1. Introduction
Overcoming acquired resistance is one of the greatest challenges in the management of cancer patients. Although patients often have an initial positive response to cancer drugs, many will eventually stop responding to treatment. Through the years, different views on how tumor cells acquire drug resistance have been adopted. Since tumorigenesis is largely driven by genetic mutations, drug resistance was initially proposed as being driven by the acquisition of mutations that allow tumor cells to overcome the drugs’ actions [
1]. This is indeed true in many cases, for example in the acquisition/enrichment of KRAS mutations in colorectal cancer during treatment with cetuximab [
2]; or with de novo mutations occurring in EGFR during treatment with tyrosine kinase inhibitors in lung cancer [
3]. In recent years, however, several examples have been described of drug resistance mediated through non-genetic mechanisms [
4,
5,
6,
7]. In general, non-genetic drug acquired resistance involves major transcriptional changes driven by epigenetic reprogramming in a very small fraction of tumor cells [
8,
9]. A more recent emerging concept with the potential to change the drug resistance framework is drug tolerance [
10]. Originally observed during the treatment of bacteria with antibiotics [
11], drug tolerance refers to a transitory state in which a small percentage of cells can survive high concentrations of a drug by entering into a so-called slow-cycling phase; such cells are often referred to as drug-tolerant persister (DTP) cells [
12,
13].
The concepts of drug tolerance and DTP have intriguing implications for biomarker discovery since they both tolerate (survive) the drug treatment, yet also reflect the initial drug response in a pharmacodynamic sense. Thus, DTPs represent both positive (initial response) and negative (transition toward full resistance) therapeutic aspects, i.e., the Yin and Yang of response to drug treatment.
Eribulin is a fully synthetic macrocyclic ketone analog of the structurally complex marine sponge natural product halichondrin B [
14]. As a microtubule targeting agent (MTA), eribulin (in its clinical formulation of eribulin mesylate) has gained regulatory approval in many countries worldwide for the treatment of certain patients with advanced breast cancer, liposarcoma, or soft tissue sarcoma (according to country-specific approval indications). Eribulin acts by binding to terminal β-tubulin subunits at the growing plus (+) ends of microtubules, thus inhibiting the growth phase of microtubule dynamics while having no direct effect on microtubule shortening, a mode of action distinct from other MTAs which likely contributes to its clinical efficacy and tolerability profile [
15,
16,
17,
18,
