, J.; Zhou, W.; Tang, J.; Chen, W.; Li, J.; Yang, R. Azoreductase-Responsive Nanoprobe for Hypoxia-Induced Mitophagy Imaging. Anal. Chem. 2019, 91, 1360–1367. [Google Scholar] [CrossRef]
- Piao, W.; Tsuda, S.; Tanaka, Y.; Maeda, S.; Liu, F.; Takahashi, S.; Kushida, Y.; Komatsu, T.; Ueno, T.; Terai, T.; et al. Development of Azo-Based Fluorescent Probes to Detect Different Levels of Hypoxia. Angew. Chem. Int. Ed. 2013, 52, 13028–13032. [Google Scholar] [CrossRef]
- Ross, D.; Siegel, D. NAD(P)H:Quinone Oxidoreductase 1 (NQO1, DT-Diaphorase), Functions and Pharmaco-genetics. Methods Enzymol. 2004, 382, 115–144. [Google Scholar] [CrossRef]
- Yang, Y.J.; Dai, M.; Reo, Y.J.; Song, C.W.; Sarkar, S.; Ahn, K.H. NAD(P)H Quinone Oxidoreductase-1 in Organ and Tumor Tissues: Distinct Activity Levels Observed with a Benzo-rosol-Based Dual-Excitation and Du-al-Emission Probe. Anal. Chem. 2021, 93, 7523–7531. [Google Scholar] [CrossRef]
- AbuKhader, M.; Heap, J.; de Matteis, C.; Kellam, B.; Doughty, S.W.; Minton, N.; Paoli, M. Binding of the anti-cancer prodrug CB1954 to the activating enzyme NQO2 revealed by the crystal structure of their complex. J. Med. Chem. 2005, 48, 7714–7719. [Google Scholar] [CrossRef]
- Workman, P. Bioreductive Mechanisms. Int. J. Radiat. Onco.l Biol. Phys. 1992, 22, 631–637. [Google Scholar] [CrossRef]
- Pandey, A.V.; Flück, C.E. NADPH P450 oxidoreductase: Structure, function, and pathology of diseases. Pharmacol. Ther. 2013, 138, 229–254. [Google Scholar] [CrossRef] [PubMed]
- Guise, C.P.; Wang, A.T.; Theil, A.; Bridewell, D.J.; Wilson, W.R.; Patterson, A.V. Identification of human reduc-tases that activate the dinitrobenzamide mustard prodrug PR-104A: A role for NADPH:cytochrome P450 oxidoreductase under hypoxia. Biochem. Pharm. 2007, 74, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.; Hoey, B.M. The one-electron reduction potential of several substrates can be related to their reduc-tion rates by cytochrome P-450 reductase. Biochim. Biophys. Acta 1993, 1161, 73–78. [Google Scholar] [CrossRef]
- Bailey, S.M.; Lewis, A.D.; Patterson, L.H.; Fisher, G.R.; Knox, R.J.; Workman, P. Involvement of NADPH: Cyto-chrome P450 reductase in the activation of indoloquinone EO9 to free radical and DNA damaging species. Biochem. Pharm. 2001, 62, 461–468. [Google Scholar] [CrossRef]
- Cenas, N.; Anusevicius, Z.; Bironaite, D.; Bachmanova, G.I.; Archakov, A.I.; Ollinger, K. The electron transfer reactions of NADPH: Cytochrome P450 reductase with nonphysiological oxidants. Arch Biochem. Biophys. 1994, 315, 400–406. [Google Scholar] [CrossRef]
- Patterson, A.v.; Saunders, M.P.; Chinje, E.C.; Talbot, D.C.; Harris, A.L.; Stratford, I.J. Overexpression of human NADPH:cytochrome c (P450) reductase confers enhanced sensitivity to both tirapazamine (SR 4233) and RSU 1069. Br. J. Cancer 1997, 76, 1338–1347. [Google Scholar] [CrossRef]
- Wang, J.; Guise, C.P.; Dachs, G.U.; Phung, Y.; Hsu, A.H.L.; Lambie, N.K.; Patterson, A.V.; Wilson, W.R. Identifica-tion of one-electron reductases that activate both the hypoxia prodrug SN30000 and diagnostic probe EF5. Biochem. Pharm. 2014, 91, 436–446. [Google Scholar] [CrossRef]
- Hunter, F.W.; Young, R.J.; Shalev, Z.; Vellanki, R.N.; Wang, J.; Gu, Y.; Joshi, N.; Sreebhavan, S.; Weinreb, I.; Goldstein, D.P.; et al. Identification of P450 Oxidoreductase as a Major Determinant of Sensitivity to Hypoxia-Activated Prodrugs. Cancer Res. 2015, 75, 4211–4223. [Google Scholar] [CrossRef] [Green Version]
