
Nuclear Receptor Coregulators in Hormone-Dependent Cancers


Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Nuclear Receptors and Hormone-Dependent Cancers
1.2. A Primer on Nuclear Receptors and Their Interaction with Regulatory Complexes
1.3. Functions Played by Nuclear Receptor Coregulators
2. Examples of Nuclear Receptor Interactions with Coactivators
2.1. SWI/SNF (BAF) Complex
2.2. NCOA/p160/SRC
2.3. Transforming Acidic Coiled-Coil Containing Protein 1 (TACC1)
2.4. PPARG Coactivator 1 Alpha (PPARGC1A)
3. Examples of Nuclear Receptor Interactions with Corepressors
3.1. RE1-Silencing Transcription Corepressor (REST) Complex 1 (RCOR1)
3.2. NCOR1 and NCOR2 Corepressor Complex
3.3. SIN3 Transcription Regulator Family Member A (SIN3A)
3.4. Ligand-Dependent Nuclear Receptor Corepressor (LCOR)
4. Cointegrators
5. Disrupted Coregulators Function in Therapy-Resistant Hormone-Dependent Cancers
6. Technology and Computational Approaches to Discover Coregulator and Nuclear Interactions
6.1. Two-Hybrid Assay
6.2. Affinity Purification Followed by Mass Spectrometry (MS)
6.3. Rapid Immunoprecipitation Mass Spectrometry of Endogenous Proteins (RIME)
6.4. Protein–Protein Monitoring Methods
6.5. Bioinformatics Approaches to Identifying Coregulators
Author Contributions
Funding
Conflicts of Interest
References
- Siddappa, M.; Wani, S.A.; Long, M.D.; Leach, D.A.; Mathé, E.A.; Bevan, C.L.; Campbell, M.J. Identification of transcription factor co-regulators that drive prostate cancer progression. Sci. Rep. 2020, 10, 20332. [Google Scholar] [CrossRef] [PubMed]
- Abugessaisa, I.; Ramilowski, J.A.; Lizio, M.; Severin, J.; Hasegawa, A.; Harshbarger, J.; Kondo, A.; Noguchi, S.; Yip, C.W.; Ooi, J.L.C.; et al. FANTOM enters 20th year: Expansion of transcriptomic atlases and functional annotation of non-coding RNAs. Nucleic Acids Res. 2021, 49, D892–D898. [Google Scholar] [CrossRef] [PubMed]
- Wingender, E.; Schoeps, T.; Haubrock, M.; Krull, M.; Donitz, J. TFClass: Expanding the classification of human transcription factors to their mammalian orthologs. Nucleic Acids Res. 2018, 46, D343–D347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, H. Daniel Whistler: English physician who published the first book on rickets in 1645. Br. J. Hosp. Med. 2019, 80, 51. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C.; Campbell, M.J. Vitamin D receptor signaling mechanisms: Integrated actions of a well-defined transcription factor. Steroids 2013, 78, 127–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schutz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef] [Green Version]
- Paul, J. Sir George Beatson and the Royal Beatson Memorial Hospital. Med. Hist 1981, 25, 200–201. [Google Scholar]
- Huggins, C.; Stevens, R.; Hodges, C.V. Studies on prostatic cancer: II. The effects of castration on advanced carcinoma of the prostate gland. Arch. Surg. 1941, 43, 209–223. [Google Scholar] [CrossRef]
- Kumar, S.; Freelander, A.; Lim, E. Type 1 Nuclear Receptor Activity in Breast Cancer: Translating Preclinical Insights to the Clinic. Cancers 2021, 13, 4972. [Google Scholar] [CrossRef]
- Porter, B.A.; Ortiz, M.A.; Bratslavsky, G.; Kotula, L. Structure and Function of the Nuclear Receptor Superfamily and Current Targeted Therapies of Prostate Cancer. Cancers 2019, 11, 1852. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Prakash, C.; Zuniga, B.; Song, C.S.; Jiang, S.; Cropper, J.; Park, S.; Chatterjee, B. Nuclear Receptors in Drug Metabolism, Drug Response and Drug Interactions. Nucl. Recept. Res. 2015, 2, 101178. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, G.; Metivier, R.; Lin, C.-Y.; Denger, S.; Ibberson, D.; Ivacevic, T.; Brand, H.; Benes, V.; Liu, E.T.; Gannon, F. Multiple mechanisms induce transcriptional silencing of a subset of genes, including oestrogen receptor α, in response to deacetylase inhibition by valproic acid and trichostatin A. Oncogene 2005, 24, 4894–4907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waghray, A.; Schober, M.; Feroze, F.; Yao, F.; Virgin, J.; Chen, Y. Identification of differentially expressed genes by serial analysis of gene expression in human prostate cancer. Cancer Res. 2001, 61, 4283–4286. [Google Scholar]
- Wang, Q.; Li, W.; Zhang, Y.; Yuan, X.; Xu, K.; Yu, J.; Chen, Z.; Beroukhim, R.; Wang, H.; Lupien, M.; et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 2009, 138, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Pomerantz, M.M.; Qiu, X.; Zhu, Y.; Takeda, D.Y.; Pan, W.; Baca, S.C.; Gusev, A.; Korthauer, K.D.; Severson, T.M.; Ha, G.; et al. Prostate cancer reactivates developmental epigenomic programs during metastatic progression. Nat. Genet. 2020, 52, 790–799. [Google Scholar] [CrossRef]
- Toropainen, S.; Niskanen, E.A.; Malinen, M.; Sutinen, P.; Kaikkonen, M.U.; Palvimo, J.J. Global analysis of transcription in castration-resistant prostate cancer cells uncovers active enhancers and direct androgen receptor targets. Sci. Rep. 2016, 6, 33510. [Google Scholar] [CrossRef]
- Massie, C.E.; Adryan, B.; Barbosa-Morais, N.L.; Lynch, A.G.; Tran, M.G.; Neal, D.E.; Mills, I.G. sNew androgen receptor genomic targets show an interaction with the ETS1 transcription factor. EMBO Rep. 2007, 8, 871–878. [Google Scholar] [CrossRef] [Green Version]
- Copeland, B.T.; Pal, S.K.; Bolton, E.C.; Jones, J.O. The androgen receptor malignancy shift in prostate cancer. Prostate 2018, 78, 521–531. [Google Scholar] [CrossRef]
- Chattopadhyay, I.; Wang, J.; Qin, M.; Gao, L.; Holtz, R.; Vessella, R.L.; Leach, R.W.; Gelman, I.H. Src promotes castration-recurrent prostate cancer through androgen receptor-dependent canonical and non-canonical transcriptional signatures. Oncotarget 2017, 8, 10324–10347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malinen, M.; Niskanen, E.A.; Kaikkonen, M.U.; Palvimo, J.J. Crosstalk between androgen and pro-inflammatory signaling remodels androgen receptor and NF-kappaB cistrome to reprogram the prostate cancer cell transcriptome. Nucleic Acids Res. 2017, 45, 619–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volante, M.; Tota, D.; Giorcelli, J.; Bollito, E.; Napoli, F.; Vatrano, S.; Buttigliero, C.; Molinaro, L.; Gontero, P.; Porpiglia, F.; et al. Androgen deprivation modulates gene expression profile along prostate cancer progression. Hum. Pathol 2016, 56, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, M.M.; Li, F.; Takeda, D.Y.; Lenci, R.; Chonkar, A.; Chabot, M.; Cejas, P.; Vazquez, F.; Cook, J.; Shivdasani, R.A.; et al. The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat. Genet. 2015, 47, 1346–1351. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Lonergan, P.E.; Nacusi, L.P.; Wang, L.; Schmidt, L.J.; Sun, Z.; Van der Steen, T.; Boorjian, S.A.; Kosari, F.; Vasmatzis, G.; et al. The cistrome and gene signature of androgen receptor splice variants in castration resistant prostate cancer cells. J. Urol. 2015, 193, 690–698. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chi, P.; Rockowitz, S.; Iaquinta, P.J.; Shamu, T.; Shukla, S.; Gao, D.; Sirota, I.; Carver, B.S.; Wongvipat, J.; et al. ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis in response to PTEN loss. Nat. Med. 2013, 19, 1023–1029. [Google Scholar] [CrossRef] [Green Version]
- Beltran, H.; Hruszkewycz, A.; Scher, H.I.; Hildesheim, J.; Isaacs, J.; Yu, E.Y.; Kelly, K.; Lin, D.; Dicker, A.P.; Arnold, J.T.; et al. The role of lineage plasticity in prostate cancer therapy resistance. Clin. Cancer Res. 2019, 25, 6916–6924. [Google Scholar] [CrossRef] [Green Version]
- Berger, A.; Brady, N.J.; Bareja, R.; Robinson, B.; Conteduca, V.; Augello, M.A.; Puca, L.; Ahmed, A.; Dardenne, E.; Lu, X.; et al. N-Myc-mediated epigenetic reprogramming drives lineage plasticity in advanced prostate cancer. J. Clin. Investig. 2019, 130, 3924–3940. [Google Scholar] [CrossRef]
- Sheahan, A.V.; Ellis, L. Epigenetic reprogramming: A key mechanism driving therapeutic resistance. Urol. Oncol. 2018, 36, 375–379. [Google Scholar] [CrossRef]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbe, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Bohrer, L.R.; Chen, S.; Hallstrom, T.C.; Huang, H. Androgens suppress EZH2 expression via retinoblastoma (RB) and p130-dependent pathways: A potential mechanism of androgen-refractory progression of prostate cancer. Endocrinology 2010, 151, 5136–5145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banwell, C.M.; MacCartney, D.P.; Guy, M.; Miles, A.E.; Uskokovic, M.R.; Mansi, J.; Stewart, P.M.; O’Neill, L.P.; Turner, B.M.; Colston, K.W.; et al. Altered nuclear receptor corepressor expression attenuates vitamin D receptor signaling in breast cancer cells. Clin. Cancer Res. 2006, 12, 2004–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battaglia, S.; Maguire, O.; Thorne, J.L.; Hornung, L.B.; Doig, C.L.; Liu, S.; Sucheston, L.E.; Bianchi, A.; Khanim, F.L.; Gommersall, L.M.; et al. Elevated NCOR1 disrupts PPARalpha/gamma signaling in prostate cancer and forms a targetable epigenetic lesion. Carcinogenesis 2010, 31, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Brzostek, S.; Lee, S.R.; Hollenberg, A.N.; Balk, S.P. Inhibition of the dihydrotestosterone-activated androgen receptor by nuclear receptor corepressor. Mol. Endocrinol. 2002, 16, 1492–1501. [Google Scholar] [CrossRef]
- Doig, C.L.; Singh, P.K.; Dhiman, V.K.; Thorne, J.L.; Battaglia, S.; Sobolewski, M.; Maguire, O.; O’Neill, L.P.; Turner, B.M.; McCabe, C.J. Recruitment of NCOR1 to VDR target genes is enhanced in prostate cancer cells and associates with altered DNA methylation patterns. Carcinogenesis 2013, 34, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Fereshteh, M.P.; Tilli, M.T.; Kim, S.E.; Xu, J.; O’Malley, B.W.; Wellstein, A.; Furth, P.A.; Riegel, A.T. The nuclear receptor coactivator amplified in breast cancer-1 is required for Neu (ErbB2/HER2) activation, signaling, and mammary tumorigenesis in mice. Cancer Res. 2008, 68, 3697–3706. [Google Scholar] [CrossRef] [Green Version]