19]. Moreover, preclinical studies indicate that eribulin also exerts effects unrelated to its anti-mitotic effects, including vascular remodeling, reversal of the epithelial-to-mesenchymal (EMT) transition, inhibition of cancer cell migration/invasion and alterations in cellular phenotype, with growing clinical evidence supporting the concept that these non-cytotoxic effects likely contribute to eribulin’s clinical efficacy [
20,
21]. However, the complex interplay of both cytotoxic and non-cytotoxic effects may be different in different patients, therefore the need for the development of non-invasive circulating biomarkers to both monitor the response to therapy as well as identify patients who either do not respond to eribulin or are headed toward the development of primary resistance.
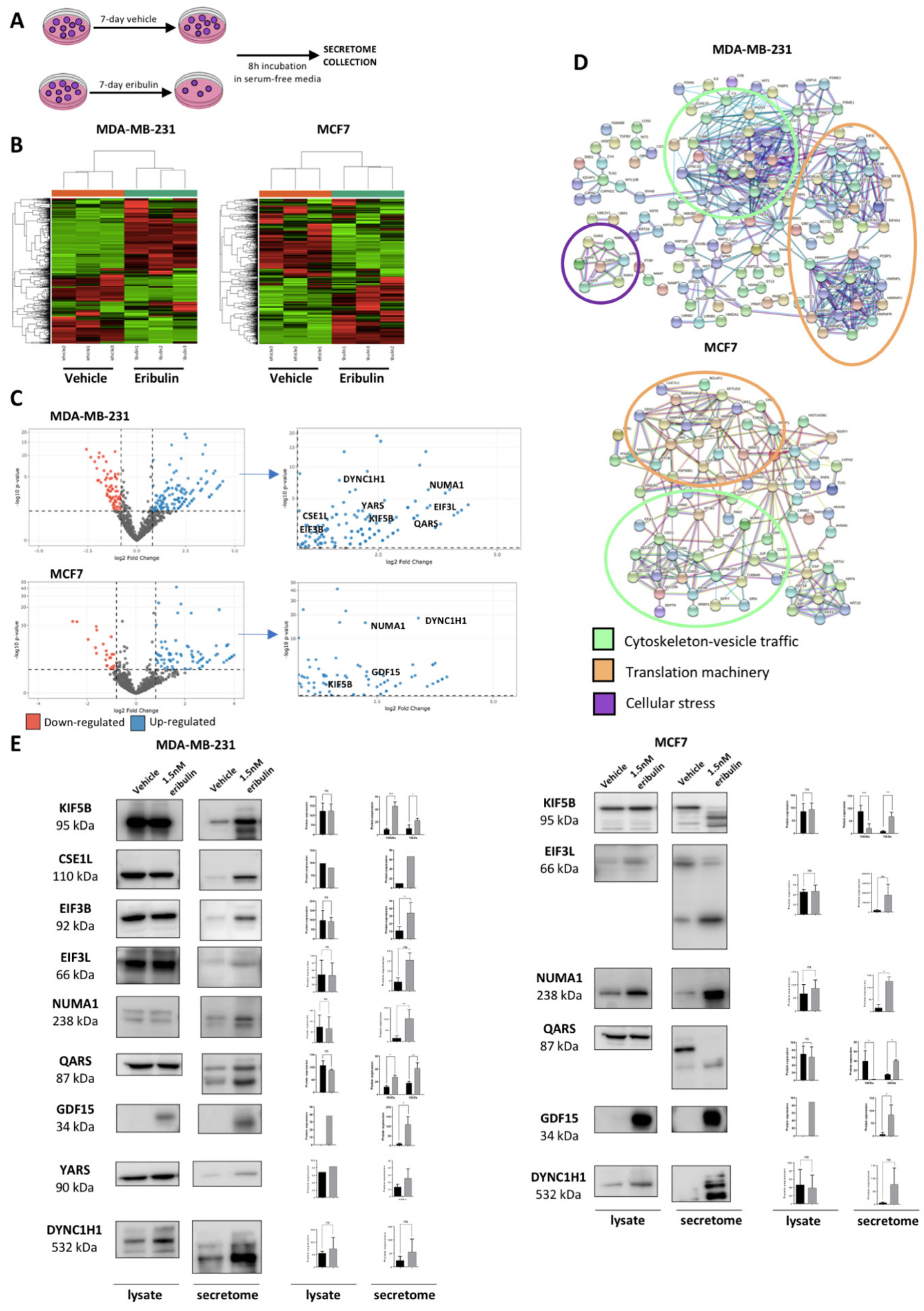
Since eribulin disrupts the cytoskeleton, and hence intracellular vesicle trafficking [
22], we hypothesized that eribulin treatment would alter the secretome of breast cancer cells. Thus, in this study, we sought to employ quantitative proteomics to characterize the secretome of breast cancer cells following eribulin treatment. Here, we show that eribulin induces a population of metastable DTP cells which are characterized by a specific secretome. Among the many proteins significantly over-secreted upon eribulin treatment, growth differentiation factor 15 (GDF15; originally called macrophage inhibitory cytokine-1 or MIC-1), seemed particularly promising and was selected for further validation. GDF15 is a cell stress cytokine belonging to the TGF-β superfamily. Different cell stressors and pathological conditions trigger increased serum levels of GDF15 [
23]. Elevated levels of circulating GDF15 cause anorexia and weight loss through binding to its receptor GDNF family receptor alpha-like (GFRAL), which in turn enables signaling through the proto-oncogene tyrosine kinase RET [
24,
25,
26]. In cancer, GDF15 mediates cachexia, causing extreme weight loss and muscle wastage in late-stage cancer patients [
27,
28,
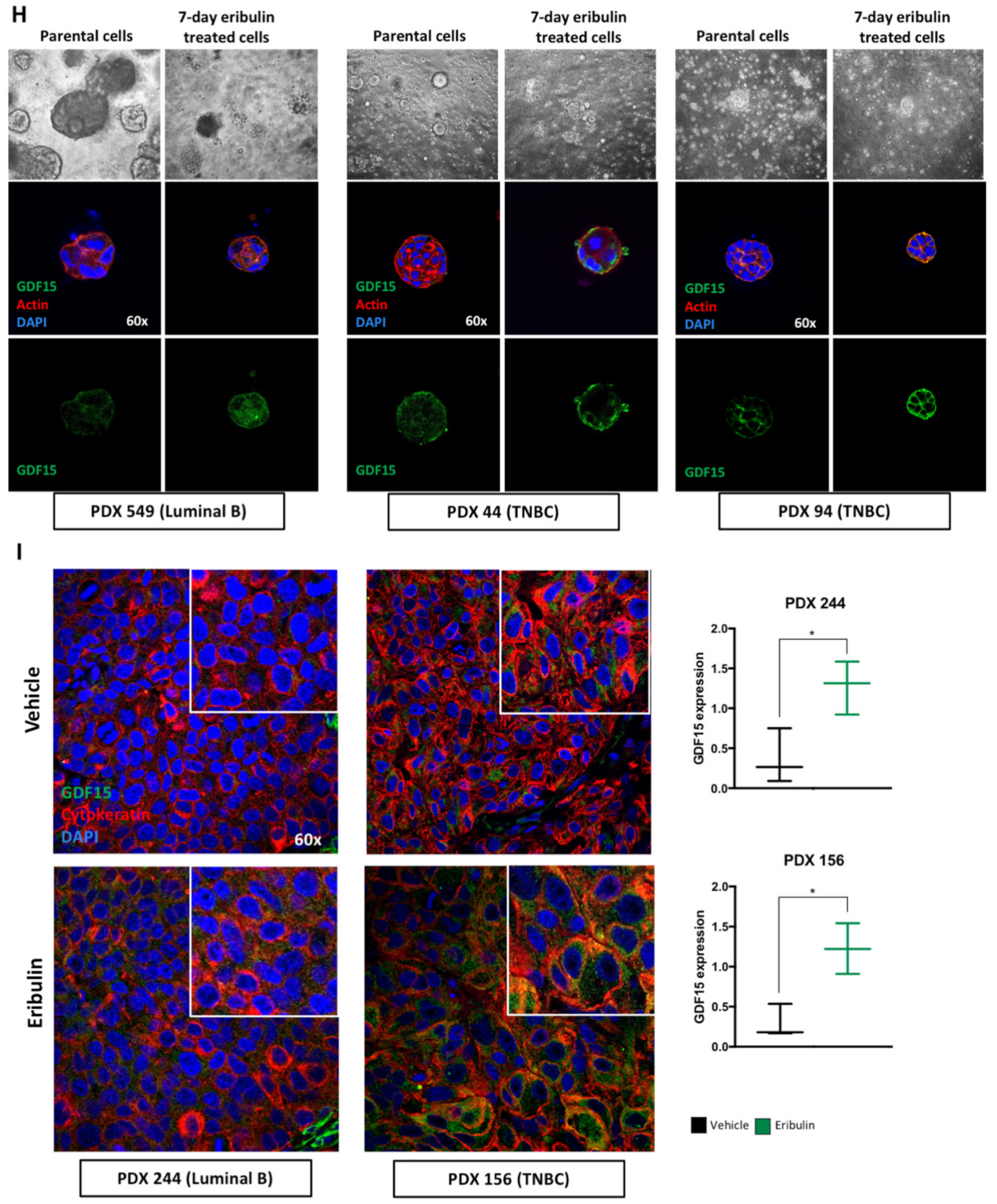
29]. Intriguingly, in our studies, GDF15 was only detected during the metastable DTP state following eribulin treatment, yet disappears after the acquisition of full drug resistance following prolonged eribulin treatment. Results obtained in cell lines grown in two-dimensional culture were further confirmed using breast cancer three-dimensional organoid derived from both established cell lines and PDX models. Finally, we tested and confirmed the hypothesis that GDF15 itself plays a role in maintaining the DTP state by conducting GDF15 loss of function experiments, results of which led to a proof-of-concept study combining eribulin with an anti-GDF15 antibody to specifically inhibit the generation of DTP cells in both TNBC and luminal breast cancer cell lines.
2. Material and Methods
2.1. Cell Culture and Breast Cancer PDX Tumor Cells Isolation