- Pewklang, T.; Wet-Osot, S.; Wangngae, S.; Ngivprom, U.; Chansaenpak, K.; Duangkamol, C.; Lai, R.Y.; Noisa, P.; Sukwattanasinitt, M.; Kamkaew, A. Flavylium-Based Hypoxia-Responsive Probe for Cancer Cell Imaging. Molecules 2021, 26, 4938. [Google Scholar] [CrossRef]
- Melo, T.; Ballinger, J.R.; Rauth, A.M. Role of NADPH:cytochrome P450 reductase in the hypoxic accumulation and metabolism of BRU59-21, a technetium-99m-nitroimidazole for imaging tumor hypoxia. Biochem. Pharmacol. 2000, 60, 625–634. [Google Scholar] [CrossRef]
- Gao, Y.; Lin, Y.; Liu, T.; Chen, H.; Yang, X.; Tian, C.; Du, L.; Li, M. Bioluminescent Probe for Tumor Hypoxia De-tection via CYP450 Reductase in Living Animals. Anal. Chem. 2017, 89, 12488–12493. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, S.; Huang, J.; Cui, L.; Hu, J.; Tan, S. Novel designed azo substituted semi-cyanine fluorescent probe for cytochrome P450 reductase detection and hypoxia imaging in cancer cells. RSC Adv. 2019, 9, 21572–21577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishino, T.; Okamoto, K.; Eger, B.T.; Pai, E.F.; Nishino, T. Mammalian xanthine oxidoreductase–mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J. 2008, 275, 3278–3289. [Google Scholar] [CrossRef]
- Kostić, D.A.; Dimitrijević, D.S.; Stojanović, G.S.; Palić, I.R.; Dordević, A.S.; Ickovski, J.D. Xanthine oxidase: Isola-tion, assays of activity, and inhibition. J. Chem. 2015, 2015, 294858. [Google Scholar] [CrossRef] [Green Version]
- Hille, R.; Hall, J.; Basu, P. The Mononuclear Molybdenum Enzymes. Chem. Rev. 2014, 114, 3963–4038. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Pauff, J.M.; Hille, R. Substrate orientation and catalytic specificity in the action of xanthine oxidase: The sequential hydroxylation of hypoxanthine to uric acid. J. Biol. Chem. 2010, 285, 28044–28053. [Google Scholar] [CrossRef] [Green Version]
- Kelley, E.E.; Khoo, N.K.H.; Hundley, N.J.; Malik, U.Z.; Freeman, B.A.; Tarpey, M.M. Hydrogen Peroxide is the Major Oxidant Product of Xanthine Oxidase. Free. Radic. Biol. Med. 2010, 48, 493. [Google Scholar] [CrossRef] [Green Version]
- Battelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine oxidoreductase-derived reactive species: Physi-ological and pathological effects. Oxid. Med. Cell. Longev. 2016, 2016, 3527579. [Google Scholar] [CrossRef] [Green Version]
- Tatsumi, K.; Inoue, A.; Yoshimura, H. Mode of reactions between xanthine oxidase and aromatic nitro com-pounds. J. Pharm. 1981, 4, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Thakur, M.; Thakur, A.; Balasubramanian, K. QSAR and SAR Studies on the Reduction of Some Aromatic Ni-tro Compounds by Xanthine Oxidase. J. Chem. Inf. Model. 2006, 46, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Ueda, O.; Kitamura, S.; Ohashi, K.; Sugihara, K.; Ohta, S. Xanthine Oxidase-Catalyzed Metabolism of 2-Nitrofluorene, a Carcinogenic Air Pollutant, in Rat Skin. Drug Metab. Dispos. 2003, 31, 367–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dopp, J.M.; Philippi, N.R.; Marcus, N.J.; Olson, E.B.; Bird, C.E.; Moran, J.J.M.; Mueller, S.W.; Morgan, B.J. Xanthine Oxidase Inhibition Attenuates Endothelial Dysfunction Caused by Chronic Intermittent Hypoxia in Rats. Respiration 2011, 82, 458–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, E.D.; Goulding, K.H.; Wardman, P. Nitroimidazoles as anaerobic electron acceptors for xanthine oxi-dase. Biochem. Pharmacol. 1982, 31, 3237–3242. [Google Scholar] [CrossRef]
- Rajapakse, A.; Linder, C.; Morrison, R.D.; Sarkar, U.; Leigh, N.D.; Barnes, C.L.; Daniels, J.S.; Gates, K.S. Enzymatic conversion of 6-nitroquinoline to the fluorophore 6-aminoquinoline selectively under hypoxic conditions. Chem. Res. Toxicol. 2013, 26, 555–563. [Google Scholar] [CrossRef] [Green Version]