- Lavinsky, R.M.; Jepsen, K.; Heinzel, T.; Torchia, J.; Mullen, T.M.; Schiff, R.; Del-Rio, A.L.; Ricote, M.; Ngo, S.; Gemsch, J.; et al. Diverse signaling pathways modulate nuclear receptor recruitment of N-CoR and SMRT complexes. Proc. Natl. Acad. Sci. USA 1998, 95, 2920–2925. [Google Scholar] [CrossRef] [Green Version]
- Lopez, S.M.; Agoulnik, A.I.; Zhang, M.; Peterson, L.E.; Suarez, E.; Gandarillas, G.A.; Frolov, A.; Li, R.; Rajapakshe, K.; Coarfa, C. Nuclear receptor corepressor 1 expression and output declines with prostate cancer progression. Clin. Cancer Res. 2016, 22, 3937–3949. [Google Scholar] [CrossRef] [Green Version]
- Hodgson, M.C.; Shen, H.C.; Hollenberg, A.N.; Balk, S.P. Structural basis for nuclear receptor corepressor recruitment by antagonist-liganded androgen receptor. Mol. Cancer Ther. 2008, 7, 3187–3194. [Google Scholar] [CrossRef] [Green Version]
- McKenna, N.J.; Lanz, R.B.; O’Malley, B.W. Nuclear receptor coregulators: Cellular and molecular biology. Endocr. Rev. 1999, 20, 321–344. [Google Scholar]
- Lazar, M.A. Maturing of the nuclear receptor family. J. Clin. Investig. 2017, 127, 1123–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aranda, A.; Pascual, A. Nuclear hormone receptors and gene expression. Physiol. Rev. 2001, 81, 1269–1304. [Google Scholar] [CrossRef] [PubMed]
- Gronemeyer, H.; Gustafsson, J.A.; Laudet, V. Principles for modulation of the nuclear receptor superfamily. Nat. Rev. Drug Discov. 2004, 3, 950–964. [Google Scholar] [CrossRef] [PubMed]
- Nettles, K.W.; Greene, G.L. Ligand control of coregulator recruitment to nuclear receptors. Annu. Rev. Physiol. 2005, 67, 309–333. [Google Scholar] [CrossRef]
- Weston, A.D.; Blumberg, B.; Underhill, T.M. Active repression by unliganded retinoid receptors in development: Less is sometimes more. J. Cell Biol. 2003, 161, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Foulds, C.E.; Panigrahi, A.K.; Coarfa, C.; Lanz, R.B.; O’Malley, B.W. Long Noncoding RNAs as Targets and Regulators of Nuclear Receptors. Curr. Top. Microbiol. Immunol. 2016, 394, 143–176. [Google Scholar]
- Lanz, R.B.; Razani, B.; Goldberg, A.D.; O’Malley, B.W. Distinct RNA motifs are important for coactivation of steroid hormone receptors by steroid receptor RNA activator (SRA). Proc. Natl. Acad. Sci. USA 2002, 99, 16081–16086. [Google Scholar] [CrossRef] [Green Version]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef] [Green Version]
- Umesono, K.; Evans, R.M. Determinants of target gene specificity for steroid/thyroid hormone receptors. Cell 1989, 57, 1139–1146. [Google Scholar] [CrossRef]
- Bourguet, W.; Ruff, M.; Chambon, P.; Gronemeyer, H.; Moras, D. Crystal structure of the ligand-binding domain of the human nuclear receptor RXR-alpha. Nature 1995, 375, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, A.M.; Pike, A.C.; Dauter, Z.; Hubbard, R.E.; Bonn, T.; Engström, O.; Ohman, L.; Greene, G.L.; Gustafsson, J.A.; Carlquist, M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Darimont, B.D.; Wagner, R.L.; Apriletti, J.W.; Stallcup, M.R.; Kushner, P.J.; Baxter, J.D.; Fletterick, R.J.; Yamamoto, K.R. Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev. 1998, 12, 3343–3356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moras, D.; Gronemeyer, H. The nuclear receptor ligand-binding domain: Structure and function. Curr. Opin. Cell Biol. 1998, 10, 384–391. [Google Scholar] [CrossRef]
- Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.; Rosenfeld, M.G.; Willson, T.M.; Glass, C.K.; Milburn, M.V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 1998, 395, 137–143. [Google Scholar] [CrossRef]
- Renaud, J.P.; Rochel, N.; Ruff, M.; Vivat, V.; Chambon, P.; Gronemeyer, H.; Moras, D. Crystal structure of the RAR-gamma ligand-binding domain bound to all-trans retinoic acid. Nature 1995, 378, 681–689. [Google Scholar] [CrossRef]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef] [Green Version]
- Wagner, R.L.; Apriletti, J.W.; McGrath, M.E.; West, B.L.; Baxter, J.D.; Fletterick, R.J. A structural role for hormone in the thyroid hormone receptor. Nature 1995, 378, 690–697. [Google Scholar] [CrossRef]
- Arnal, J.F.; Lenfant, F.; Metivier, R.; Flouriot, G.; Henrion, D.; Adlanmerini, M.; Fontaine, C.; Gourdy, P.; Chambon, P.; Katzenellenbogen, B.; et al. Membrane and Nuclear Estrogen Receptor Alpha Actions: From Tissue Specificity to Medical Implications. Physiol. Rev. 2017, 97, 1045–1087. [Google Scholar] [CrossRef]
- Perissi, V.; Rosenfeld, M.G. Controlling nuclear receptors: The circular logic of cofactor cycles. Nat. Rev. Mol. Cell Biol. 2005, 6, 542–554. [Google Scholar] [CrossRef]
- Mangelsdorf, D.J.; Evans, R.M. The RXR heterodimers and orphan receptors. Cell 1995, 83, 841–850. [Google Scholar] [CrossRef] [Green Version]
- Ptashne, M.; Gann, A.A. Activators and targets. Nature 1990, 346, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Long, M.D.; Jacobi, J.J.; Singh, P.K.; Llimos, G.; Wani, S.A.; Rowsam, A.M.; Rosario, S.R.; Hoogstraat, M.; Linder, S.; Kirk, J.; et al. Reduced NCOR2 expression accelerates androgen deprivation therapy failure in prostate cancer. Cell Rep. 2021, 37, 110109. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Lazar, M.A. The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature 1999, 402, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Nagy, L.; Kao, H.Y.; Chakravarti, D.; Lin, R.J.; Hassig, C.A.; Ayer, D.E.; Schreiber, S.L.; Evans, R.M. Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell 1997, 89, 373–380. [Google Scholar] [CrossRef] [Green Version]
- Perissi, V.; Aggarwal, A.; Glass, C.K.; Rose, D.W.; Rosenfeld, M.G. A corepressor/coactivator exchange complex required for transcriptional activation by nuclear receptors and other regulated transcription factors. Cell 2004, 116, 511–526. [Google Scholar] [CrossRef] [Green Version]
- Webb, P.; Anderson, C.M.; Valentine, C.; Nguyen, P.; Marimuthu, A.; West, B.L.; Baxter, J.D.; Kushner, P.J. The nuclear receptor corepressor (N-CoR) contains three isoleucine motifs (I/LXXII) that serve as receptor interaction domains (IDs). Mol. Endocrinol. 2000, 14, 1976–1985. [Google Scholar] [CrossRef]
- Soutourina, J. Transcription regulation by the Mediator complex. Nat. Rev. Mol. Cell Biol. 2018, 19, 262–274. [Google Scholar] [CrossRef]
- Su, Q.; Hu, S.; Gao, H.; Ma, R.; Yang, Q.; Pan, Z.; Wang, T.; Li, F. Role of AIB1 for tamoxifen resistance in estrogen receptor-positive breast cancer cells. Oncology 2008, 75, 159–168. [Google Scholar] [CrossRef]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Glass, C.K. Nuclear receptors and inflammation control: Molecular mechanisms and pathophysiological relevance. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1542–1549. [Google Scholar] [CrossRef] [PubMed]
- Melekhova, A.; Baniahmad, A. ING Tumour Suppressors and ING Splice Variants as Coregulators of the Androgen Receptor Signalling in Prostate Cancer. Cells 2021, 10, 2599. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Parra, M.A.; Gronemeyer, H. Integrative genomics to dissect retinoid functions. Subcell Biochem. 2014, 70, 181–202. [Google Scholar] [PubMed]
- Moore, N.L.; Hickey, T.E.; Butler, L.M.; Tilley, W.D. Multiple nuclear receptor signaling pathways mediate the actions of synthetic progestins in target cells. Mol. Cell Endocrinol. 2012, 357, 60–70. [Google Scholar] [CrossRef]
- Panigrahi, A.; O’Malley, B.W. Mechanisms of enhancer action: The known and the unknown. Genome Biol. 2021, 22, 108. [Google Scholar] [CrossRef]
- Yang, P.B.; Hou, P.P.; Liu, F.Y.; Hong, W.B.; Chen, H.Z.; Sun, X.Y.; Li, P.; Zhang, Y.; Ju, C.Y.; Luo, L.J.; et al. Blocking PPARgamma interaction facilitates Nur77 interdiction of fatty acid uptake and suppresses breast cancer progression. Proc. Natl. Acad. Sci. USA 2020, 117, 27412–27422. [Google Scholar] [CrossRef]
- Jeong, K.W.; Lee, Y.H.; Stallcup, M.R. Recruitment of the SWI/SNF chromatin remodeling complex to steroid hormone-regulated promoters by nuclear receptor coactivator flightless-I. J. Biol. Chem. 2009, 284, 29298–29309. [Google Scholar] [CrossRef] [Green Version]
- Perez-Calero, C.; Bayona-Feliu, A.; Xue, X.; Barroso, S.I.; Munoz, S.; Gonzalez-Basallote, V.M.; Sung, P.; Aguilera, A. Corrigendum: UAP56/DDX39B is a major cotranscri.iptional RNA-DNA helicase that unwinds harmful R loops genome-wide. Genes Dev. 2021, 35, 573. [Google Scholar] [CrossRef]
- Zhou, X.; Khan, S.G.; Tamura, D.; Ueda, T.; Boyle, J.; Compe, E.; Egly, J.M.; DiGiovanna, J.J.; Kraemer, K.H. Abnormal XPD-induced nuclear receptor transactivation in DNA repair disorders: Trichothiodystrophy and xeroderma pigmentosum. Eur. J. Hum. Genet. 2013, 21, 831–837. [Google Scholar] [CrossRef] [Green Version]
- Lonard, D.M.; O’Malley, B.W. Nuclear receptor coregulators: Modulators of pathology and therapeutic targets. Nat. Rev. Endocrinol. 2012, 8, 598–604. [Google Scholar] [CrossRef]
- D’Santos, C.; Taylor, C.; Carroll, J.S.; Mohammed, H. RIME proteomics of estrogen and progesterone receptors in breast cancer. Data Brief 2015, 5, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Foulds, C.E.; Feng, Q.; Ding, C.; Bailey, S.; Hunsaker, T.L.; Malovannaya, A.; Hamilton, R.A.; Gates, L.A.; Zhang, Z.; Li, C.; et al. Proteomic analysis of coregulators bound to ERα on DNA and nucleosomes reveals coregulator dynamics. Mol. Cell 2013, 51, 185–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddappa, M.H.; Wani, S.A.; Tang, H.; Gray, J.S.; Jafari, H.; Wu, H.; Long, M.D.; Elhussin, I.; Karanam, B.; Wang, H.; et al. Vitamin D Receptor Cistrome-Transcriptome Analyses Establishes Quantitatively Distinct Receptor Genomic Interactions in African American Prostate Cancer Regulated by BAZ1A. 2022. Available online: https://www.biorxiv.org/content/10.1101/2022.01.31.478573v1 (accessed on 28 April 2022).
