
Shared Gene Expression and Immune Pathway Changes Associated with Progression from Nevi to Melanoma

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Acquisition and Processing
2.2. Differential Expression Analysis
2.3. Regularized Regression Analysis
2.4. Assessment of Progression of Genes
2.5. Reactome Over-Representation Analysis
3. Results
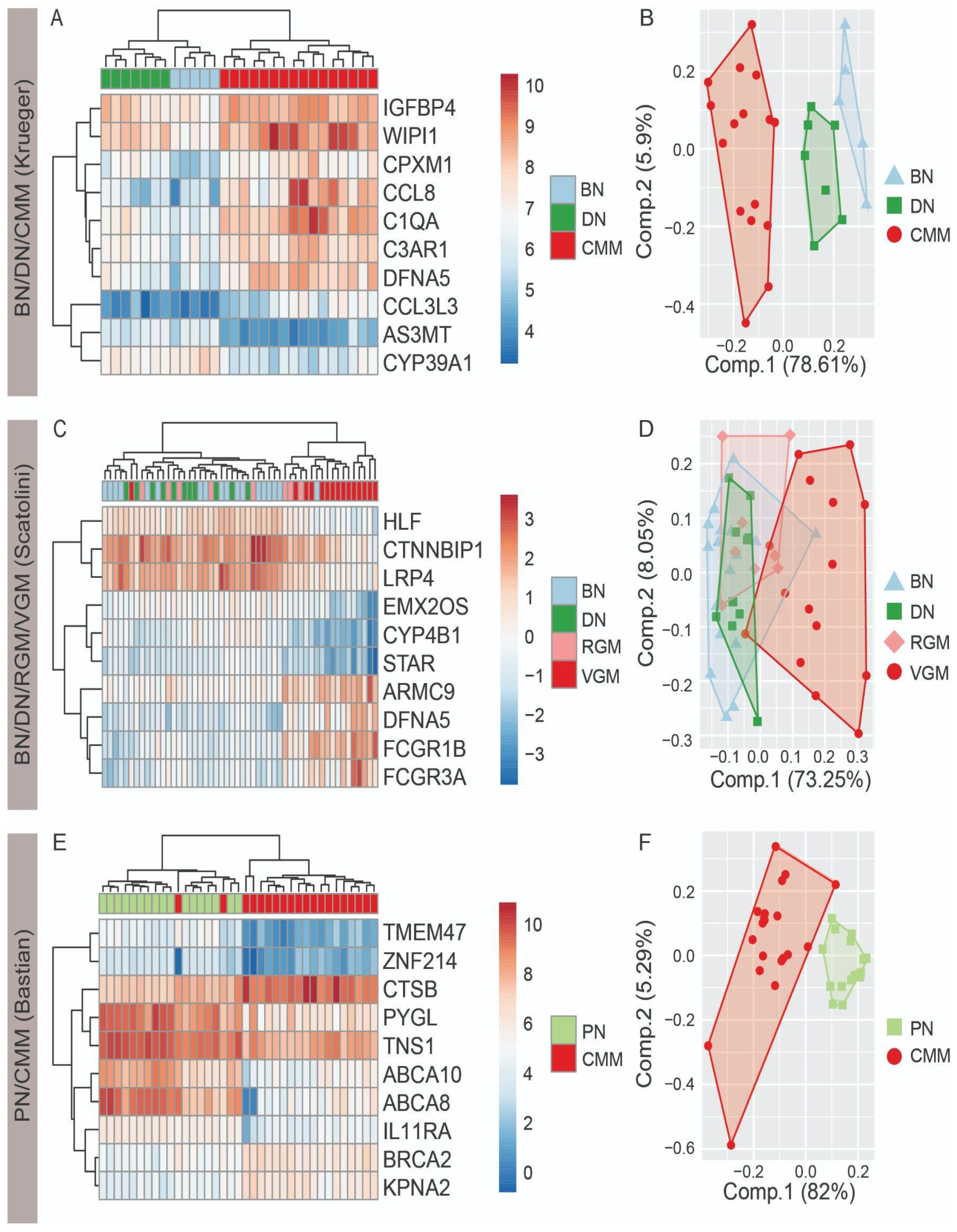
3.1. Top Ten Differentially Expressed Genes Differentiate Nevi and Melanoma