- Bejot, R.; Kersemans, V.; Kelly, C.; Carroll, L.; King, R.C.; Gouverneur, V. Pre-clinical evaluation of a 3-nitro-1,2,4-triazole analogue of [18F]FMISO as hypoxia-selective tracer for PET. Nucl. Med. Biol. 2010, 37, 565–575. [Google Scholar] [CrossRef]
- Seow, H.A.; Penketh, P.G.; Shyam, K.; Rockwell, S.; Sartorelli, A.C. 1,2-Bis(methylsulfonyl)-1-(2-chloroethyl)-2-[[1-(4-nitrophenyl)ethoxy]carbonyl]hydrazine: An anticancer agent targeting hypoxic cells. Proc. Natl. Acad. Sci. USA 2005, 102, 9282–9287. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Sun, G.; Fan, T.; Liu, J.; Zhang, N.; Zhao, L.; Zhong, R. Reductive Activity and Mechanism of Hypoxia- Targeted AGT Inhibitors: An Experimental and Theoretical Investigation. Int. J. Mol. Sci. 2019, 20, 6308. [Google Scholar] [CrossRef] [Green Version]
- Linder, K.E.; Chan, Y.W.; Cyr, J.E.; Malley, M.F.; Nowotnik, D.P.; Nunn, A.D. TcO(PnA.O-1-(2-nitroimidazole)) [BMS-181321], a new technetium-containing nitroimidazole complex for imaging hypoxia: Synthesis, char-acterization, and xanthine oxidase-catalyzed reduction. J. Med. Chem. 1994, 37, 9–17. [Google Scholar] [CrossRef]
- Penketh, P.G.; Shyam, K.; Baumann, R.P.; Ishiguro, K.; Patridge, E.V.; Zhu, R.; Sartorelli, A.C. A strategy for se-lective O(6)-alkylguanine-DNA alkyltransferase depletion under hypoxic conditions. Chem. Biol. Drug Des. 2012, 80, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Zhu, R.; Liu, M.C.; Luo, M.Z.; Penketh, P.G.; Baumann, R.P.; Shyam, K.; Sartorelli, A.C. 4-Nitrobenzyloxycarbonyl Derivatives of O6-Benzylguanine as Hypoxia-Activated Prodrug Inhibitors of O6-Alkylguanine-DNA Alkyl-transferase (AGT), Which Produces Resistance to Agents Targeting the O-6 Position of DNA Guanine. J. Med. Chem. 2011, 54, 7720–7728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Y.; Lai, X.; Li, J.; Yu, R.; Zhuang, Z.; Sun, G.; Cui, X.; Zhang, N.; Zhao, L.; Upadhyaya, P.; et al. NBGNU: A hypoxia-activated tripartite combi-nitrosourea prodrug overcoming AGT-mediated chemoresistance. Future Med. Chem. 2019, 11, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Elahian, F.; Sepehrizadeh, Z.; Moghimi, B.; Mirzaei, S.A. Human cytochrome b5 reductase: Structure, function, and potential applications. Crit. Rev. Biotechnol. 2014, 34, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Passon, P.G.; Hultquist, D.E. Soluble cytochrome b5 reductase from human erythrocytes. Biochim. Biophys. Acta Bioenerg. 1972, 275, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Marín, A.; de Cerain, A.L.; Hamilton, E.; Lewis, A.D.; Martinez-Peñuela, J.M.; Idoate, M.A.; Bello, J. DT-diaphorase and cytochrome B5 reductase in human lung and breast tumours. Br. J. Cancer 1997, 76, 923–929. [Google Scholar] [CrossRef] [Green Version]
- Holtz, K.M.; Rockwell, S.; Tomasz, M.; Sartorelli, A.C. Nuclear overexpression of NADH:cytochrome b5 re-ductase activity increases the cytotoxicity of mitomycin C (MC) and the total number of MC-DNA adducts in Chinese hamster ovary cells. J. Biol. Chem. 2003, 278, 5029–5034. [Google Scholar] [CrossRef] [Green Version]
- Hodnick, W.F.; Sartorelli, A.C. Reductive Activation of Mitomycin C by NADH:Cytochrome b5 Reductase. Cancer Res. 1993, 53, 4907–4912. [Google Scholar]
- Guise, C.P.; Abbattista, M.R.; Tipparaju, S.R.; Lambie, N.K.; Su, J.; Li, D.; Wilson, W.R.; Dachs, G.U.; Patterson, A.V. Diflavin oxidoreductases activate the bioreductive prodrug PR-104A under hypoxia. Mol. Pharm. 2012, 81, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Vikram, D.S.; Zweier, J.L.; Kuppusamy, P. Methods for noninvasive imaging of tissue hypoxia. Antioxid. Redox. Signal. 2007, 9, 1745–1756. [Google Scholar] [CrossRef]