Breast cancer cell lines MDA-MB-231, MCF7, HS578T, BT549, HCC1937 and MCF10A were purchased from the American Type Culture Collection (ATCC). The cells were cultured in 5% CO2 and 95% humidified atmosphere air at 37 °C. The cell lines were authenticated by short tandem repeat profiling (IdentiCell, Aarthus University Hospital). All cell lines were routinely tested by PCR for Mycoplasma and passaged until passage number 15. MDA-MB-231, MCF7 and HS578T cells were maintained in DMEM: Nutrient Mixture F12 (DMEM/F12; Invitrogen, Waltham, MA, USA) supplemented with 10% FBS (Invitrogen), 1% Pen/Strep (Thermo Fisher Scientific, Waltham, MA, USA) and 2 mmol/L l-Glutamine (Invitrogen). BT549 and HCC1937 cells were maintained in RPMI (Invitrogen) supplemented with 10% FBS (Invitrogen), 1% Pen/Strep (Thermo Fisher Scientific) and 2 mmol/L l-Glutamine (Invitrogen). MCF10A cells were maintained in DMEM: Nutrient Mixture F12 (DMEM/F12; Invitrogen) supplemented with 10% FBS (Invitrogen), 1% Pen/Strep (Thermo Fisher Scientific), 2 mmol/L l-Glutamine (Invitrogen), 20 ng/mL human EGF (#AF-100-15, Peprotech, Cranbury, NJ, USA), 10 μg/mL insulin (#I9278, Sigma-Aldrich, St. Louis, MO, USA) and 500 ng/mL hydrocortisone (#H0888, Sigma-Aldrich). Breast cancer PDXs were kindly provided by the Experimental Therapeutics Group headed by Violeta Serra at the VHIO institute. Breast cancer samples were taken from resected tumors and underwent multiple washes with PBS before minced into small pieces using a scalpel and incubated with human collagenase (3 mg/mL, Sigma) and hyaluronidase (1 mg/mL, Sigma) for 1 h at 37 °C with shacking at 200–300 rpm. After incubation, the mixture was resuspended in DMEM-HEPES 1% and centrifuged at 1500 rpm for 5 min to remove debris and residual collagenase and hyaluronidase. After the wash, the cell pellet was resuspended in DMEM-HEPES 1% and filtered through a 40 μm cell strainer to remove large undigested fragments. The cell suspension was centrifuged at 200× g for 3 min. The cell pellet was resuspended in Trypsin-EDTA (Sigma) and gently pipetted up and down with a p1000 pipette for 3 min at RT. The effect of the Trypsin was blocked adding cold Hank’ Balanced Salt Solution (Biowest, Riverside, MO, USA) supplemented with 2% FBS and 2% HEPES and the cell suspension was centrifuged at 1500 rpm for 5 min. After removal of the supernatant, pre-warmed Dispase (5 mg/mL; Sigma) and DNase I (1 mg/mL; Sigma) were added. The samples were pipetted for 3 min with a p1000 pipette for further dissociation of cell clumps. After washing in cold HF-2% HEPES 2% FBS, cell pellets were resuspended in RBC lysis buffer (eBioscience, San Diego, CA, USA) and incubated for 3 min at RT to lysis erythrocytes. After a final washing in cold HF-2% HEPES 2% FBS, cells were resuspended in DMEM/F12 medium supplemented with 10% FBS and counted. The isolated breast cancer PDX cells were resuspended at the concentration of 50,000 cells/drop of Matrigel matrix and plated. Detailed protocol for breast cancer organoids is provided in the specific session. Cellular images were taken at different enlargements with the microscope Leica MCF170 HD.