- Papachristou, E.K.; Kishore, K.; Holding, A.N.; Harvey, K.; Roumeliotis, T.I.; Chilamakuri, C.S.R.; Omarjee, S.; Chia, K.M.; Swarbrick, A.; Lim, E.; et al. A quantitative mass spectrometry-based approach to monitor the dynamics of endogenous chromatin-associated protein complexes. Nat. Commun. 2018, 9, 2311. [Google Scholar] [CrossRef]
- Browne, A.L.; Charmsaz, S.; Vareslija, D.; Fagan, A.; Cosgrove, N.; Cocchiglia, S.; Purcell, S.; Ward, E.; Bane, F.; Hudson, L.; et al. Network analysis of SRC-1 reveals a novel transcription factor hub which regulates endocrine resistant breast cancer. Oncogene 2018, 37, 2008–2021. [Google Scholar] [CrossRef] [PubMed]
- Stashi, E.; York, B.; O’Malley, B.W. Steroid receptor coactivators: Servants and masters for control of systems metabolism. Trends Endocrinol. Metab. 2014, 25, 337–347. [Google Scholar] [CrossRef] [Green Version]
- Bagheri-Yarmand, R.; Talukder, A.H.; Wang, R.A.; Vadlamudi, R.K.; Kumar, R. Metastasis-associated protein 1 deregulation causes inappropriate mammary gland development and tumorigenesis. Development 2004, 131, 3469–3479. [Google Scholar] [CrossRef] [Green Version]
- O’Malley, B.W.; Kumar, R. Nuclear receptor coregulators in cancer biology. Cancer Res. 2009, 69, 8217–8222. [Google Scholar] [CrossRef] [Green Version]
- Dou, Y.; Milne, T.A.; Tackett, A.J.; Smith, E.R.; Fukuda, A.; Wysocka, J.; Allis, C.D.; Chait, B.T.; Hess, J.L.; Roeder, R.G. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell 2005, 121, 873–885. [Google Scholar] [CrossRef] [Green Version]
- Lindroth, A.M.; Shultis, D.; Jasencakova, Z.; Fuchs, J.; Johnson, L.; Schubert, D.; Patnaik, D.; Pradhan, S.; Goodrich, J.; Schubert, I.; et al. Dual histone H3 methylation marks at lysines 9 and 27 required for interaction with CHROMOMETHYLASE3. EMBO J. 2004, 23, 4286–4296. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Li, Y.; Ishizuka, T.; Guenther, M.G.; Lazar, M.A. A SANT motif in the SMRT corepressor interprets the histone code and promotes histone deacetylation. EMBO J. 2003, 22, 3403–3410. [Google Scholar] [CrossRef] [Green Version]
- Khan, O.Y.; Fu, G.; Ismail, A.; Srinivasan, S.; Cao, X.; Tu, Y.; Lu, S.; Nawaz, Z. Multifunction steroid receptor coactivator, E6-associated protein, is involved in development of the prostate gland. Mol. Endocrinol. 2006, 20, 544–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Ma, H.; Hong, H.; Koh, S.S.; Huang, S.M.; Schurter, B.T.; Aswad, D.W.; Stallcup, M.R. Regulation of transcription by a protein methyltransferase. Science 1999, 284, 2174–2177. [Google Scholar] [CrossRef] [PubMed]
- Poukka, H.; Karvonen, U.; Janne, O.A.; Palvimo, J.J. Covalent modification of the androgen receptor by small ubiquitin-like modifier 1 (SUMO-1). Proc. Natl. Acad. Sci. USA 2000, 97, 14145–14150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.; Cai, C.; Omwancha, J.; Chen, S.Y.; Baslan, T.; Shemshedini, L. SUMO-3 enhances androgen receptor transcriptional activity through a sumoylation-independent mechanism in prostate cancer cells. J. Biol. Chem. 2006, 281, 4002–4012. [Google Scholar] [CrossRef] [Green Version]
- Ishitani, K.; Yoshida, T.; Kitagawa, H.; Ohta, H.; Nozawa, S.; Kato, S. p54nrb acts as a transcriptional coactivator for activation function 1 of the human androgen receptor. Biochem. Biophys. Res. Commun. 2003, 306, 660–665. [Google Scholar] [CrossRef]
- Galbraith, L.C.A.; Mui, E.; Nixon, C.; Hedley, A.; Strachan, D.; MacKay, G.; Sumpton, D.; Sansom, O.J.; Leung, H.Y.; Ahmad, I. PPAR-gamma induced AKT3 expression increases levels of mitochondrial biogenesis driving prostate cancer. Oncogene 2021, 40, 2355–2366. [Google Scholar] [CrossRef]
- Liu, S.; Lin, S.J.; Li, G.; Kim, E.; Chen, Y.T.; Yang, D.R.; Tan, M.H.; Yong, E.L.; Chang, C. Differential roles of PPARgamma vs TR4 in prostate cancer and metabolic diseases. Endocr. Relat. Cancer 2014, 21, R279–R300. [Google Scholar] [CrossRef] [Green Version]
- Kaikkonen, S.; Paakinaho, V.; Sutinen, P.; Levonen, A.L.; Palvimo, J.J. Prostaglandin 15d-PGJ(2) inhibits androgen receptor signaling in prostate cancer cells. Mol. Endocrinol. 2013, 27, 212–223. [Google Scholar] [CrossRef] [Green Version]
- Govindarajan, R.; Ratnasinghe, L.; Simmons, D.L.; Siegel, E.R.; Midathada, M.V.; Kim, L.; Kim, P.J.; Owens, R.J.; Lang, N.P. Thiazolidinediones and the risk of lung, prostate, and colon cancer in patients with diabetes. J. Clin. Oncol 2007, 25, 1476–1481. [Google Scholar] [CrossRef] [Green Version]
- Butler, R.; Mitchell, S.H.; Tindall, D.J.; Young, C.Y. Nonapoptotic cell death associated with S-phase arrest of prostate cancer cells via the peroxisome proliferator-activated receptor gamma ligand, 15-deoxy-delta12,14-prostaglandin J2. Cell Growth Differ. 2000, 11, 49–61. [Google Scholar]
- Zheng, K.; Chen, S.; Hu, X. Peroxisome Proliferator Activated Receptor Gamma Coactivator-1 Alpha: A Double-Edged Sword in Prostate Cancer. Curr. Cancer Drug Targets 2022. [Google Scholar] [CrossRef] [PubMed]
- Torrano, V.; Valcarcel-Jimenez, L.; Cortazar, A.R.; Liu, X.; Urosevic, J.; Castillo-Martin, M.; Fernandez-Ruiz, S.; Morciano, G.; Caro-Maldonado, A.; Guiu, M.; et al. The metabolic co-regulator PGC1alpha suppresses prostate cancer metastasis. Nat. Cell Biol. 2016, 18, 645–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayeur, G.L.; Kung, W.J.; Martinez, A.; Izumiya, C.; Chen, D.J.; Kung, H.J. Ku is a novel transcriptional recycling coactivator of the androgen receptor in prostate cancer cells. J. Biol. Chem. 2005, 280, 10827–10833. [Google Scholar] [CrossRef] [Green Version]
- Fan, C.Y.; Ren, H.Y.; Lee, P.; Caplan, A.J.; Cyr, D.M. The type I Hsp40 zinc finger-like region is required for Hsp70 to capture non-native polypeptides from Ydj1. J. Biol. Chem. 2005, 280, 695–702. [Google Scholar] [CrossRef] [Green Version]
- Takeshita, Y.; Fujinaga, R.; Zhao, C.; Yanai, A.; Shinoda, K. Huntingtin-associated protein 1 (HAP1) interacts with androgen receptor (AR) and suppresses SBMA-mutant-AR-induced apoptosis. Hum. Mol. Genet. 2006, 15, 2298–2312. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.L.; Schneider, M.C.; Zheng, Y.; Zhang, X.; Richie, J.P. Caveolin-1 interacts with androgen receptor. A positive modulator of androgen receptor mediated transactivation. J. Biol. Chem. 2001, 276, 13442–13451. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, N.; Yeh, S.; Kang, H.Y.; Inui, S.; Chang, H.C.; Mizokami, A.; Chang, C. Cloning and characterization of androgen receptor coactivator, ARA55, in human prostate. J. Biol. Chem. 1999, 274, 8316–8321. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yang, Y.; Guo, X.; Sampson, E.R.; Hsu, C.L.; Tsai, M.Y.; Yeh, S.; Wu, G.; Guo, Y.; Chang, C. Suppression of androgen receptor transactivation by Pyk2 via interaction and phosphorylation of the ARA55 coregulator. J. Biol. Chem. 2002, 277, 15426–15431. [Google Scholar] [CrossRef] [Green Version]
- Kasai, M.; Guerrero-Santoro, J.; Friedman, R.; Leman, E.S.; Getzenberg, R.H.; DeFranco, D.B. The Group 3 LIM domain protein paxillin potentiates androgen receptor transactivation in prostate cancer cell lines. Cancer Res. 2003, 63, 4927–4935. [Google Scholar]
- Gangisetty, O.; Lauffart, B.; Sondarva, G.V.; Chelsea, D.M.; Still, I.H. The transforming acidic coiled coil proteins interact with nuclear histone acetyltransferases. Oncogene 2004, 23, 2559–2563. [Google Scholar] [CrossRef] [Green Version]
- Wilde, J.J.; Siegenthaler, J.A.; Dent, S.Y.; Niswander, L.A. Diencephalic Size Is Restricted by a Novel Interplay Between GCN5 Acetyltransferase Activity and Retinoic Acid Signaling. J. Neurosci 2017, 37, 2565–2579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guyot, R.; Vincent, S.; Bertin, J.; Samarut, J.; Ravel-Chapuis, P. The transforming acidic coiled coil (TACC1) protein modulates the transcriptional activity of the nuclear receptors TR and RAR. BMC Mol. Biol 2010, 11, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Moy, L.Y.; Sanada, K.; Zhou, Y.; Buchman, J.J.; Tsai, L.H. Cep120 and TACCs control interkinetic nuclear migration and the neural progenitor pool. Neuron 2007, 56, 79–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cully, M.; Shiu, J.; Piekorz, R.P.; Muller, W.J.; Done, S.J.; Mak, T.W. Transforming acidic coiled coil 1 promotes transformation and mammary tumorigenesis. Cancer Res. 2005, 65, 10363–10370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Still, I.H.; Hamilton, M.; Vince, P.; Wolfman, A.; Cowell, J.K. Cloning of TACC1, an embryonically expressed, potentially transforming coiled coil containing gene, from the 8p11 breast cancer amplicon. Oncogene 1999, 18, 4032–4038. [Google Scholar] [CrossRef] [Green Version]
- Fish, L.; Pencheva, N.; Goodarzi, H.; Tran, H.; Yoshida, M.; Tavazoie, S.F. Muscleblind-like 1 suppresses breast cancer metastatic colonization and stabilizes metastasis suppressor transcripts. Genes Dev. 2016, 30, 386–398. [Google Scholar] [CrossRef] [Green Version]
- Long, M.D.; Singh, P.K.; Russell, J.R.; Llimos, G.; Rosario, S.; Rizvi, A.; van den Berg, P.R.; Kirk, J.; Sucheston-Campbell, L.E.; Smiraglia, D.J.; et al. The miR-96 and RARgamma signaling axis governs androgen signaling and prostate cancer progression. Oncogene 2019, 38, 421–444. [Google Scholar] [CrossRef] [Green Version]
- Hodges, C.; Kirkland, J.G.; Crabtree, G.R. The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026930. [Google Scholar] [CrossRef] [Green Version]
- Jelinic, P.; Mueller, J.J.; Olvera, N.; Dao, F.; Scott, S.N.; Shah, R.; Gao, J.; Schultz, N.; Gonen, M.; Soslow, R.A.; et al. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat. Genet. 2014, 46, 424–426. [Google Scholar] [CrossRef]
- Ramos, P.; Karnezis, A.N.; Craig, D.W.; Sekulic, A.; Russell, M.L.; Hendricks, W.P.; Corneveaux, J.J.; Barrett, M.T.; Shumansky, K.; Yang, Y.; et al. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat. Genet. 2014, 46, 427–429. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.S.; Jin, V.X.; Fan, M.; Smith, L.T.; Liyanarachchi, S.; Yan, P.S.; Leu, Y.W.; Chan, M.W.; Plass, C.; Nephew, K.P.; et al. Combinatorial analysis of transcription factor partners reveals recruitment of c-MYC to estrogen receptor-alpha responsive promoters. Mol. Cell 2006, 21, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, J. The novel function of HINFP as a co-activator in sterol-regulated transcription of PCSK9 in HepG2 cells. Biochem. J. 2012, 443, 757–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unno, A.; Takada, I.; Takezawa, S.; Oishi, H.; Baba, A.; Shimizu, T.; Tokita, A.; Yanagisawa, J.; Kato, S. TRRAP as a hepatic coactivator of LXR and FXR function. Biochem. Biophys. Res. Commun. 2005, 327, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Liu, Y.; Ford, O.H., 3rd; Mohler, J.L.; Whang, Y.E. Involvement of arginine methyltransferase CARM1 in androgen receptor function and prostate cancer cell viability. Prostate 2006, 66, 1292–1301. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.L.; Li, W.J.; Ding, J.C.; He, Y.H.; Ran, T.; Xie, B.L.; Wang, Z.R.; Shen, H.F.; Xiao, R.Q.; Gao, W.W.; et al. A hypermethylation strategy utilized by enhancer-bound CARM1 to promote estrogen receptor α-dependent transcriptional activation and breast carcinogenesis. Theranostics 2020, 10, 3451–3473. [Google Scholar] [CrossRef]