- Daimiel, I. Insights into Hypoxia: Non-invasive Assessment through Imaging Modalities and Its Application in Breast Cancer. J. Breast Cancer 2019, 22, 155. [Google Scholar] [CrossRef]
- Carlin, S.; Humm, J.L. PET of Hypoxia: Current and Future Perspectives. J. Nucl. Med. 2012, 53, 1171–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, I.N.; Manavaki, R.; Blower, P.J.; West, C.; Williams, K.J.; Harris, A.L.; Domarkas, J.; Lord, S.; Baldry, C.; Gilbert, F.J. Imaging tumour hypoxia with positron emission tomography. Br. J. Cancer 2014, 112, 238–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Foehrenbacher, A.; Su, J.; Patel, R.; Hay, M.P.; Hicks, K.O.; Wilson, W.R. The 2-nitroimidazole EF5 is a biomarker for oxidoreductases that activate the bioreductive prodrug CEN-209 under hypoxia. Clin. Cancer Res. 2012, 18, 1684–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmet, C.M.; Lafosse, A.; Vériter, S.; Porporato, P.E.; Sonveaux, P.; Dufrane, D.; Levêque, P.; Gallez, B. Applica-tion of Electron Paramagnetic Resonance (EPR) Oximetry to Monitor Oxygen in Wounds in Diabetic Models. PLoS ONE 2015, 10, e0144914. [Google Scholar] [CrossRef]
- Sandhu, S.; Kydd, L.; Jaworski, J. Luminescent Probe Based Techniques for Hypoxia Imaging. J. Nanomed. Res. 2017, 6, 00160. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Park, S.; Yoon, J.; Shin, I. Recent progress in the development of near-infrared fluorescent probes for bioimaging applications. Chem. Soc. Rev. 2013, 43, 16–29. [Google Scholar] [CrossRef]
- Liu, J.N.; Bu, W.; Shi, J. Chemical Design and Synthesis of Functionalized Probes for Imaging and Treating Tumor Hypoxia. Chem. Rev. 2017, 117, 6160–6224. [Google Scholar] [CrossRef]
- Li, Z.; He, X.; Wang, Z.; Yang, R.; Shi, W.; Ma, H. in vivo imaging and detection of nitroreductase in zebrafish by a new near-infrared fluorescence off–on probe. Biosens. Bioelectron. 2015, 63, 112–116. [Google Scholar] [CrossRef]
- Li, X.F.; Carlin, S.; Urano, M.; Russell, J.; Ling, C.C.; O’Donoghue, J.A. Visualization of hypoxia in microscopic tumors by immunofluorescent microscopy. Cancer Res. 2007, 67, 7646–7653. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Study Type | Target | Trial | Reference |
|---|---|---|---|---|
| Tirapazamine | Phase II | Squamous cell carcinoma of the head and neck | NCT00094081 | [39] |
| NCT00002774 | [40] | |||
| Phase III | NCT00174837 | [41] | ||
| TH-302 (evofosfamide) | Phase III | Pancreatic cancer | NCT01746979 | [42] |
| Phase III | Soft tissue carcinoma | NCT01440088 | [42] | |
| Phase III | Esophageal carcinoma | NCT02598687 | [43] | |
| Nimorazole (radiosensitizer) | Phase II | Squamous cell carcinoma of the head and neck | DAHANCA | [44,45] |
| Phase III | NCT01950689 | [46] | ||
| TH-4000 (tarloxotinib) | Phase II | Non-small cell lung cancer | NCT02454842 | - |
| Phase II | Squamous cell carcinoma of the head and neck | NCT02449681 | [47] | |
| PR-104 | Phase II | Small cell lung cancer | NCT00544674 | [48] |
| Phase II | Non-small cell lung cancer | NCT00862134 | ||
| AQ4N | Phase I | Esophageal carcinoma | NCT00394628 | [49] |
| Phase II | Glioblastoma | |||
| EO9 (apaziquone) | Phase III | Bladder cancer | NCT00598806 | - |
| NCT01475266 | ||||
| NCT02563561 | ||||
| Porfiromycin | Phase III | Squamous cell carcinoma of the head and neck | NCT00002507 | [50] |