2.2. Three-Dimensional Cell Culture Organoids
Cell suspension (from breast cancer cell lines or PDXs derived cells) was resuspended in serum-free DMEM/F12 media to wash the cells from FBS that could interfere in organoid formation and centrifuged at 1500 rpm for 5 min. Cells were resuspended in serum-free DMEM/F12 media, counted and divided into 50,000 cells/Eppendorf for each condition. Cells were resuspended in a solution 60% BD-Matrigel matrix (Cultek S.L.U.) and 40% growth medium-organoid medium consisting of DMEM/F12, B27 Supplement (1:50; Gibco), and insulin (250 ng/mL; Life Technologies)- and plated as 50 μL droplets in a 24-well plate. The Matrigel drops were incubated for 30 min at 37 °C and then organoid media was carefully added to each well. Media with fresh growth factors was changed every 3 days.
2.3. Drug Treatment (Eribulin, Vinorelbine and Paclitaxel)—CellTiter, Cell Counting and Crystal Violet Staining
Eribulin was provided by Eisai Inc. Vinorelbine and Paclitaxel was provided by the Pharmaceutical Centre of Vall d’Hebron Hospital. IC50 values (the concentration required to kill and/or inhibit growth of cells by 50% as compared with untreated control wells) of these drugs were estimated from concentration–response curves by using CellTiter colorimetric assay (CellTiter-Blue Cell Viability Assay; Promega) analysis. All the cell lines were plated in 96-well plates (5000 cells/well in 100 μL media). After overnight incubation, Eribulin (0.1, 0.5, 1, 2 nmol/L), Vinorelbine (5, 10, 50, 100 nmol/L), and Paclitaxel (1, 2, 4, 10 nmol/L) were added in six replicates to each population. Cell viability was measured after 7 days using the CellTiter assay. A calibration curve was prepared using the data obtained from wells that contained a known number of cells. Cell proliferation was assessed by Trypan Blue (Thermo Fisher Scientific, Waltham, MA, USA) staining and cell counting following treatment with the indicated concentrations of each drug. Crystal violet staining was performed to collect representative images of drug treatment effect on cell viability. Specifically, vehicle and drug-treated cells were washed twice with cold PBS and fixed for 10 min with ice-cold 100% methanol. The methanol was aspired and cells incubated with 0.5% crystal violet solution in 25% methanol for 20 min. The cells were washed several times and left to dry overnight. Crystal violet staining was quantified dissolving the dye in 10% acid acetic and measuring the absorbance at 595 nM using a luminometer. All the drug treatments were 7 days with retreatment every 72 h due to the half-lives of the drugs.
2.4. Immunofluorescence and Confocal Microscopy
Cells were seeded on Collagen-coated (Collagen I-Rat Tail; Life Technologies, Carlsbad, CA, USA) glass coverslips. After growing them for the appropriate time, cells were treated according to eribulin treatment scheme and cells were fixed with paraformaldehyde 4% for 30 min and permeabilized with 0.5% Triton-X10 for 15 min. For Tubulin staining, cells were fixed in cold methanol for 10 min on dry ice. Unspecific binding sites were blocked by incubating with 3% BSA (Sigma-Aldrich) for 1 h. Phalloidin (Phalloidin–Tetramethylrhodamine B isothiocyanate; Sigma-Aldrich) was used to stain actin cytoskeleton of cells (1:1000, incubation 15 min at room temperature). Primary antibodies were prepared in 3% BSA and incubated O/N at 4 °C: mouse anti-tubulin (1:500, Sigma-Aldrich), rabbit anti-GDF15 (1:200, ThermoFisher). Primary antibody incubations were followed by PBS washes and incubation for 1 h at room temperature with the appropriate secondary antibody (goat anti-mouse and goat anti-rabbit) conjugated to Alexa Fluor 594 and Alexa Fluor 488 (Invitrogen). Nuclei were stained with Hoechst 33342 (5 μg/mL; Sigma-Aldrich). The coverslips were mounted in slides using Prolong Diamond Antifade Mountant solution (Invitrogen).
Immunostaining of 3D-organoid cells was performed on glass coverslips where cells were grown in Matrigel matrix. Matrigel matrix was disaggregated with incubation for 20 min in ice with Corning Cell Recovery Solution (Cultek). After this incubation, the previously described protocol was followed using 1% of Triton-X10 during the permeabilization incubation.