- Purcell, D.J.; Chauhan, S.; Jimenez-Stinson, D.; Elliott, K.R.; Tsewang, T.D.; Lee, Y.H.; Marples, B.; Lee, D.Y. Novel CARM1-Interacting Protein, DZIP3, Is a Transcriptional Coactivator of Estrogen Receptor-α. Mol. Endocrinol. 2015, 29, 1708–1719. [Google Scholar] [CrossRef] [Green Version]
- Yadav, N.; Cheng, D.; Richard, S.; Morel, M.; Iyer, V.R.; Aldaz, C.M.; Bedford, M.T. CARM1 promotes adipocyte differentiation by coactivating PPARgamma. EMBO Rep. 2008, 9, 193–198. [Google Scholar] [CrossRef]
- Giraud, S.; Bienvenu, F.; Avril, S.; Gascan, H.; Heery, D.M.; Coqueret, O. Functional interaction of STAT3 transcription factor with the coactivator NcoA/SRC1a. J. Biol. Chem. 2002, 277, 8004–8011. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M. Functional proteomics of the epigenetic regulators ASXL1, ASXL2 and ASXL3: A convergence of proteomics and epigenetics for translational medicine. Expert Rev. Proteom. 2015, 12, 317–328. [Google Scholar] [CrossRef]
- Wang, N.; Zou, Q.; Xu, J.; Zhang, J.; Liu, J. Ligand binding and heterodimerization with retinoid X receptor α (RXRα) induce farnesoid X receptor (FXR) conformational changes affecting coactivator binding. J. Biol. Chem. 2018, 293, 18180–18191. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Yang, C.; Wang, Y.; Liao, Y.; Huang, K. PARP-1 suppresses adiponectin expression through poly(ADP-ribosyl)ation of PPAR gamma in cardiac fibroblasts. Cardiovasc. Res. 2009, 81, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeyakumar, M.; Liu, X.F.; Erdjument-Bromage, H.; Tempst, P.; Bagchi, M.K. Phosphorylation of thyroid hormone receptor-associated nuclear receptor corepressor holocomplex by the DNA-dependent protein kinase enhances its histone deacetylase activity. J. Biol. Chem. 2007, 282, 9312–9322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavri, R.; Lewis, B.; Kim, T.K.; Dilworth, F.J.; Erdjument-Bromage, H.; Tempst, P.; de Murcia, G.; Evans, R.; Chambon, P.; Reinberg, D. PARP-1 determines specificity in a retinoid signaling pathway via direct modulation of mediator. Mol. Cell 2005, 18, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, Y.; Wang, L.; Luo, X.; Huang, K.; Wang, C.; Du, M.; Liu, F.; Luo, T.; Huang, D.; et al. Poly(ADP-ribose) polymerase 1 is a key regulator of estrogen receptor α-dependent gene transcription. J. Biol. Chem. 2013, 288, 11348–11357. [Google Scholar] [CrossRef] [Green Version]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.W.S.; Anjum, B.; Shen, H.M.; Ghosh, S.; Yen, P.M.; Sinha, R.A. Lysosomal inhibition attenuates peroxisomal gene transcription via suppression of PPARA and PPARGC1A levels. Autophagy 2019, 15, 1455–1459. [Google Scholar] [CrossRef]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [CrossRef]
- He, S.; Wu, Z.; Tian, Y.; Yu, Z.; Yu, J.; Wang, X.; Li, J.; Liu, B.; Xu, Y. Structure of nucleosome-bound human BAF complex. Science 2020, 367, 875–881. [Google Scholar] [CrossRef]
- Seth-Vollenweider, T.; Joshi, S.; Dhawan, P.; Sif, S.; Christakos, S. Novel mechanism of negative regulation of 1,25-dihydroxyvitamin D3-induced 25-hydroxyvitamin D3 24-hydroxylase (Cyp24a1) Transcription: Epigenetic modification involving cross-talk between protein-arginine methyltransferase 5 and the SWI/SNF complex. J. Biol. Chem. 2014, 289, 33958–33970. [Google Scholar] [CrossRef] [Green Version]
- Roberts, C.W.; Orkin, S.H. The SWI/SNF complex—Chromatin and cancer. Nat. Rev. Cancer 2004, 4, 133–142. [Google Scholar] [CrossRef]
- Wu, Q.; Lian, J.B.; Stein, J.L.; Stein, G.S.; Nickerson, J.A.; Imbalzano, A.N. The BRG1 ATPase of human SWI/SNF chromatin remodeling enzymes as a driver of cancer. Epigenomics 2017, 9, 919–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Lu, F.; Zhang, Y. Loss of HDAC-Mediated Repression and Gain of NF-kappaB Activation Underlie Cytokine Induction in ARID1A- and PIK3CA-Mutation-Driven Ovarian Cancer. Cell Rep. 2016, 17, 275–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, M.D.; Bycroft, M.; Zinzalla, G. Structure of the BRK domain of the SWI/SNF chromatin remodeling complex subunit BRG1 reveals a potential role in protein-protein interactions. Protein Sci. 2020, 29, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Allis, C.D. SWI/SNF complex in cancer. Nat. Genet. 2017, 49, 178–179. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, A.F.; Vakoc, C.R. A rationale to target the SWI/SNF complex for cancer therapy. Trends Genet. 2014, 30, 356–363. [Google Scholar] [CrossRef] [Green Version]
- Medina, P.P.; Sanchez-Cespedes, M. Involvement of the chromatin-remodeling factor BRG1/SMARCA4 in human cancer. Epigenetics 2008, 3, 64–68. [Google Scholar] [CrossRef] [Green Version]
- Trotter, K.W.; Archer, T.K. The BRG1 transcriptional coregulator. Nucl. Recept. Signal. 2008, 6, e004. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.Y.; Beliakoff, J.; Li, X.; Lee, J.; Li, X.; Sharma, M.; Lim, B.; Sun, Z. hZimp7, a novel PIAS-like protein, enhances androgen receptor-mediated transcription and interacts with SWI/SNF-like BAF complexes. Mol. Endocrinol. 2005, 19, 2915–2929. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.L.; Kim, Y.W.; Jeong, K.W. BAF53A regulates androgen receptor-mediated gene expression and proliferation in LNCaP cells. Biochem. Biophys. Res. Commun. 2018, 505, 618–623. [Google Scholar] [CrossRef]
- Tropee, R.; de la Pena Avalos, B.; Gough, M.; Snell, C.; Duijf, P.H.G.; Dray, E. The SWI/SNF subunit SMARCD3 regulates cell cycle progression and predicts survival outcome in ER+ breast cancer. Breast Cancer Res. Treat. 2021, 185, 601–614. [Google Scholar] [CrossRef]
- Yates, L.R.; Knappskog, S.; Wedge, D.; Farmery, J.H.R.; Gonzalez, S.; Martincorena, I.; Alexandrov, L.B.; Van Loo, P.; Haugland, H.K.; Lilleng, P.K.; et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 2017, 32, 169–184.e167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trotter, K.W.; King, H.A.; Archer, T.K. Glucocorticoid Receptor Transcriptional Activation via the BRG1-Dependent Recruitment of TOP2beta and Ku70/86. Mol. Cell Biol. 2015, 35, 2799–2817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsia, E.Y.; Goodson, M.L.; Zou, J.X.; Privalsky, M.L.; Chen, H.W. Nuclear receptor coregulators as a new paradigm for therapeutic targeting. Adv. Drug Deliv. Rev. 2010, 62, 1227–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onate, S.A.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science 1995, 270, 1354–1357. [Google Scholar] [CrossRef]
- Carapeti, M.; Aguiar, R.C.; Chase, A.; Goldman, J.M.; Cross, N.C. Assignment of the steroid receptor coactivator-1 (SRC-1) gene to human chromosome band 2p23. Genomics 1998, 52, 242–244. [Google Scholar] [CrossRef] [PubMed]
- Anzick, S.L.; Kononen, J.; Walker, R.L.; Azorsa, D.O.; Tanner, M.M.; Guan, X.Y.; Sauter, G.; Kallioniemi, O.P.; Trent, J.M.; Meltzer, P.S. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 1997, 277, 965–968. [Google Scholar] [CrossRef] [Green Version]
- Guan, X.Y.; Xu, J.; Anzick, S.L.; Zhang, H.; Trent, J.M.; Meltzer, P.S. Hybrid selection of transcribed sequences from microdissected DNA: Isolation of genes within amplified region at 20q11-q13.2 in breast cancer. Cancer Res. 1996, 56, 3446–3450. [Google Scholar]
- Dasgupta, S.; O’Malley, B.W. Transcriptional coregulators: Emerging roles of SRC family of coactivators in disease pathology. J. Mol. Endocrinol. 2014, 53, R47–R59. [Google Scholar] [CrossRef] [Green Version]
- York, B.; Sagen, J.V.; Tsimelzon, A.; Louet, J.F.; Chopra, A.R.; Reineke, E.L.; Zhou, S.; Stevens, R.D.; Wenner, B.R.; Ilkayeva, O.; et al. Research resource: Tissue- and pathway-specific metabolomic profiles of the steroid receptor coactivator (SRC) family. Mol. Endocrinol. 2013, 27, 366–380. [Google Scholar] [CrossRef] [Green Version]
- Hsia, E.Y.; Zou, J.X.; Chen, H.W. The roles and action mechanisms of p160/SRC coactivators and the ANCCA coregulator in cancer. Prog Mol. Biol. Transl. Sci. 2009, 87, 261–298. [Google Scholar]
- Sharif, G.M.; Campbell, M.J.; Nasir, A.; Sengupta, S.; Graham, G.T.; Kushner, M.H.; Kietzman, W.B.; Schmidt, M.O.; Pearson, G.W.; Loudig, O.; et al. An AIB1 Isoform Alters Enhancer Access and Enables Progression of Early-Stage Triple-Negative Breast Cancer. Cancer Res. 2021, 81, 4230–4241. [Google Scholar] [CrossRef] [PubMed]
- Agoulnik, I.U.; Vaid, A.; Nakka, M.; Alvarado, M.; Bingman, W.E., 3rd; Erdem, H.; Frolov, A.; Smith, C.L.; Ayala, G.E.; Ittmann, M.M.; et al. Androgens modulate expression of transcription intermediary factor 2, an androgen receptor coactivator whose expression level correlates with early biochemical recurrence in prostate cancer. Cancer Res. 2006, 66, 10594–10602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-azawi, D.; Ilroy, M.M.; Kelly, G.; Redmond, A.M.; Bane, F.T.; Cocchiglia, S.; Hill, A.D.; Young, L.S. Ets-2 and p160 proteins collaborate to regulate c-Myc in endocrine resistant breast cancer. Oncogene 2008, 27, 3021–3031. [Google Scholar] [CrossRef] [Green Version]
- Louie, M.C.; Zou, J.X.; Rabinovich, A.; Chen, H.W. ACTR/AIB1 functions as an E2F1 coactivator to promote breast cancer cell proliferation and antiestrogen resistance. Mol. Cell Biol. 2004, 24, 5157–5171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.J.; Yan, J.; Luo, W.; Ayala, G.; Lin, S.H.; Erdem, H.; Ittmann, M.; Tsai, S.Y.; Tsai, M.J. SRC-3 is required for prostate cancer cell proliferation and survival. Cancer Res. 2005, 65, 7976–7983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, J.X.; Zhong, Z.; Shi, X.B.; Tepper, C.G.; de Vere White, R.W.; Kung, H.J.; Chen, H. ACTR/AIB1/SRC-3 and androgen receptor control prostate cancer cell proliferation and tumor growth through direct control of cell cycle genes. Prostate 2006, 66, 1474–1486. [Google Scholar] [CrossRef]
- Lonard, D.M.; O’Malley, B.W. Nuclear receptor coregulators: Judges, juries, and executioners of cellular regulation. Mol. Cell 2007, 27, 691–700. [Google Scholar] [CrossRef]
- Fleming, F.J.; Myers, E.; Kelly, G.; Crotty, T.B.; McDermott, E.W.; O’Higgins, N.J.; Hill, A.D.; Young, L.S. Expression of SRC-1, AIB1, and PEA3 in HER2 mediated endocrine resistant breast cancer; a predictive role for SRC-1. J. Clin. Pathol. 2004, 57, 1069–1074. [Google Scholar] [CrossRef] [Green Version]