| Specific Oxidoreductases | Detection Method | Cell Line | Enzyme Changes (↑ Increase Under Hypoxia) | Comment | Ref. |
|---|---|---|---|---|---|
| Nitroreductases (NTR) | NTR ELISA KIT | HepG-2 | 4 U/L (normoxia and hypoxia) | No significant change in NTR concentrations between cancer cells cultured at different oxygen concentrations | [80] |
| A549 | 3 U/L (normoxia and hypoxia) | ||||
| SKOV-3 | 2.5 U/L (normoxia and hypoxia) | ||||
| HepG-2 (in vivo) | 1.8 U/g (6 mm tumor diameter) | ||||
| 2.0 U/g (14 mm tumor diameter) | |||||
| NTR ELISA KIT | A2058 | 180 pg/mL (normoxia) | Hypoxia led to the enhancement of NTR expression | [79] | |
| 300 pg/mL (hypoxia) | |||||
| NTR ELISA KIT | HeLa | ~ 2× at 10% O2 | Hypoxia led to clear enhancement of NTR expression | [81] | |
| ~ 5× at 5% O2 | |||||
| ~ 10× at 1% O2 | |||||
| compared to 20% O2 | |||||
| Western blot | A549 (in vivo) | No quantitative analysis (7 mm tumor diameter) | NTR expression only in tumor tissue | [82] | |
| Western blot detection of carbonic anhydrase 9 (CA9) | U87 | ↑ ~ 2× at 2% O2 | Indirectly assessing NTR activity by determination of CAIX | [83] | |
| U251 | ↑ ~ 4× at 2% O2 | ||||
| GBM2 | ↑ ~ 4× at 2% O2 | ||||
| GBM39 | ↑ ~ 8× at 2% O2 | ||||
| NAD(P)H quinone dehydrogenase 1 (NQO1) | Western blot | A549 H460 | No quantitative analysis | Confirmed NQO1 expression, but not in normal cells (IMR90, HUVEC) No tests in hypoxia | [84] |
| A549 (in vivo) | Confirmed NQO1 expression in tumor lysates, but not in other organs | ||||
| Western blot | H460, HT-29, DU145, A549, FaDu, 9L, 9L/2B11, Colo-205, PC3, MCF-7, MB231, T47D, U251, BxPC-3, KM12, H522 | No quantitative analysis | NQO1 levels were similar in cells grown under hypoxia (0.2% O2) and normoxia | [85] | |
| Northern blot | HT29 | ↑ ~ 4× at 1 ppm O2 | Hypoxia caused a marked increase in NQO1 level | [86] | |
| Cytochrome p450 reductase (POR) | Western blot | UT-SCC-14 | No quantitative analysis | Low expression of hypoxia and normoxia | [55] |
| A549 | No quantitative analysis | No change in expression of hypoxia vs. normoxia | |||
| Xanthine oxidoreductase (XOR) | Western blot Northern blot (+XOR activity) | BEAS-2B | ↑ ~ 3× XOR activity | No changes in protein and mRNA expression under hypoxia | [87] |
| PCR Western blot (+XOR activity determined by HPLC) | Rat lungs (in vivo) | ↑ ~ 2× (mRNA and XOR activity) | [88] | ||
| Western blot (+XOR activity) | RPMEC (endothelial cells) | ↑ ~ 2.3× XOR activity | 50-fold increase in phosphorylation, but without changing the OXR expression under hypoxia | [89] | |
| Western blot (+XOR activity) | BEAC bovine aortic endothelial cell | ↑ ~ 2× XOR activity | No changes in XOR mRNA expression under hypoxia | [90] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janczy-Cempa, E.; Mazuryk, O.; Kania, A.; Brindell, M. Significance of Specific Oxidoreductases in the Design of Hypoxia-Activated Prodrugs and Fluorescent Turn off–on Probes for Hypoxia Imaging. Cancers 2022, 14, 2686. https://doi.org/10.3390/cancers14112686
Janczy-Cempa E, Mazuryk O, Kania A, Brindell M. Significance of Specific Oxidoreductases in the Design of Hypoxia-Activated Prodrugs and Fluorescent Turn off–on Probes for Hypoxia Imaging. Cancers. 2022; 14(11):2686. https://doi.org/10.3390/cancers14112686
Chicago/Turabian StyleJanczy-Cempa, Ewelina, Olga Mazuryk, Agnieszka Kania, and Małgorzata Brindell. 2022. "Significance of Specific Oxidoreductases in the Design of Hypoxia-Activated Prodrugs and Fluorescent Turn off–on Probes for Hypoxia Imaging" Cancers 14, no. 11: 2686. https://doi.org/10.3390/cancers14112686