Immunostaining breast cancer PDX tumor samples was performed on paraffin-embedded tissues. Tissue blocks were sectioned, mounted on microscope slides, and heated at 56 °C O/N. Paraffin was removed with xylene, and tissues were serially rehydrated through descending ethanol concentrations to water. Sections were stained with hematoxylin and eosin (H&E) to assess cellular morphology. For immunofluorescence, antigen retrieval was performed by boiling the samples in EDTA (Calbiochem, San Diego, CA, USA) buffer 9941 mmol/L (pH 8), using a microwave oven. Slides were then washed twice in PBS and once in PBS-1% Tween-20 (Sigma-Aldrich) for 15 min. Unspecific binding sites, for both cells and tissues, were blocked by incubating with 3% BSA (Sigma-Aldrich) for 1 h. Primary antibodies were prepared in 3% BSA and incubated O/N at 4 °C: rabbit anti-GDF15 was used at 1:200 for tissues, and mouse anti-Cytokeratin (clones AE1/AE3; Dako GmbH, Glostrup, Denmark) was used at 1:100 for tissue samples. Primary antibody incubations were followed by PBS washes and incubation for 1 h at room temperature with the appropriate secondary antibody (goat anti-mouse and goat anti-rabbit) conjugated to Alexa Fluor 594 and Alexa Fluor 488 (Invitrogen). Nuclei were stained with Hoechst 33342 (5 mg/mL; Sigma Aldrich).
A confocal microscope (Nikon Eclipse Ti) was used to visualize fluorescence and acquire images from five representative fields of each sample. GDF15 expression was measured in confocal images using Image J software.
2.5. Western Blotting
Whole-cell lysates were prepared from cell lines or 3D-organoid cells with RIPA lysis buffer supplemented with phosphatase, protease, and kinase inhibitors (all from Sigma–Aldrich). To collect cell lysates from 3D-organoid cells, the Matrigel was disaggregated as previously described (1 h of incubation). The protein concentration of the resulting supernatants was assessed using Pierce BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA), and ten to sixty micrograms of protein lysates were resolved on specific percentage of gel in agreement with the molecular weight of the proteins under study and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Burlington, MA, USA). Following transference, assessment of protein loading was visualized using Ponceau Solution (Sigma–Aldrich, St. Louis, Missouri, USA), and membranes were then blocked in 5% non-fat milk in TBST. Primary antibodies were incubated overnight at 4 °C: mouse anti-p21Cip1 (1:1000, ThermoFisher Scientific), mouse anti-actin (1:10,000, Sigma–Aldrich), rabbit anti-EIF3L (1:1000, Proteintech), rabbit anti-EIF3B (1:1000, Proteintech), rabbit anti-KIF5B (1:1000, Proteintech), rabbit anti-GDF15 (1:1000, Proteintech), rabbit anti-GFRAL (1:1000, ThermoFisher), rabbit anti-YARS (1:1000, Life Technologies), rabbit anti-QARS (1:1000, AB Clonal), rabbit anti-DYNC1H1 (1:1000, Proteintech), rabbit anti-NUMA1 (1:1000, Novus Biologicals), rabbit anti-CSE1L (1:1000, Proteintech). Membranes were then incubated with either a mouse or rabbit horseradish peroxidase (HRP)-conjugated secondary antibody at a 1:1000 (VWR) and developed using a chemiluminescent detection reagent (ProSignal Dura, Genesee Scientific, San Diego, CA, USA). All images were captured using the BioRad ChemiDoc Imaging system and analyzed using Image J (Bethesda, MD, USA).
2.6. Cell Proliferation Assays: Cell Cycle and PKH26 Staining
Cell proliferation was analyzed by flow-cytometry with cell-cycle analysis and PKH26 staining analysis. For cell-cycle analysis cells were treated with either vehicle or 1.5 nM eribulin for 7 days. The cells were then fixed in 70% ethanol, incubated with 10 μg/mL RNase A (Sigma-Aldrich) at room temperature, and stained with 2 μg/mL DAPI (Hoechst 33342, Sigma-Aldrich, St. Louis, MO, USA). The DNA content of the cells (100,000 events per experimental group) was quantified using a BD-Celesta cytofluorimeter (Beckton Dickinson) and analyzed using cell-cycle software of FlowJo 10.2 version. For PKH26 staining, the PKH26 Fluorescent Cell Linker Kits (Sigma Aldrich, St. Louis, MO, USA) were used following the manufacturer’s protocol. The PKH26 staining was quantified at times 0 h, 72 h and 7 days of 1.5 nM eribulin treatment by flow cytometry (BD-Celesta cytofluorimeter; Beckton Dickinson, Franklin Lakes, NJ, USA) and collecting pictures with epifluorescence microscope (Nikon Eclipse Ti, Tokyo, Japan).
2.7. Secretome Collection
Two-dimensional and three-dimensional secretomes were prepared using a modified protocol of the previously described protocol developed in our laboratory [
30].
Two-dimensional secretomes. Briefly, 4 × 106 cells in exponential phase were seeded in 150 cc tissue culture plates and allowed to grow for 48 h. After that, media was aspirated, and cells were washed 5 times, 2 times with PBS and the last 3 with serum-free media. Then, cells were maintained for 8 h in the presence of serum-free media before collecting the conditioned media (secretome).
Three-dimensional secretome. Three-dimensional organoids were set up as explained in the 3D cell culture organoids section. For the secretome collection, cells were collected after 8 h of incubation in serum-free media. First Matrigel matrix was disaggregated with incubation for 1 h in ice with Corning Cell Recovery Solution (Cultek). Then 3D-organoids were plated as 50 μL droplets in a 24-well plate using a solution of: Collagen I − Rat Tail (Life Technologies) + serum-free media + NaOH for titration. The 3D-organoids drops were incubated for 30 min at 37 °C and then serum-free media was carefully added to each well.