- Gnanapragasam, V.J.; Leung, H.Y.; Pulimood, A.S.; Neal, D.E.; Robson, C.N. Expression of RAC 3, a steroid hormone receptor co-activator in prostate cancer. Br. J. Cancer 2001, 85, 1928–1936. [Google Scholar] [CrossRef]
- Gregory, C.W.; He, B.; Johnson, R.T.; Ford, O.H.; Mohler, J.L.; French, F.S.; Wilson, E.M. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. 2001, 61, 4315–4319. [Google Scholar]
- Harigopal, M.; Heymann, J.; Ghosh, S.; Anagnostou, V.; Camp, R.L.; Rimm, D.L. Estrogen receptor co-activator (AIB1) protein expression by automated quantitative analysis (AQUA) in a breast cancer tissue microarray and association with patient outcome. Breast Cancer Res. Treat. 2009, 115, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Bardou, V.; Hopp, T.A.; Chamness, G.C.; Hilsenbeck, S.G.; Fuqua, S.A.; Wong, J.; Allred, D.C.; Clark, G.M.; Schiff, R. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J. Natl. Cancer Inst. 2003, 95, 353–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Erdem, H.; Li, R.; Cai, Y.; Ayala, G.; Ittmann, M.; Yu-Lee, L.Y.; Tsai, S.Y.; Tsai, M.J. Steroid receptor coactivator-3/AIB1 promotes cell migration and invasiveness through focal adhesion turnover and matrix metalloproteinase expression. Cancer Res. 2008, 68, 5460–5468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkner, S.; Bendahl, P.O.; Grabau, D.; Lövgren, K.; Stål, O.; Rydén, L.; Fernö, M. AIB1 is a predictive factor for tamoxifen response in premenopausal women. Ann. Oncol. 2010, 21, 238–244. [Google Scholar] [CrossRef]
- Iwase, H.; Omoto, Y.; Toyama, T.; Yamashita, H.; Hara, Y.; Sugiura, H.; Zhang, Z. Clinical significance of AIB1 expression in human breast cancer. Breast Cancer Res. Treat. 2003, 80, 339–345. [Google Scholar] [CrossRef]
- Jiang, L.; Ren, L.; Chen, H.; Pan, J.; Zhang, Z.; Kuang, X.; Chen, X.; Bao, W.; Lin, C.; Zhou, Z.; et al. NCAPG confers trastuzumab resistance via activating SRC/STAT3 signaling pathway in HER2-positive breast cancer. Cell Death Dis. 2020, 11, 547. [Google Scholar] [CrossRef]
- Shou, J.; Massarweh, S.; Osborne, C.K.; Wakeling, A.E.; Ali, S.; Weiss, H.; Schiff, R. Mechanisms of tamoxifen resistance: Increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J. Natl. Cancer Inst. 2004, 96, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Zhang, Q.; Kang, X.; Jin, S.; Lou, C. AIB1 is required for the acquisition of epithelial growth factor receptor-mediated tamoxifen resistance in breast cancer cells. Biochem. Biophys. Res. Commun. 2009, 380, 699–704. [Google Scholar] [CrossRef]
- Agoulnik, I.U.; Vaid, A.; Bingman, W.E., 3rd; Erdeme, H.; Frolov, A.; Smith, C.L.; Ayala, G.; Ittmann, M.M.; Weigel, N.L. Role of SRC-1 in the promotion of prostate cancer cell growth and tumor progression. Cancer Res. 2005, 65, 7959–7967. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Chen, J.; Lu, D.; Jain, P.; Yu, Y.; Cardenas, D.; Peng, X.; Yu, X.; Xu, J.; Wang, J.; et al. Development of improved SRC-3 inhibitors as breast cancer therapeutic agents. Endocr. Relat. Cancer 2021, 28, 657–670. [Google Scholar] [CrossRef]
- Cheng, S.; Douglas-Jones, A.; Yang, X.; Mansel, R.E.; Jiang, W.G. Transforming acidic coiled-coil-containing protein 2 (TACC2) in human breast cancer, expression pattern and clinical/prognostic relevance. Cancer Genom. Proteom. 2010, 7, 67–73. [Google Scholar]
- Piekorz, R.P.; Hoffmeyer, A.; Duntsch, C.D.; McKay, C.; Nakajima, H.; Sexl, V.; Snyder, L.; Rehg, J.; Ihle, J.N. The centrosomal protein TACC3 is essential for hematopoietic stem cell function and genetically interfaces with p53-regulated apoptosis. EMBO J. 2002, 21, 653–664. [Google Scholar] [CrossRef] [Green Version]
- Conte, N.; Charafe-Jauffret, E.; Delaval, B.; Adélaïde, J.; Ginestier, C.; Geneix, J.; Isnardon, D.; Jacquemier, J.; Birnbaum, D. Carcinogenesis and translational controls: TACC1 is down-regulated in human cancers and associates with mRNA regulators. Oncogene 2002, 21, 5619–5630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conte, N.; Delaval, B.; Ginestier, C.; Ferrand, A.; Isnardon, D.; Larroque, C.; Prigent, C.; Séraphin, B.; Jacquemier, J.; Birnbaum, D. TACC1-chTOG-Aurora A protein complex in breast cancer. Oncogene 2003, 22, 8102–8116. [Google Scholar] [CrossRef] [Green Version]
- Delaval, B.; Ferrand, A.; Conte, N.; Larroque, C.; Hernandez-Verdun, D.; Prigent, C.; Birnbaum, D. Aurora B -TACC1 protein complex in cytokinesis. Oncogene 2004, 23, 4516–4522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabillard, J.C.; Ulisse, S.; Baldini, E.; Sorrenti, S.; Cremet, J.Y.; Coccaro, C.; Prigent, C.; D’Armiento, M.; Arlot-Bonnemains, Y. Aurora-C interacts with and phosphorylates the transforming acidic coiled-coil 1 protein. Biochem. Biophys. Res. Commun. 2011, 408, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.; Sabiha, B.; Jan, H.U.; Haider, S.A.; Khan, A.A.; Ali, S.S. Genetic etiology of oral cancer. Oral Oncol 2017, 70, 23–28. [Google Scholar] [CrossRef]
- Ding, A.; Zhao, W.; Shi, X.; Yao, R.; Zhou, F.; Yue, L.; Liu, S.; Qiu, W. Impact of NPM, TFF3 and TACC1 on the prognosis of patients with primary gastric cancer. PLoS ONE 2013, 8, e82136. [Google Scholar] [CrossRef]
- Xiang, L.; Li, J.; Jiang, W.; Shen, X.; Yang, W.; Wu, X.; Yang, H. Comprehensive analysis of targetable oncogenic mutations in chinese cervical cancers. Oncotarget 2015, 6, 4968–4975. [Google Scholar] [CrossRef] [Green Version]
- Petrie, K.; Urban-Wojciuk, Z.; Sbirkov, Y.; Graham, A.; Hamann, A.; Brown, G. Retinoic acid receptor gamma is a therapeutically targetable driver of growth and survival in prostate cancer. Cancer Rep. 2020, 3, e1284. [Google Scholar]
- Cai, K.; Gudas, L.J. Retinoic acid receptors and GATA transcription factors activate the transcription of the human lecithin:retinol acyltransferase gene. Int. J. Biochem. Cell Biol. 2009, 41, 546–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Knutti, D.; Kaul, A.; Kralli, A. A tissue-specific coactivator of steroid receptors, identified in a functional genetic screen. Mol. Cell Biol. 2000, 20, 2411–2422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerin, C.; Rodgers, J.T.; Kalume, D.E.; Kim, S.H.; Pandey, A.; Puigserver, P. GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1alpha. Cell Metab. 2006, 3, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Lawson, D.E.; Charman, M.; Wilson, P.W.; Edelstein, S. Some characteristics of new tissue-binding proteins for metabolites of vitamin D other than 1,25-dihydroxyvitamin D. Biochim. Biophys. Acta 1976, 437, 403–415. [Google Scholar] [CrossRef]
- Shiota, M.; Yokomizo, A.; Tada, Y.; Inokuchi, J.; Tatsugami, K.; Kuroiwa, K.; Uchiumi, T.; Fujimoto, N.; Seki, N.; Naito, S. Peroxisome proliferator-activated receptor gamma coactivator-1alpha interacts with the androgen receptor (AR) and promotes prostate cancer cell growth by activating the AR. Mol. Endocrinol. 2010, 24, 114–127. [Google Scholar] [CrossRef]
- LaGory, E.L.; Wu, C.; Taniguchi, C.M.; Ding, C.C.; Chi, J.T.; von Eyben, R.; Scott, D.A.; Richardson, A.D.; Giaccia, A.J. Suppression of PGC-1alpha Is Critical for Reprogramming Oxidative Metabolism in Renal Cell Carcinoma. Cell Rep. 2015, 12, 116–127. [Google Scholar] [CrossRef] [Green Version]
- Zu, Y.; Chen, X.F.; Li, Q.; Zhang, S.T.; Si, L.N. PGC-1α activates SIRT3 to modulate cell proliferation and glycolytic metabolism in breast cancer. Neoplasma 2021, 68, 352–361. [Google Scholar] [CrossRef]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, R.; Manzar, N.; Bhatia, V.; Yadav, A.; Nengroo, M.A.; Datta, D.; Carskadon, S.; Gupta, N.; Sigouros, M.; Khani, F.; et al. Androgen deprivation upregulates SPINK1 expression and potentiates cellular plasticity in prostate cancer. Nat. Commun. 2020, 11, 384. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.S.; Evgrafov, O.V.; Souaiaia, T.; Bonyad, A.; Herstein, J.; Lee, J.Y.; Kim, J.; Ning, Y.; Sixto, M.; Weitz, A.C.; et al. Non-coding RNAs derived from an alternatively spliced REST transcript (REST-003) regulate breast cancer invasiveness. Sci. Rep. 2015, 5, 11207. [Google Scholar] [CrossRef] [Green Version]
- Svensson, C.; Ceder, J.; Iglesias-Gato, D.; Chuan, Y.C.; Pang, S.T.; Bjartell, A.; Martinez, R.M.; Bott, L.; Helczynski, L.; Ulmert, D.; et al. REST mediates androgen receptor actions on gene repression and predicts early recurrence of prostate cancer. Nucleic Acids Res. 2014, 42, 999–1015. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, P.; Westbrook, T.F.; Ottinger, M.; Pavlova, N.; Chang, B.; Macia, E.; Shi, Y.J.; Barretina, J.; Liu, J.; Howley, P.M.; et al. CDYL bridges REST and histone methyltransferases for gene repression and suppression of cellular transformation. Mol. Cell 2008, 32, 718–726. [Google Scholar] [CrossRef]
- Lempiainen, J.K.; Niskanen, E.A.; Vuoti, K.M.; Lampinen, R.E.; Goos, H.; Varjosalo, M.; Palvimo, J.J. Agonist-specific Protein Interactomes of Glucocorticoid and Androgen Receptor as Revealed by Proximity Mapping. Mol. Cell Proteom. 2017, 16, 1462–1474. [Google Scholar] [CrossRef] [Green Version]
- Tong, D.; Zhang, J.; Wang, X.; Li, Q.; Liu, L.Y.; Yang, J.; Guo, B.; Ni, L.; Zhao, L.; Huang, C. MeCP2 facilitates breast cancer growth via promoting ubiquitination-mediated P53 degradation by inhibiting RPL5/RPL11 transcription. Oncogenesis 2020, 9, 56. [Google Scholar] [CrossRef]
- Luo, D.; Ge, W. MeCP2 Promotes Colorectal Cancer Metastasis by Modulating ZEB1 Transcription. Cancers 2020, 12, 758. [Google Scholar] [CrossRef] [Green Version]
- Bian, E.; Chen, X.; Xu, Y.; Ji, X.; Cheng, M.; Wang, H.; Fang, Z.; Zhao, B. A central role for MeCP2 in the epigenetic repression of miR-200c during epithelial-to-mesenchymal transition of glioma. J. Exp. Clin. Cancer Res. 2019, 38, 366. [Google Scholar] [CrossRef]
- Yang, C.; Wu, J.; Liu, X.; Wang, Y.; Liu, B.; Chen, X.; Wu, X.; Yan, D.; Han, L.; Liu, S.; et al. Circadian Rhythm Is Disrupted by ZNF704 in Breast Carcinogenesis. Cancer Res. 2020, 80, 4114–4128. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Huang, W.; Qiu, R.; Liu, R.; Zeng, Y.; Gao, J.; Zheng, Y.; Hou, Y.; Wang, S.; Yu, W.; et al. LSD1 coordinates with the SIN3A/HDAC complex and maintains sensitivity to chemotherapy in breast cancer. J. Mol. Cell Biol. 2018, 10, 285–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Zhang, Z.; Liu, X.; Cheng, X.; Zhang, Y.; Han, X.; Zhang, Y.; Liu, S.; Yang, J.; Xu, B.; et al. The FOXN3-NEAT1-SIN3A repressor complex promotes progression of hormonally responsive breast cancer. J. Clin. Investig. 2017, 127, 3421–3440. [Google Scholar] [CrossRef] [PubMed]
- Neupane, M.; Clark, A.P.; Landini, S.; Birkbak, N.J.; Eklund, A.C.; Lim, E.; Culhane, A.C.; Barry, W.T.; Schumacher, S.E.; Beroukhim, R.; et al. MECP2 Is a Frequently Amplified Oncogene with a Novel Epigenetic Mechanism That Mimics the Role of Activated RAS in Malignancy. Cancer Discov. 2016, 6, 45–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.; Sun, Z. 5’TG3’ interacting factor interacts with Sin3A and represses AR-mediated transcription. Mol. Endocrinol. 2001, 15, 1918–1928. [Google Scholar] [PubMed]