The conditioned media of both 2D and 3D cell cultures was spun down at 200 g for 5 min, and the supernatants were collected and filtered through a Millex-GP 0.22 μm pore syringe-driven filter (Millipore). Then, secretomes were concentrated using a 10,000 MWCO Millipore Amicon Ultra (Millipore). Protein concentration was determined with a Pierce BCA protein assay kit (Thermo Scientific).
Concentrated secretome samples (15 μg of total protein) were taken to 40 μL of 6 M Urea, 50 mM ammonium bicarbonate for tryptic digestion. Samples were first reduced with DTT to a final concentration of 10 mM, for 1 h at RT, and then alkylated with 20 mM of iodoacetamide for 30 min at RT in the dark. Carbamidomethylation reaction was quenched by addition of N-acetyl-L-cysteine to final concentration of 35 mM followed by incubation for 15 min at RT in the dark. Samples were diluted with 50 mM ammonium bicarbonate to a final concentration of 1M Urea, modified porcine trypsin (Promega, Madison, WI, USA) was added in a ratio of 1:10 (w/w), and the mixture was incubated overnight at 37 °C. The reaction was stopped with formic acid (FA) at a final concentration of 0.5%. The digests were then purified using SCX micro columns (PolyLC, Columbia, MD, USA). Purified samples were evaporated to dryness, dissolved in 5% acetonitrile-0.1% formic acid, and kept frozen at −20 °C until analyzed.
2.8. Liquid Chromatography–Mass Spectrometry Analysis (LC–MS) and Protein Identification
Tryptic digests were analyzed using a linear ion trap Velos-Orbitrap mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). Instrument control was performed using Xcalibur software package, version 2.2.0 (Thermo Fisher Scientific, Bremen, Germany). Peptide mixtures were fractionated by online nanoflow liquid chromatography using an EASY-nLC 1000 system (Proxeon Biosystems, Thermo Fisher Scientific, Waltham, MA, USA) with a two-linear-column system. Samples were first loaded onto a trapping guard column (Acclaim PepMap 100 nanoviper, 2 cm long, ID 75 μm and packed with C18, 3 μm particle size from Thermo Fisher Scientific) at 4 μL/min. Then, samples were separated on the analytical column (Dr Maisch, 25 cm long, ID 75 μm, packed with Reprosil Pur C18-AQ, 3 μm particle size). Elution was performed using 0.1% formic acid in water (mobile phase A) and acetonitrile with 0.1% formic acid (mobile phase B), with a linear gradient from 0 to 35% of mobile phase B for 120 min at a flow rate of 300 nL/min. Ions were generated applying a voltage of 1.9 kV to a stainless-steel nano-bore emitter (Proxeon, Thermo Fisher Scientific), coupled to end of the analytical column, on a Proxeon nano-spray flex ion source.
The LTQ Orbitrap Velos mass spectrometer was operated in data-dependent mode. A scan cycle was initiated with a full-scan MS spectrum (from m/z 300 to 1600) acquired in the Orbitrap with a resolution of 30,000. The 20 most abundant ions were selected for collision-induced dissociation fragmentation in the linear ion trap when their intensity exceeded a minimum threshold of 1000 counts, excluding singly charged ions. Accumulation of ions for both MS and MS/MS scans was performed in the linear ion trap, and the AGC target values were set to 1 × 106 ions for survey MS and 5000 ions for MS/MS experiments. The maximum ion accumulation time was 500 and 200 ms in the MS and MS/MS modes, respectively. The normalized collision energy was set to 35%, and one microscan was acquired per spectrum. Ions subjected to MS/MS with a relative mass window of 10 ppm were excluded from further sequencing for 20 s. For all precursor masses, a window of 20 ppm and isolation width of 2 Da were defined. Orbitrap measurements were performed enabling the lock mass option (m/z 445.120024) for survey scans to improve mass accuracy.
2.9. Protein Identification and Quantitative Differential Analysis
LC–MS/MS data were analyzed using the Proteome Discoverer v. 2.1 software (Thermo Fisher Scientific, Waltham, MA, USA). Proteins were identified using Mascot v. 2.5 (Matrix Science, London, UK) to search the SwissProt database (2018_11, taxonomy restricted to human proteins, 20,413 sequences). MS/MS spectra were searched with a precursor mass tolerance of 10 ppm, fragment tolerance of 0.7 Da, trypsin specificity with a maximum of 2 missed cleavages, cysteine carbamidomethylation set as fixed modification and methionine oxidation as variable modification.
Files generated from Mascot (DAT files) were loaded into Scaffold (version 3.00.07; Proteome software, Inc., Portland, OR, USA), resulting in a no redundant list of identified proteins per sample. Peptide identification was given as valid as long as a PeptideProphet probability greater than 95% was determined. Those proteins whose identification could be established with a probability higher than 95% and contained at least two identified spectra were accepted. Using these filters, a false protein discovery rate (FDR) below 1.0%, as estimated by a database search, was achieved. The generated “scaffold” files containing all the “spectral counts” (SpC) for each sample and their replicates were exported to the POMAcounts 1.1.0 software for normalization and statistical analysis (see statistical analysis section).