- Kemper, J.K.; Kim, H.; Miao, J.; Bhalla, S.; Bae, Y. Role of an mSin3A-Swi/Snf chromatin remodeling complex in the feedback repression of bile acid biosynthesis by SHP. Mol. Cell Biol. 2004, 24, 7707–7719. [Google Scholar] [CrossRef] [Green Version]
- Sharma, D.; Saxena, N.K.; Davidson, N.E.; Vertino, P.M. Restoration of tamoxifen sensitivity in estrogen receptor-negative breast cancer cells: Tamoxifen-bound reactivated ER recruits distinctive corepressor complexes. Cancer Res. 2006, 66, 6370–6378. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.O.; Kang, H.J. Role of coactivators and corepressors in the induction of the RARbeta gene in human colon cancer cells. Biol. Pharm. Bull. 2002, 25, 1298–1302. [Google Scholar] [CrossRef] [Green Version]
- Asim, M.; Hafeez, B.B.; Siddiqui, I.A.; Gerlach, C.; Patz, M.; Mukhtar, H.; Baniahmad, A. Ligand-dependent corepressor acts as a novel androgen receptor corepressor, inhibits prostate cancer growth, and is functionally inactivated by the Src protein kinase. J. Biol. Chem. 2011, 286, 37108–37117. [Google Scholar] [CrossRef] [Green Version]
- White, J.H.; Fernandes, I.; Mader, S.; Yang, X.J. Corepressor recruitment by agonist-bound nuclear receptors. Vitam Horm. 2004, 68, 123–143. [Google Scholar]
- Klotz-Noack, K.; Klinger, B.; Rivera, M.; Bublitz, N.; Uhlitz, F.; Riemer, P.; Luthen, M.; Sell, T.; Kasack, K.; Gastl, B.; et al. SFPQ Depletion Is Synthetically Lethal with BRAF(V600E) in Colorectal Cancer Cells. Cell Rep. 2020, 32, 108184. [Google Scholar] [CrossRef] [PubMed]
- Mitobe, Y.; Iino, K.; Takayama, K.I.; Ikeda, K.; Suzuki, T.; Aogi, K.; Kawabata, H.; Suzuki, Y.; Horie-Inoue, K.; Inoue, S. PSF Promotes ER-Positive Breast Cancer Progression via Posttranscriptional Regulation of ESR1 and SCFD2. Cancer Res. 2020, 80, 2230–2242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Silva, H.C.; Lin, M.Z.; Phillips, L.; Martin, J.L.; Baxter, R.C. IGFBP-3 interacts with NONO and SFPQ in PARP-dependent DNA damage repair in triple-negative breast cancer. Cell Mol. Life Sci. 2019, 76, 2015–2030. [Google Scholar] [CrossRef] [PubMed]
- Mathur, M.; Tucker, P.W.; Samuels, H.H. PSF is a novel corepressor that mediates its effect through Sin3A and the DNA binding domain of nuclear hormone receptors. Mol. Cell Biol. 2001, 21, 2298–2311. [Google Scholar] [CrossRef] [Green Version]
- Andres, M.E.; Burger, C.; Peral-Rubio, M.J.; Battaglioli, E.; Anderson, M.E.; Grimes, J.; Dallman, J.; Ballas, N.; Mandel, G. CoREST: A functional corepressor required for regulation of neural-specific gene expression. Proc. Natl. Acad. Sci. USA 1999, 96, 9873–9878. [Google Scholar] [CrossRef] [Green Version]
- Metzger, E.; Wissmann, M.; Yin, N.; Muller, J.M.; Schneider, R.; Peters, A.H.; Gunther, T.; Buettner, R.; Schule, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef]
- Lee, M.G.; Wynder, C.; Cooch, N.; Shiekhattar, R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 2005, 437, 432–435. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.; Goldman, D.C.; Nechiporuk, T.; Kawane, S.; McWeeney, S.K.; Tyner, J.W.; Fan, G.; Kerenyi, M.A.; Orkin, S.H.; Fleming, W.H.; et al. Corepressor Rcor1 is essential for murine erythropoiesis. Blood 2014, 123, 3175–3184. [Google Scholar] [CrossRef] [Green Version]
- Horlein, A.J.; Naar, A.M.; Heinzel, T.; Torchia, J.; Gloss, B.; Kurokawa, R.; Ryan, A.; Kamei, Y.; Soderstrom, M.; Glass, C.K.; et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature 1995, 377, 397–404. [Google Scholar] [CrossRef]
- Chen, J.D.; Evans, R.M. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature 1995, 377, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Jepsen, K.; Hermanson, O.; Onami, T.M.; Gleiberman, A.S.; Lunyak, V.; McEvilly, R.J.; Kurokawa, R.; Kumar, V.; Liu, F.; Seto, E.; et al. Combinatorial roles of the nuclear receptor corepressor in transcription and development. Cell 2000, 102, 753–763. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Williams, E.G.; Mouchiroud, L.; Canto, C.; Fan, W.; Downes, M.; Heligon, C.; Barish, G.D.; Desvergne, B.; Evans, R.M.; et al. NCoR1 is a conserved physiological modulator of muscle mass and oxidative function. Cell 2011, 147, 827–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jepsen, K.; Solum, D.; Zhou, T.; McEvilly, R.J.; Kim, H.J.; Glass, C.K.; Hermanson, O.; Rosenfeld, M.G. SMRT-mediated repression of an H3K27 demethylase in progression from neural stem cell to neuron. Nature 2007, 450, 415–419. [Google Scholar] [CrossRef]
- Mottis, A.; Mouchiroud, L.; Auwerx, J. Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev. 2013, 27, 819–835. [Google Scholar] [CrossRef] [Green Version]
- Perissi, V.; Jepsen, K.; Glass, C.K.; Rosenfeld, M.G. Deconstructing repression: Evolving models of co-repressor action. Nat. Rev. Genet. 2010, 11, 109–123. [Google Scholar] [CrossRef]
- Stanya, K.J.; Kao, H.Y. New insights into the functions and regulation of the transcriptional corepressors SMRT and N-CoR. Cell Div. 2009, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Ordentlich, P.; Downes, M.; Evans, R.M. Corepressors and nuclear hormone receptor function. Curr. Top. Microbiol. Immunol. 2001, 254, 101–116. [Google Scholar]
- Yang, W.M.; Tsai, S.C.; Wen, Y.D.; Fejer, G.; Seto, E. Functional domains of histone deacetylase-3. J. Biol. Chem. 2002, 277, 9447–9454. [Google Scholar] [CrossRef] [Green Version]
- Tiefenbach, J.; Novac, N.; Ducasse, M.; Eck, M.; Melchior, F.; Heinzel, T. SUMOylation of the corepressor N-CoR modulates its capacity to repress transcription. Mol. Biol. Cell 2006, 17, 1643–1651. [Google Scholar] [CrossRef] [Green Version]
- Privalsky, M.L.; Goodson, M.L. Evolution of NCoR-1 and NCoR-2 corepressor alternative mRNA splicing in placental mammals. BMC Res. Notes 2019, 12, 343. [Google Scholar] [CrossRef] [PubMed]
- Short, S.; Peterkin, T.; Guille, M.; Patient, R.; Sharpe, C. Short linear motif acquisition, exon formation and alternative splicing determine a pathway to diversity for NCoR-family co-repressors. Open Biol. 2015, 5, 150063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muscat, G.E.; Burke, L.J.; Downes, M. The corepressor N-CoR and its variants RIP13a and RIP13Delta1 directly interact with the basal transcription factors TFIIB, TAFII32 and TAFII70. Nucleic Acids Res. 1998, 26, 2899–2907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodson, M.L.; Jonas, B.A.; Privalsky, M.L. Alternative mRNA splicing of SMRT creates functional diversity by generating corepressor isoforms with different affinities for different nuclear receptors. J. Biol. Chem. 2005, 280, 7493–7503. [Google Scholar] [CrossRef] [Green Version]
- Jonas, B.A.; Varlakhanova, N.; Hayakawa, F.; Goodson, M.; Privalsky, M.L. Response of SMRT (silencing mediator of retinoic acid and thyroid hormone receptor) and N-CoR (nuclear receptor corepressor) corepressors to mitogen-activated protein kinase kinase kinase cascades is determined by alternative mRNA splicing. Mol. Endocrinol. 2007, 21, 1924–1939. [Google Scholar] [CrossRef] [PubMed]
- Jonas, B.A.; Privalsky, M.L. SMRT and N-CoR corepressors are regulated by distinct kinase signaling pathways. J. Biol. Chem. 2004, 279, 54676–54686. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.H.; Privalsky, M.L. The SMRT corepressor is regulated by a MEK-1 kinase pathway: Inhibition of corepressor function is associated with SMRT phosphorylation and nuclear export. Mol. Cell Biol. 2000, 20, 6612–6625. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Wong, C.W.; Privalsky, M.L. Signaling by tyrosine kinases negatively regulates the interaction between transcription factors and SMRT (silencing mediator of retinoic acid and thyroid hormone receptor) corepressor. Mol. Endocrinol. 1998, 12, 1161–1171. [Google Scholar] [CrossRef]
- Sternberg, P.W.; Stern, M.J.; Clark, I.; Herskowitz, I. Activation of the yeast HO gene by release from multiple negative controls. Cell 1987, 48, 567–577. [Google Scholar] [CrossRef]
- Nasmyth, K.; Stillman, D.; Kipling, D. Both positive and negative regulators of HO transcription are required for mother-cell-specific mating-type switching in yeast. Cell 1987, 48, 579–587. [Google Scholar] [CrossRef]
- Wang, H.; Stillman, D.J. In vitro regulation of a SIN3-dependent DNA-binding activity by stimulatory and inhibitory factors. Proc. Natl. Acad. Sci. USA 1990, 87, 9761–9765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Radhakrishnan, I. Solution NMR studies of apo-mSin3A and -mSin3B reveal that the PAH1 and PAH2 domains are structurally independent. Protein Sci. 2008, 17, 171–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dannenberg, J.H.; David, G.; Zhong, S.; van der Torre, J.; Wong, W.H.; Depinho, R.A. mSin3A corepressor regulates diverse transcriptional networks governing normal and neoplastic growth and survival. Genes Dev. 2005, 19, 1581–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Guezennec, X.; Vermeulen, M.; Stunnenberg, H.G. Molecular characterization of Sin3 PAH-domain interactor specificity and identification of PAH partners. Nucleic Acids Res. 2006, 34, 3929–3937. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Sweet, J.; Challis, J.R.; Brown, T.; Lye, S.J. Transcriptional activity of androgen receptor is modulated by two RNA splicing factors, PSF and p54nrb. Mol. Cell Biol. 2007, 27, 4863–4875. [Google Scholar] [CrossRef] [Green Version]
- Awad, T.A.; Bigler, J.; Ulmer, J.E.; Hu, Y.J.; Moore, J.M.; Lutz, M.; Neiman, P.E.; Collins, S.J.; Renkawitz, R.; Lobanenkov, V.V.; et al. Negative transcriptional regulation mediated by thyroid hormone response element 144 requires binding of the multivalent factor CTCF to a novel target DNA sequence. J. Biol. Chem. 1999, 274, 27092–27098. [Google Scholar] [CrossRef] [Green Version]
- Bigler, J.; Eisenman, R.N. Novel location and function of a thyroid hormone response element. EMBO J. 1995, 14, 5710–5723. [Google Scholar] [CrossRef]
- Lutz, M.; Baniahmad, A.; Renkawitz, R. Modulation of thyroid hormone receptor silencing function by co-repressors and a synergizing transcription factor. Biochem. Soc. Trans. 2000, 28, 386–389. [Google Scholar] [CrossRef]
- Ellison-Zelski, S.J.; Alarid, E.T. Maximum growth and survival of estrogen receptor-alpha positive breast cancer cells requires the Sin3A transcriptional repressor. Mol. Cancer 2010, 9, 263. [Google Scholar] [CrossRef] [Green Version]
- Ellison-Zelski, S.J.; Solodin, N.M.; Alarid, E.T. Repression of ESR1 through actions of estrogen receptor alpha and Sin3A at the proximal promoter. Mol. Cell Biol. 2009, 29, 4949–4958. [Google Scholar] [CrossRef] [Green Version]
- Moehren, U.; Dressel, U.; Reeb, C.A.; Vaisanen, S.; Dunlop, T.W.; Carlberg, C.; Baniahmad, A. The highly conserved region of the co-repressor Sin3A functionally interacts with the co-repressor Alien. Nucleic Acids Res. 2004, 32, 2995–3004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polly, P.; Herdick, M.; Moehren, U.; Baniahmad, A.; Heinzel, T.; Carlberg, C. VDR-Alien: A novel, DNA-selective vitamin D(3) receptor-corepressor partnership. FASEB J. 2000, 14, 1455–1463. [Google Scholar] [PubMed]
- Nagase, T.; Nakayama, M.; Nakajima, D.; Kikuno, R.; Ohara, O. Prediction of the coding sequences of unidentified human genes. XX. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res. 2001, 8, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalom-Barak, T.; Liersemann, J.; Memari, B.; Flechner, L.; Devor, C.E.; Bernardo, T.M.; Kim, S.; Matsumoto, N.; Friedman, S.L.; Evans, R.M.; et al. Ligand-Dependent Corepressor (LCoR) Is a Rexinoid-Inhibited Peroxisome Proliferator-Activated Receptor gamma-Retinoid X Receptor alpha Coactivator. Mol. Cell Biol. 2018, 38, e00107-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palijan, A.; Fernandes, I.; Verway, M.; Kourelis, M.; Bastien, Y.; Tavera-Mendoza, L.E.; Sacheli, A.; Bourdeau, V.; Mader, S.; White, J.H. Ligand-dependent corepressor LCoR is an attenuator of progesterone-regulated gene expression. J. Biol. Chem. 2009, 284, 30275–30287. [Google Scholar] [CrossRef] [Green Version]
- Palijan, A.; Fernandes, I.; Bastien, Y.; Tang, L.; Verway, M.; Kourelis, M.; Tavera-Mendoza, L.E.; Li, Z.; Bourdeau, V.; Mader, S.; et al. Function of histone deacetylase 6 as a cofactor of nuclear receptor coregulator LCoR. J. Biol. Chem. 2009, 284, 30264–30274. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, I.; Bastien, Y.; Wai, T.; Nygard, K.; Lin, R.; Cormier, O.; Lee, H.S.; Eng, F.; Bertos, N.R.; Pelletier, N.; et al. Ligand-dependent nuclear receptor corepressor LCoR functions by histone deacetylase-dependent and -independent mechanisms. Mol. Cell 2003, 11, 139–150. [Google Scholar] [CrossRef]
- Jalaguier, S.; Teyssier, C.; Nait Achour, T.; Lucas, A.; Bonnet, S.; Rodriguez, C.; Elarouci, N.; Lapierre, M.; Cavailles, V. Complex regulation of LCoR signaling in breast cancer cells. Oncogene 2017, 36, 4790–4801. [Google Scholar] [CrossRef]
- Calderon, M.R.; Verway, M.; An, B.S.; DiFeo, A.; Bismar, T.A.; Ann, D.K.; Martignetti, J.A.; Shalom-Barak, T.; White, J.H. Ligand-dependent corepressor (LCoR) recruitment by Kruppel-like factor 6 (KLF6) regulates expression of the cyclin-dependent kinase inhibitor CDKN1A gene. J. Biol. Chem. 2012, 287, 8662–8674. [Google Scholar] [CrossRef] [Green Version]
- Kwok, R.P.; Lundblad, J.R.; Chrivia, J.C.; Richards, J.P.; Bachinger, H.P.; Brennan, R.G.; Roberts, S.G.; Green, M.R.; Goodman, R.H. Nuclear protein CBP is a coactivator for the transcription factor CREB. Nature 1994, 370, 223–226. [Google Scholar] [CrossRef]
- Smith, C.L.; Onate, S.A.; Tsai, M.J.; O’Malley, B.W. CREB binding protein acts synergistically with steroid receptor coactivator-1 to enhance steroid receptor-dependent transcription. Proc. Natl. Acad. Sci. USA 1996, 93, 8884–8888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heery, D.M.; Kalkhoven, E.; Hoare, S.; Parker, M.G. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 1997, 387, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Kamei, Y.; Xu, L.; Heinzel, T.; Torchia, J.; Kurokawa, R.; Gloss, B.; Lin, S.C.; Heyman, R.A.; Rose, D.W.; Glass, C.K.; et al. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell 1996, 85, 403–414. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Cummings, R.; Li, Y.; Mitra, S.; Wilkinson, H.A.; Elbrecht, A.; Hermes, J.D.; Schaeffer, J.M.; Smith, R.G.; Moller, D.E. Nuclear receptors have distinct affinities for coactivators: Characterization by fluorescence resonance energy transfer. Mol. Endocrinol. 1998, 12, 1594–1604. [Google Scholar] [CrossRef]
- McKenna, N.J.; Nawaz, Z.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. Distinct steady-state nuclear receptor coregulator complexes exist in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 11697–11702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goo, Y.H.; Na, S.Y.; Zhang, H.; Xu, J.; Hong, S.; Cheong, J.; Lee, S.K.; Lee, J.W. Interactions between activating signal cointegrator-2 and the tumor suppressor retinoblastoma in androgen receptor transactivation. J. Biol. Chem. 2004, 279, 7131–7135. [Google Scholar] [CrossRef] [Green Version]
- Altwegg, K.A.; Vadlamudi, R.K. Role of estrogen receptor coregulators in endocrine resistant breast cancer. Explor. Target. Antitumor Ther. 2021, 2, 385–400. [Google Scholar] [CrossRef]
- Turner, N.C.; Neven, P.; Loibl, S.; Andre, F. Advances in the treatment of advanced oestrogen-receptor-positive breast cancer. Lancet 2017, 389, 2403–2414. [Google Scholar] [CrossRef]
- Musgrove, E.A.; Sutherland, R.L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 2009, 9, 631–643. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Z.; Li, M.; Tang, W.; Pratap, U.P.; Luo, Y.; Altwegg, K.A.; Li, X.; Zou, Y.; Zhu, H.; et al. Interaction of transcription factor AP-2 gamma with proto-oncogene PELP1 promotes tumorigenesis by enhancing RET signaling. Mol. Oncol. 2021, 15, 1146–1161. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, Z.; Cenciarini, M.E.; Proietti, C.J.; Amasino, M.; Hong, T.; Yang, M.; Liao, Y.; Chiang, H.C.; Kaklamani, V.G.; et al. Tamoxifen Resistance in Breast Cancer Is Regulated by the EZH2-ERalpha-GREB1 Transcriptional Axis. Cancer Res. 2018, 78, 671–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Wei, X.; Tufan, T.; Kuscu, C.; Unlu, H.; Farooq, S.; Demirtas, E.; Paschal, B.M.; Adli, M. Recurrent mutations at estrogen receptor binding sites alter chromatin topology and distal gene expression in breast cancer. Genome Biol. 2018, 19, 190. [Google Scholar] [CrossRef] [PubMed]
- Toy, W.; Weir, H.; Razavi, P.; Lawson, M.; Goeppert, A.U.; Mazzola, A.M.; Smith, A.; Wilson, J.; Morrow, C.; Wong, W.L.; et al. Activating ESR1 Mutations Differentially Affect the Efficacy of ER Antagonists. Cancer Discov. 2017, 7, 277–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeselsohn, R.; Buchwalter, G.; De Angelis, C.; Brown, M.; Schiff, R. ESR1 mutations-a mechanism for acquired endocrine resistance in breast cancer. Nat. Rev. Clin. Oncol. 2015, 12, 573–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Coons, L.A.; Houtman, R.; Carlson, K.E.; Martin, T.A.; Mayne, C.G.; Melchers, D.; Jefferson, T.B.; Ramsey, J.T.; Katzenellenbogen, J.A.; et al. A mutant form of ERalpha associated with estrogen insensitivity affects the coupling between ligand binding and coactivator recruitment. Sci. Signal. 2020, 13, eaaw4653. [Google Scholar] [CrossRef] [PubMed]
- Fanning, S.W.; Mayne, C.G.; Dharmarajan, V.; Carlson, K.E.; Martin, T.A.; Novick, S.J.; Toy, W.; Green, B.; Panchamukhi, S.; Katzenellenbogen, B.S.; et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. Elife 2016, 5, e12792. [Google Scholar] [CrossRef]
- Magne Nde, C.B.; Casas Gimeno, G.; Docanto, M.; Knower, K.C.; Young, M.J.; Buehn, J.; Sayed, E.; Clyne, C.D. Timeless Is a Novel Estrogen Receptor Co-activator Involved in Multiple Signaling Pathways in MCF-7 Cells. J. Mol. Biol. 2018, 430, 1531–1543. [Google Scholar] [CrossRef]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Tang, Q.; Sun, M.; Chun, J.Y.; Evans, C.P.; Gao, A.C. Interleukin-6 increases prostate cancer cells resistance to bicalutamide via TIF2. Mol. Cancer Ther. 2009, 8, 665–671. [Google Scholar] [CrossRef] [Green Version]
- Watson, R.W.; Azam, H.; Aura, C.; Russell, N.; McCormack, J.; Corey, E.; Morrissey, C.; Crown, J.; Gallagher, W.M.; Prencipe, M. Inhibition of Serum Response Factor Improves Response to Enzalutamide in Prostate Cancer. Cancers 2020, 12, 3540. [Google Scholar] [CrossRef]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hara, Y.; Kobayashi, S.; Iwase, H. NCOR1 mRNA is an independent prognostic factor for breast cancer. Cancer Lett. 2006, 237, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Girault, I.; Lerebours, F.; Amarir, S.; Tozlu, S.; Tubiana-Hulin, M.; Lidereau, R.; Bieche, I. Expression analysis of estrogen receptor alpha coregulators in breast carcinoma: Evidence that NCOR1 expression is predictive of the response to tamoxifen. Clin. Cancer Res. 2003, 9, 1259–1266. [Google Scholar] [PubMed]
- Kim, J.Y.; Son, Y.L.; Lee, Y.C. Involvement of SMRT corepressor in transcriptional repression by the vitamin D receptor. Mol. Endocrinol. 2009, 23, 251–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanim, F.L.; Gommersall, L.M.; Wood, V.H.; Smith, K.L.; Montalvo, L.; O’Neill, L.P.; Xu, Y.; Peehl, D.M.; Stewart, P.M.; Turner, B.M.; et al. Altered SMRT levels disrupt vitamin D3 receptor signalling in prostate cancer cells. Oncogene 2004, 23, 6712–6725. [Google Scholar] [CrossRef] [Green Version]
- Trtkova, K.; Paskova, L.; Matijescukova, N.; Kolar, Z. Formation of AR-SMRT binding in prostate cancer cells treated with natural histone deacetylase inhibitor. Cancer Biomark. 2010, 7, 79–90. [Google Scholar] [CrossRef]
- Quigley, D.A.; Dang, H.X.; Zhao, S.G.; Lloyd, P.; Aggarwal, R.; Alumkal, J.J.; Foye, A.; Kothari, V.; Perry, M.D.; Bailey, A.M.; et al. Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 2018, 174, 758–769.e759. [Google Scholar] [CrossRef] [Green Version]
- Viswanathan, S.R.; Ha, G.; Hoff, A.M.; Wala, J.A.; Carrot-Zhang, J.; Whelan, C.W.; Haradhvala, N.J.; Freeman, S.S.; Reed, S.C.; Rhoades, J.; et al. Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing. Cell 2018, 174, 433–447.e419. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Perez, A.A.; Hazelett, D.J.; Coetzee, G.A.; Rhie, S.K.; Farnham, P.J. CRISPR-mediated deletion of prostate cancer risk-associated CTCF loop anchors identifies repressive chromatin loops. Genome Biol. 2018, 19, 160. [Google Scholar] [CrossRef] [PubMed]
- Rhie, S.K.; Perez, A.A.; Lay, F.D.; Schreiner, S.; Shi, J.; Polin, J.; Farnham, P.J. A high-resolution 3D epigenomic map reveals insights into the creation of the prostate cancer transcriptome. Nat. Commun. 2019, 10, 4154. [Google Scholar] [CrossRef] [Green Version]