2.10. siRNA Experiment
siRNA sequences directed against GDF15 and GFRAL were purchased from Dharmacon (Horizon Discovery) and used to knockdown GDF15 and GFRAL in MCF7 and MDA-MB-231 cell lines. Briefly, cells were seeded into 100 cc tissue culture plates at a density of 1.5 × 106 cells/plate and the following day were treated for 72 h with 1.5 nM eribulin. After treatment, plasmid carrying, respectively, siRNA GDF15 or siRNA GFRAL and siRNA non-targeting (250 ng/plate) was transfected into the cells using RNAiMAX lipofectamine (Thermo Fisher). The media was changed after 24 h of transfection and the cells were plated according to the experimental plan to perform 7 days of 1.5 nM eribulin treatment. siGDF15 or siGFRAL cells were compared to the cells transfected with non-target siRNA plasmid.
- -
ON-TARGETplus Human GDF15, SMARTpool:
UGGUUUACAUGUCGACUAA
UGGUUUACAUGUUGUGUGA
UGGUUUACAUGUUUUCUGA
UGGUUUACAUGUUUUCCUA
- -
ON-TARGETplus Human GFRAL, SMARTpool:
AAACAUGCUUGGAGAGUAA
GUGAGGAAUCUUUGUGUAA
GCAACCACGUCAAGACAAC
CAGUUGGCCUCUUACCUUA
2.11. Anti-GDF15 Experiment
In vitro treatment with combination of eribulin and human antibody anti-GDF15 was performed using the commercialized rabbit anti-GDF15 from ThermoFisher (#42-1700). Briefly, cells were seeded into 96-well tissue culture plates at a density of 5000 cells/well and the following day were treated for 72 h with 1.5 nM eribulin. After treatment, eribulin (1.5 nM), and anti-GDF15 (2.5 μg/mL) were added in six replicates to each population. Cell viability was measured after 7 days using the CellTiter (Promega) assay for 2D cell-culture settings and CellTiter-Glo (Promega) for 3D cell-culture settings. Crystal violet staining was performed to collect representative images of combinational drug treatment effect on cell viability performing the experiment in 24-well plates. A rabbit IgG isotype control was used as control.
2.12. In Vivo Experiment
PDX tumor tissues were collected after 1 month of eribulin treatment in vivo according to the following protocol: eribulin mesylate weekly on days 1, 3, 5 intravenously (0.1 mg/kg, in PBS). Further details are described in the mentioned paper [
31]. Collection of tumor samples and establishment of PDXs fresh tumor samples from patients with BC were collected following an Institutional Research Board-approved protocol and the associated written informed consent. The study was compliant with the Declaration of Helsinki. Experiments were conducted following the European Union’s animal care directive (2010/63/EU) and were approved by the Ethical Committee of Animal Experimentation of the Vall d’Hebron Research Institute, the Catalan Government or the National Research Ethics Service, Cambridgeshire ([
32] and
https://caldaslab.cruk.cam.ac.uk/bcape (accessed on 14 April 2022)).
2.13. Statistical Analysis
Data were analyzed with GraphPad Prism 6.0 (GraphPad Software 6.0, Inc.). Data from replicate experiments are shown as mean values ± standard deviation. Comparisons between groups were analyzed by a two-tailed Student t-test or two-way ANOVA, as appropriate. A p-value < 0.05 was considered statistically significant.
All statistical normalizations and calculations of proteomic data were performed using the software POMAcounts 1.1.0. Exploratory data analysis was performed using principal component analysis (PCA) and hierarchical clustering of the samples in the SpC matrix to find possible outliers and patterns in the data. For statistical modeling, a GLM model based on the Poisson distribution was used as a statistical test. Adjusted p-value < 0.05, fold change > 0.8 and number spectral counts (SpC) > 4 thresholds were applied during the analysis.
4. Discussion
Until recently, tumor sensitivity to cancer drugs and acquired drug resistance were usually seen as a binary switch. In this simplistic view, tumor cells would first die due to the action of the drug and then, eventually, the residual tumor would gain or become enriched with a mutation that would cause the resumption of tumor growth [
32,
35,
36]. Recently, however, the concept of drug
tolerance, which lies between drug sensitivity and drug resistance, has changed the paradigm of how tumor cells respond to cancer drugs. Drug tolerance is induced by therapeutic stress through non-genetic reprogramming that then leads to a reversible quiescent state [
37]. In this work, we focused on the proteomic characterization of breast cancer drug-tolerant cells that survive treatment with eribulin. First, we proved that when treated with a high dose of eribulin, breast cancer cells enter into a reversible slow-cycling state, known as the drug-tolerant persister (DTP) state. Then, we performed secretome proteomic profiling of breast cancer-DTPs to identify candidate pharmacodynamic biomarkers that could inform the action of eribulin in breast cancer patients. The DTP secretome induced by eribulin showed over-secretion of different groups of proteins related to the cytoskeleton, vesicle trafficking, protein translation and cellular stress, in agreement with the biological effects of a tubulin targeting agent. Interestingly, however, two cell lines belonging to different BC molecular subtypes showed similar eribulin-induced secretomes. Moreover, our results suggest that the secretome profiles of DTPs induced by different MTAs in breast cancer cells are linked to their specific tubulin-binding modes and mechanisms [
17,
38].
One protein over-secreted in DTP cells, GDF15, attracted our particular attention since its expression and secretion in parental cells were undetectable. The existing literature indicates that GDF15 is related to a wide range of very different biological processes in human biology. For instance, GDF15 is strongly up-regulated during pregnancy and its levels remain high in neonates until they are about 2 years old [
39]. In addition, elevated serum levels of GDF15 correlate with diabetes, chronic inflammation, infections, as well as both cardiovascular and renal diseases [
23]. The mechanistic link between the roles of GDF15 in these various conditions is not well known. However, current knowledge suggests that induction of GDF15 expression is linked to either physiological or pathological stress related to energy balance [
29]. The recent discovery of the receptor for GDF15, the previous orphan receptor GFRAL [
26], will undoubtedly contribute to our understanding of the mechanisms of action of GDF15.
In cancer, GDF15 is elevated in the advanced stages of several cancer types, where it participates in the induction of cachexia affecting patients with late-stage cancer [
40,
41,
42]. Different studies have shown higher expression of GDF15 mRNA and protein in cancer biopsies compared to their corresponding normal tissues [
43,
44]. Our results, showing that GDF15 is associated with the DTP state induced by eribulin, do not immediately connect with any known biology mediated by GDF15. However, a recent report showed that levels of circulating GDF15 correlated with chemoresistance of ovarian cancer patients to platinum therapy [
45]. In light of this, our in vitro results with cell lines, PDXO and in vivo results with PDX-derived tumor tissues offer a more nuanced picture: GDF15 is a pharmacodynamic response biomarker of eribulin, even while it is also associated with the transitory DTP state between drug sensitivity and full resistance. We thus believe that the concept of drug tolerance may help solve the question of whether GDF15 is a biomarker of response or resistance; our results suggest it is both.
The down-regulation of GDF15 in cells that achieved full drug resistance, and resume proliferation, confirms that GDF15 is a stress response protein. As stated above, different cellular stresses induce the expression of GDF15 to mitigate different acute insults including infection, injuries and cancer chemotherapy. In these cases, the role of GDF15 is to block cell proliferation, protect the damaged cells and tissues, and restore homeostasis [
23,
26]. In our experimental framework, a group of tumor cells (DTPs) are able to survive a high dose of eribulin by up-regulating the expression of GDF15. Eventually, these DTP adapt to eribulin and they are able to proliferate in the presence of the drug. We cannot rule out that the down-regulation of GDF15 only correlates with acquired resistance to eribulin instead of being the cause of it. However, our results, showing that targeting GDF15 blocks the survival of DTP cells, favor the hypothesis that GDF15 is mechanistically linked to the proliferative Stop/Go phenotype shown by DTP and fully resistant cells, respectively. When we translate this hypothesis into the clinical setting, we predict that the down-regulation of GDF15 expression in a tumor responding to eribulin would anticipate the progression of the disease.
One of the most striking results of our studies is that GDF15 not only correlates with the DTP state induced by eribulin but is also required for the survival of DTP cells: silencing of both GDF15 and its receptor GFRAL led to significant increases in eribulin sensitivity in both cell lines tested. Importantly, control experiments established that modulation of GDF15 and GFRAL did not affect cell proliferation per se; therefore, up-regulation of GDF15 is required for the viability of DTP cells. Moreover, we confirmed that actual secretion of GDF15, and not just protein expression, mediates the survival phenotype in DTP cells, since GFRAL silencing had the same effect as GDF15 silencing. Intriguingly, it has recently been reported that a GDF15-neutralizing antibody reverses anorexia and body weight loss and promotes survival in mouse tumor models [
46]. Additionally, a recent study showed that treatment with a GDF15-neutralizing antibody in combination with cisplatin improves survival in a mouse tumor model [
47]. Our GDF15/GFRAL silencing experiments together with the aforementioned GDF15-neutralizing antibody studies led us to test combination therapy with eribulin plus an anti-GDF15 antibody in our experimental models. Results confirmed our suspicions that interfering with GDF15 would enhance response to eribulin: significantly increased cell death was seen when breast cancer cells were treated with the combination of anti-GDF15 antibody plus eribulin as compared to the combination using a nonspecific control IgG. Similar results to the cell line studies were seen when treating breast cancer-derived PDXO with the same combinations, supporting the idea that GDF15 neutralization in combination with drug treatment may hold considerable potential as an effective therapeutic approach for breast cancer. Perhaps most excitingly, our work suggests that combining an anti-GDF15 antibody with chemotherapy could improve the overall survival of patients by the dual mechanisms of decreasing cachexia and either killing or preventing the development of DTPs during chemotherapy. Thus, our data in the breast cancer space suggest that GDF15 should be considered as a possible therapeutic co-target to eradicate or prevent the development of DTP cells during treatment with eribulin in both TNBC and luminal breast cancer subtypes. Intriguingly, our discovery of links between GDF15 and DTP cells as a result of eribulin treatment may extend to other drugs as well: preliminary data with several other drugs including metformin, doxorubicin, carboplatin and paclitaxel point to up-regulation of GDF15 by these drugs as well, perhaps also associated with DTP states (
Supplemental Figure S6). While further work will be required to confirm this, these preliminary results suggest that the paradigms established here with eribulin may also hold generally true for at least some other chemotherapy drugs.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}