- Taberlay, P.C.; Achinger-Kawecka, J.; Lun, A.T.; Buske, F.A.; Sabir, K.; Gould, C.M.; Zotenko, E.; Bert, S.A.; Giles, K.A.; Bauer, D.C.; et al. Three-dimensional disorganization of the cancer genome occurs coincident with long-range genetic and epigenetic alterations. Genome Res. 2016, 26, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Flavahan, W.A.; Drier, Y.; Johnstone, S.E.; Hemming, M.L.; Tarjan, D.R.; Hegazi, E.; Shareef, S.J.; Javed, N.M.; Raut, C.P.; Eschle, B.K.; et al. Altered chromosomal topology drives oncogenic programs in SDH-deficient GISTs. Nature 2019, 575, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.C.; Fennell, K.A.; Chan, Y.C.; Rambow, F.; Yeung, M.M.; Vassiliadis, D.; Lara, L.; Yeh, P.; Martelotto, L.G.; Rogiers, A.; et al. Targeting enhancer switching overcomes non-genetic drug resistance in acute myeloid leukaemia. Nat. Commun. 2019, 10, 2723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labbe, D.P.; Sweeney, C.J.; Brown, M.; Galbo, P.; Rosario, S.; Wadosky, K.M.; Ku, S.Y.; Sjostrom, M.; Alshalalfa, M.; Erho, N.; et al. TOP2A and EZH2 Provide Early Detection of an Aggressive Prostate Cancer Subgroup. Clin. Cancer Res. 2017, 23, 7072–7083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotinen, M.; You, S.; Yang, J.; Coetzee, S.G.; Reis-Sobreiro, M.; Huang, W.C.; Huang, F.; Pan, X.; Yanez, A.; Hazelett, D.J.; et al. ONECUT2 is a targetable master regulator of lethal prostate cancer that suppresses the androgen axis. Nat. Med. 2018, 24, 1887–1898. [Google Scholar] [CrossRef]
- Nerlakanti, N.; Yao, J.; Nguyen, D.T.; Patel, A.K.; Eroshkin, A.M.; Lawrence, H.R.; Ayaz, M.; Kuenzi, B.M.; Agarwal, N.; Chen, Y.; et al. Targeting the BRD4-HOXB13 Coregulated Transcriptional Networks with Bromodomain-Kinase Inhibitors to Suppress Metastatic Castration-Resistant Prostate Cancer. Mol. Cancer Ther. 2018, 17, 2796–2810. [Google Scholar] [CrossRef] [Green Version]
- Stelloo, S.; Nevedomskaya, E.; Kim, Y.; Schuurman, K.; Valle-Encinas, E.; Lobo, J.; Krijgsman, O.; Peeper, D.S.; Chang, S.L.; Feng, F.Y.; et al. Integrative epigenetic taxonomy of primary prostate cancer. Nat. Commun. 2018, 9, 4900. [Google Scholar] [CrossRef]
- Patten, D.K.; Corleone, G.; Gyorffy, B.; Perone, Y.; Slaven, N.; Barozzi, I.; Erdos, E.; Saiakhova, A.; Goddard, K.; Vingiani, A.; et al. Enhancer mapping uncovers phenotypic heterogeneity and evolution in patients with luminal breast cancer. Nat. Med. 2018, 24, 1469–1480. [Google Scholar] [CrossRef]
- Hodgson, M.C.; Astapova, I.; Cheng, S.; Lee, L.J.; Verhoeven, M.C.; Choi, E.; Balk, S.P.; Hollenberg, A.N. The androgen receptor recruits nuclear receptor CoRepressor (N-CoR) in the presence of mifepristone via its N and C termini revealing a novel molecular mechanism for androgen receptor antagonists. J. Biol. Chem. 2005, 280, 6511–6519. [Google Scholar] [CrossRef] [Green Version]
- Song, L.N.; Coghlan, M.; Gelmann, E.P. Antiandrogen effects of mifepristone on coactivator and corepressor interactions with the androgen receptor. Mol. Endocrinol. 2004, 18, 70–85. [Google Scholar] [CrossRef] [Green Version]
- Masiello, D.; Chen, S.Y.; Xu, Y.; Verhoeven, M.C.; Choi, E.; Hollenberg, A.N.; Balk, S.P. Recruitment of beta-catenin by wild-type or mutant androgen receptors correlates with ligand-stimulated growth of prostate cancer cells. Mol. Endocrinol. 2004, 18, 2388–2401. [Google Scholar] [CrossRef] [Green Version]
- Yachie, N.; Petsalaki, E.; Mellor, J.C.; Weile, J.; Jacob, Y.; Verby, M.; Ozturk, S.B.; Li, S.; Cote, A.G.; Mosca, R.; et al. Pooled-matrix protein interaction screens using Barcode Fusion Genetics. Mol. Syst. Biol. 2016, 12, 863. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huo, K.; Ma, L.; Tang, L.; Li, D.; Huang, X.; Yuan, Y.; Li, C.; Wang, W.; Guan, W.; et al. Toward an understanding of the protein interaction network of the human liver. Mol. Syst. Biol. 2011, 7, 536. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.; Chen, L.Y.; Zhang, A.; Godavarthy, A.; Xia, F.; Ghosh, J.C.; Li, H.; Chen, J.D. Regulation of androgen receptor activity by the nuclear receptor corepressor SMRT. J. Biol. Chem. 2003, 278, 5052–5061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Salokas, K.; Weldatsadik, R.G.; Gawriyski, L.; Varjosalo, M. Combined proximity labeling and affinity purification-mass spectrometry workflow for mapping and visualizing protein interaction networks. Nat. Protoc. 2020, 15, 3182–3211. [Google Scholar] [CrossRef]
- Mohammed, H.; Taylor, C.; Brown, G.D.; Papachristou, E.K.; Carroll, J.S.; D’Santos, C.S. Rapid immunoprecipitation mass spectrometry of endogenous proteins (RIME) for analysis of chromatin complexes. Nat. Protoc. 2016, 11, 316–326. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Chen, Y.; Li, M.; Zhou, F.; Li, K.; Cao, H.; Ni, M.; Liu, Y.; Gu, Z.; et al. In Situ Capture of Chromatin Interactions by Biotinylated dCas9. Cell 2017, 170, 1028–1043.e1019. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.D.; Tu, L.C.; Mir, A.; Rodriguez, T.; Ding, Y.; Leszyk, J.; Dekker, J.; Shaffer, S.A.; Zhu, L.J.; Wolfe, S.A.; et al. C-BERST: Defining subnuclear proteomic landscapes at genomic elements with dCas9-APEX2. Nat. Methods 2018, 15, 433–436. [Google Scholar] [CrossRef]
- Purslow, J.A.; Khatiwada, B.; Bayro, M.J.; Venditti, V. NMR Methods for Structural Characterization of Protein-Protein Complexes. Front. Mol. Biosci. 2020, 7, 9. [Google Scholar] [CrossRef]
- Rainey, K.H.; Patterson, G.H. Photoswitching FRET to monitor protein-protein interactions. Proc. Natl. Acad. Sci. USA 2019, 116, 864–873. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Liu, J.; Huang, M.; Hu, M.; Su, Y.; Zhang, X.K. Identification of a New RXRalpha Antagonist Targeting the Coregulator-Binding Site. ACS Med. Chem. Lett. 2014, 5, 736–741. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franke, J.; Neumann, M.H. Bootstrapping neural networks. Neural Comput. 2000, 12, 1929–1949. [Google Scholar] [CrossRef] [PubMed]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Wanior, M.; Kramer, A.; Knapp, S.; Joerger, A.C. Exploiting vulnerabilities of SWI/SNF chromatin remodelling complexes for cancer therapy. Oncogene 2021, 40, 3637–3654. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Ernst, J.; Kellis, M. Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc. 2017, 12, 2478–2492. [Google Scholar] [CrossRef]
- Kim, D.H.; Sun, D.; Storck, W.K.; Welker Leng, K.; Jenkins, C.; Coleman, D.J.; Sampson, D.; Guan, X.; Kumaraswamy, A.; Rodansky, E.S.; et al. BET Bromodomain Inhibition Blocks an AR-Repressed, E2F1-Activated Treatment-Emergent Neuroendocrine Prostate Cancer Lineage Plasticity Program. Clin. Cancer Res. 2021, 27, 4923–4936. [Google Scholar] [CrossRef]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Layer, R.M.; Pedersen, B.S.; DiSera, T.; Marth, G.T.; Gertz, J.; Quinlan, A.R. GIGGLE: A search engine for large-scale integrated genome analysis. Nat. Methods 2018, 15, 123–126. [Google Scholar] [CrossRef] [Green Version]
- Long, M.D.; van Den Berg, P.; Singh, P.K.; .Battaglia, S.; Campbell, M.J. Integrative genomic analysis in K562 chronic myelogenous leukemia cells reveals that proximal NCOR1 binding positively regulates genes that govern erythroid differentiation and Imatinib sensitivity. Nucleic Acids Res. 2015, 43, 7330–7348. [Google Scholar] [CrossRef] [PubMed]


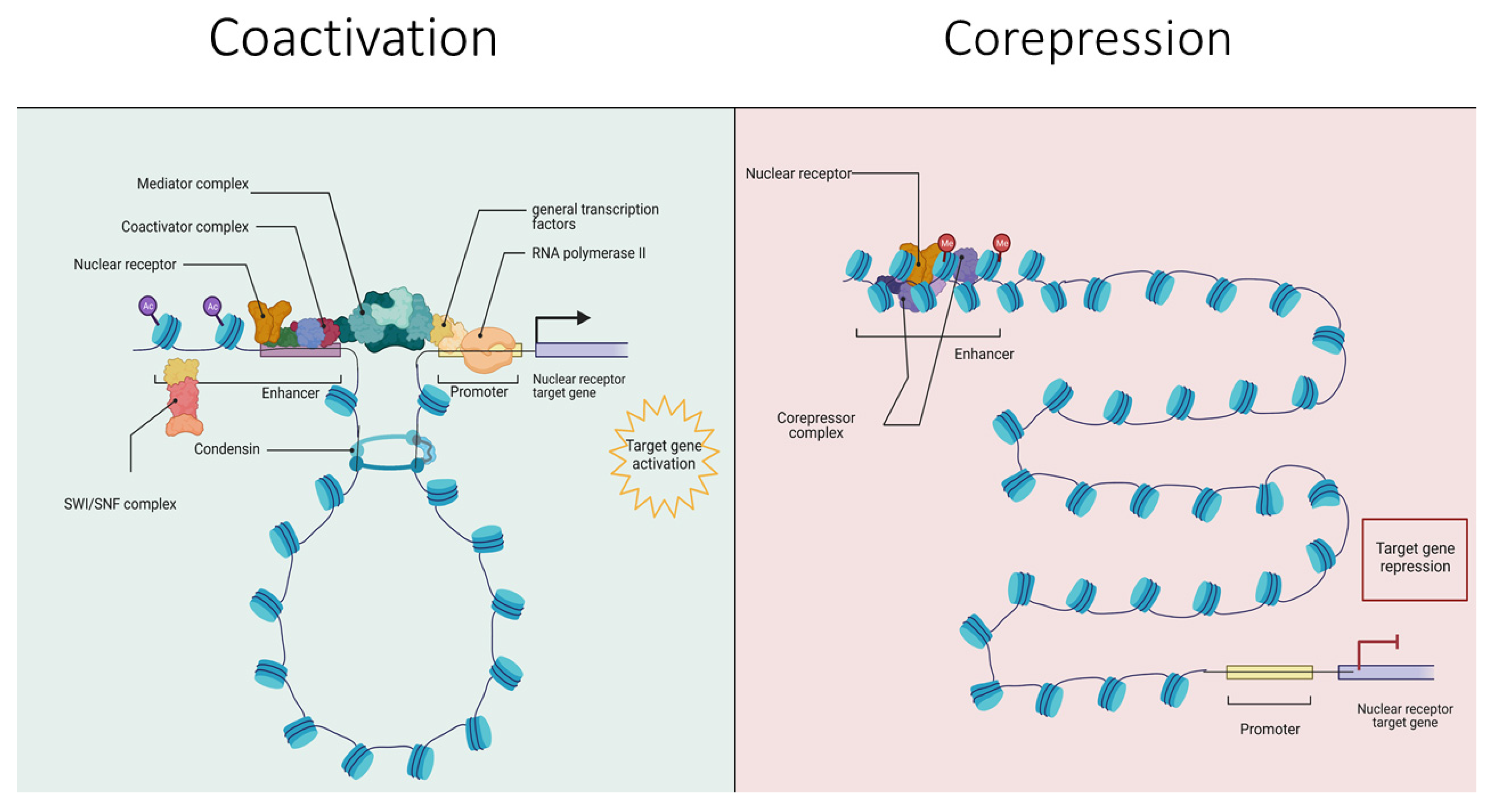{kind=link}
| Gene | Superfamily * | Functional Complex * | Example of NR Interaction ** | Gene Defects *** | References |
|---|---|---|---|---|---|
| SMARCA4 | PBAF complex | Nucleosome positioning | VDR, AR, PPARγ | BRCA—2.6% point mutations, 0.6% CNVs. CC—4.9% point mutations, 1.67% CNVs. OVCA—5.8% point mutations, 3.3% CNVs. PRAD—11.66% point mutations. | [119,120,121] |
| TRRAP | SAGA coactivator complex | Histone modifications | LXR, FXR, ERα, | BRCA—5% point mutations, 0.3% CNVs. CC—8.2% point mutations, 0.33% CNVs. OVCA—4.9% point mutations, 0.7% CNVs. PRAD—4.9% point mutations, 0.5% CNVs. | [122,123,124] |
| CARM1 | 7BS protein arginine methyltransferases | Histone modifications | ERα, AR, FXR, PPARγ, | BRCA—1.6% point mutations, 0.7% CNVs. OVCA—1.3% point mutations, 3.3% CNVs. | [125,126,127,128] |
| NCOA1 | Lysine acetyl transferase | Histone modifications | RARβ, RXRα, PPARγ, ERα, TRα, GR | BRCA—6.1% point mutations. CAN—0.1% point mutations. PRAD—6.3% point mutations. OC—5.7% point mutations, 0.4% CNVs. | [129,130,131] |
| PARP1 | Poly (ADP-ribose) polymerases | DNA repair, link to Mediator | PPARγ, ERα, NR4A4, RARβ, TR | BRCA—1.9% point mutations, 7.3% CNVs. CC—1.8% point mutations, 1.6% CNVs. OVCA—1.2% point mutations, 1.1% CNVs. PRAD—1.2% point mutations, 0.1% CNVs. | [132,133,134,135] |
| TACC1 | TACC family | Signal integrators and transducers, scaffolds, and adaptors | THRβ, THRα, PPARγ, RARα, RARγ and RXRα | BRCA—3.7% point mutations, 8.8% CNVs. CC—1.2% point mutations, 1% CNVs. OVCA—2.8% point mutations, 1.7% CNVs. PRAD—2.8% point mutations, 0.7% CNVs. | [113,116] |
| PPARGC1A | RNA-binding motif containing | Coactivator complexes for metabolic programming | Interacts with PPARγ, PPARα, TR | BRCA—3.2% point mutations, 0.1% CNVs. OVCA—4.7% point mutations, 0.3% CNVs. PRAD—4.5% point mutations. | [136,137] |
| Gene | Superfamily * | Functional Complex * | Example of NR Interaction ** | Genetic Defects *** | References |
|---|---|---|---|---|---|
| RCOR1 | Myb/SANT domain-containing | Histone modifications | GR, AR | BRCA—2.9% point mutations, 0.1% CNVs. PRAD—3.0% point mutations. THCA—0.6% point mutations. OVCA—3.3% point mutations, 0.5% CNVs. | [204,205,206,207,208] |
| MECP2 | Methyl-CpG binding domain-containing | Histone modifications | NCOR2, NCOR1 | BRCA—2.3% point mutations, 0.5% CNVs. PRAD—1.4% point mutations, 0.2% point mutations. THCA—0.8% point mutations, 0.4 CNVs. | [209,210,211] |
| SIN3A | SIN3 histone deacetylase complex subunits | Nucleosome positioning | AR, ERα, RARβ, NR0B1/SHP | BRCA—2.7% point mutations, 0.1 CNVs. PRAD—2.4% point mutations. THCA—0.8% point mutations. | [212,213,214,215,216,217,218,219] |
| NCOR1 | NCoR/SMRT transcriptional repression complex subunits | Histone modifications | AR, TR, RAR, VDR, PPARα/γ | BRCA—5.6% point mutations, 0.8% CNVs. PRAD—4.9% point mutations, 0.1% CNVs. THCA—1.8% point mutations. OVCA—3.4% point mutations, 0.1% CNVs. | [33,35,38] |
| LCOR | Histone modifications | ERα, ERβ, AR, PGR, RARα, RARβ, RARγ, RXRα VDR | BRCA—2.7% point mutations. PRAD—2.9% point mutations, 0.1% CNVs. THCA—0.3% point mutations. OVCA—2.7% point mutations, 0.1% CNVs. | [220,221] | |
| SFPQ | RNA-binding motif containing | RNA splicing and metabolism | SIN3A, THRα, RXRα | BRCA—0.8% point mutations, 0.1% CNVs. PRAD—0.7% point mutations. THCA—2.1% point mutations. | [222,223,224,225] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jafari, H.; Hussain, S.; Campbell, M.J. Nuclear Receptor Coregulators in Hormone-Dependent Cancers. Cancers 2022, 14, 2402. https://doi.org/10.3390/cancers14102402
Jafari H, Hussain S, Campbell MJ. Nuclear Receptor Coregulators in Hormone-Dependent Cancers. Cancers. 2022; 14(10):2402. https://doi.org/10.3390/cancers14102402
Chicago/Turabian StyleJafari, Hedieh, Shahid Hussain, and Moray J. Campbell. 2022. "Nuclear Receptor Coregulators in Hormone-Dependent Cancers" Cancers 14, no. 10: 2402. https://doi.org/10.3390/cancers14102402