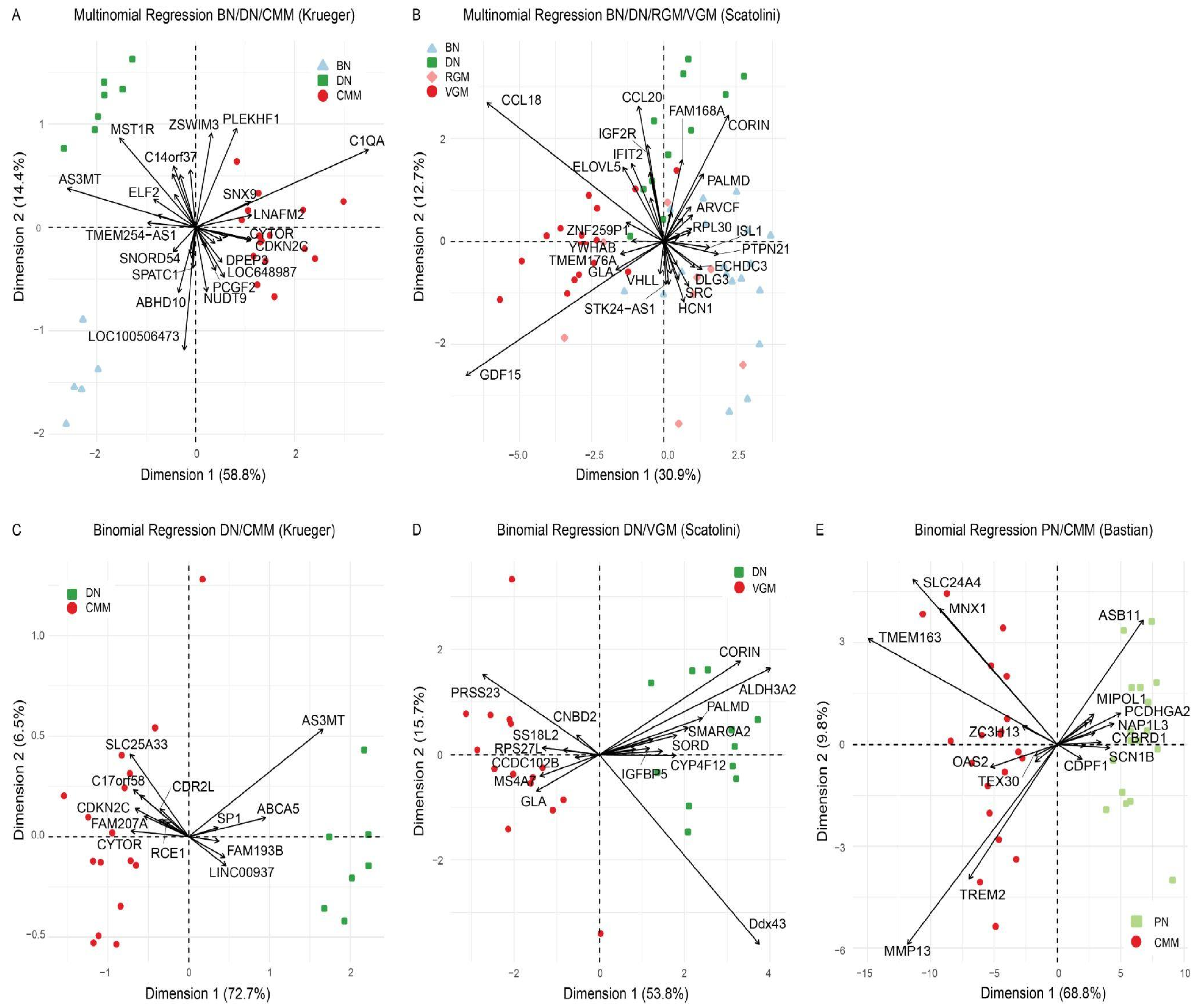
3.2. Regularized Regression Models Differentiate Nevi and Melanoma
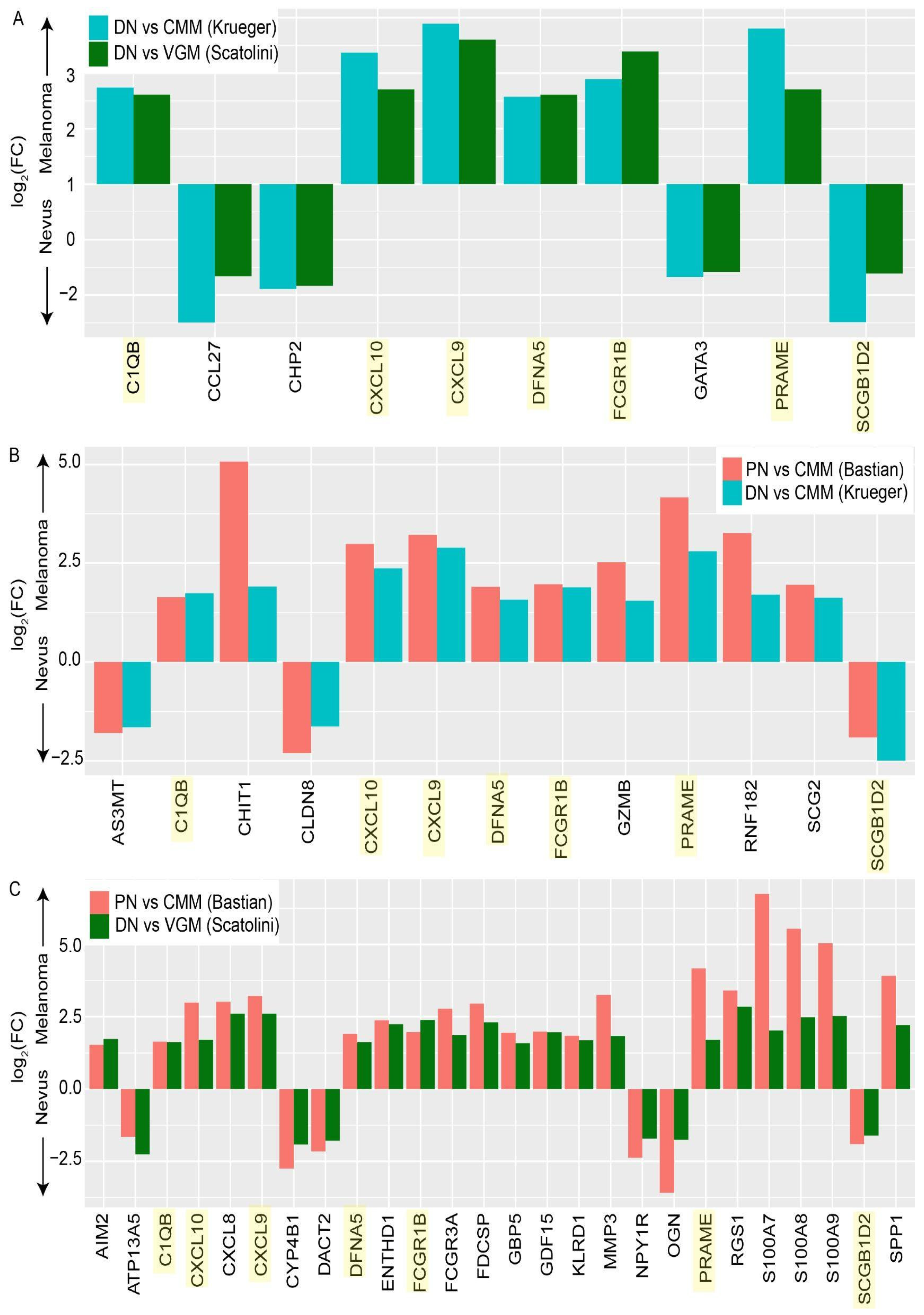
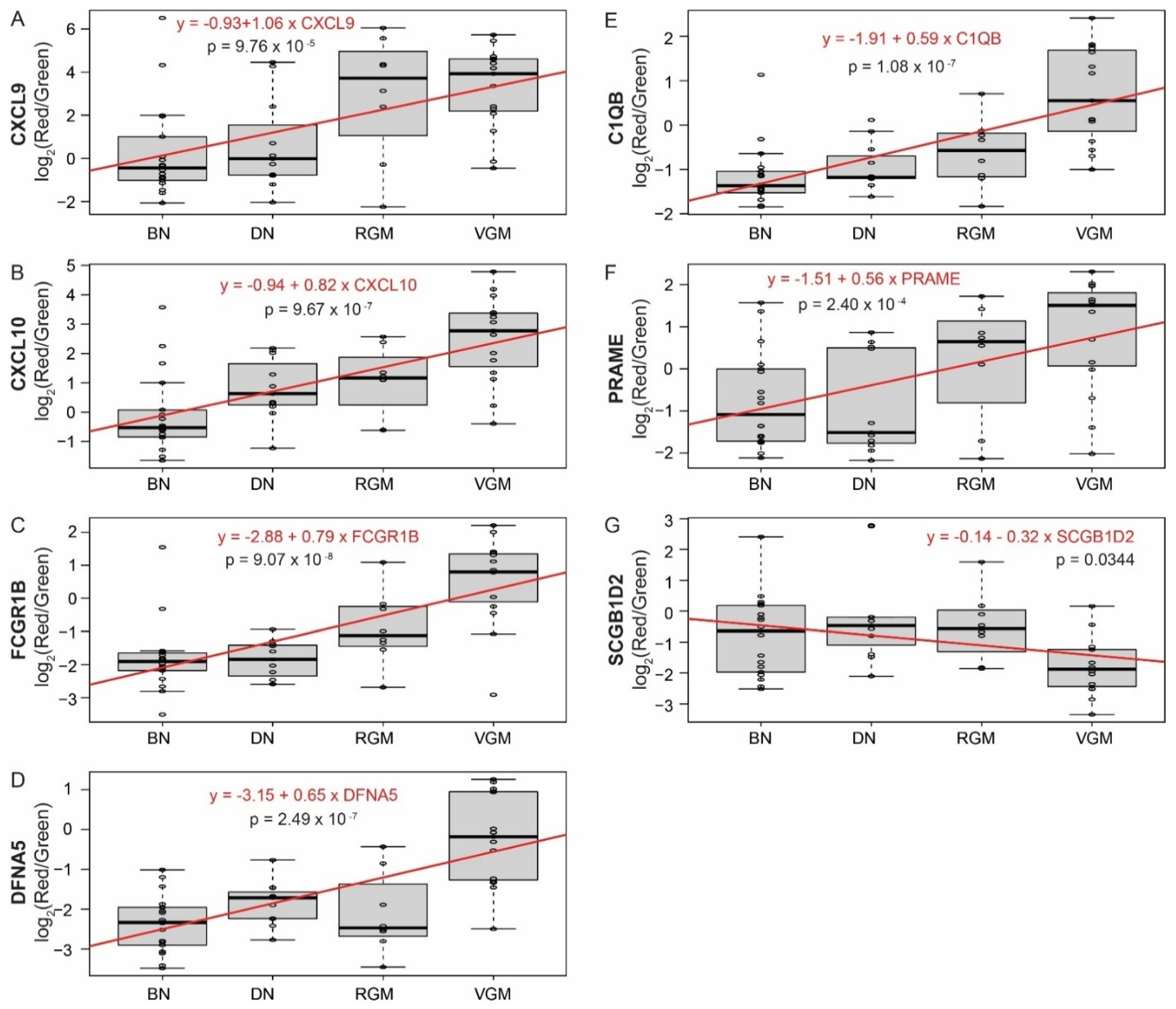
3.3. Differentially Expressed Genes between Stages of Melanoma Progression Overlap between Multiple Datasets
3.4. Genes Associated with Melanoma Progression Show Significant Enrichment of Multiple Pathways Related to the Immune System
4. Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
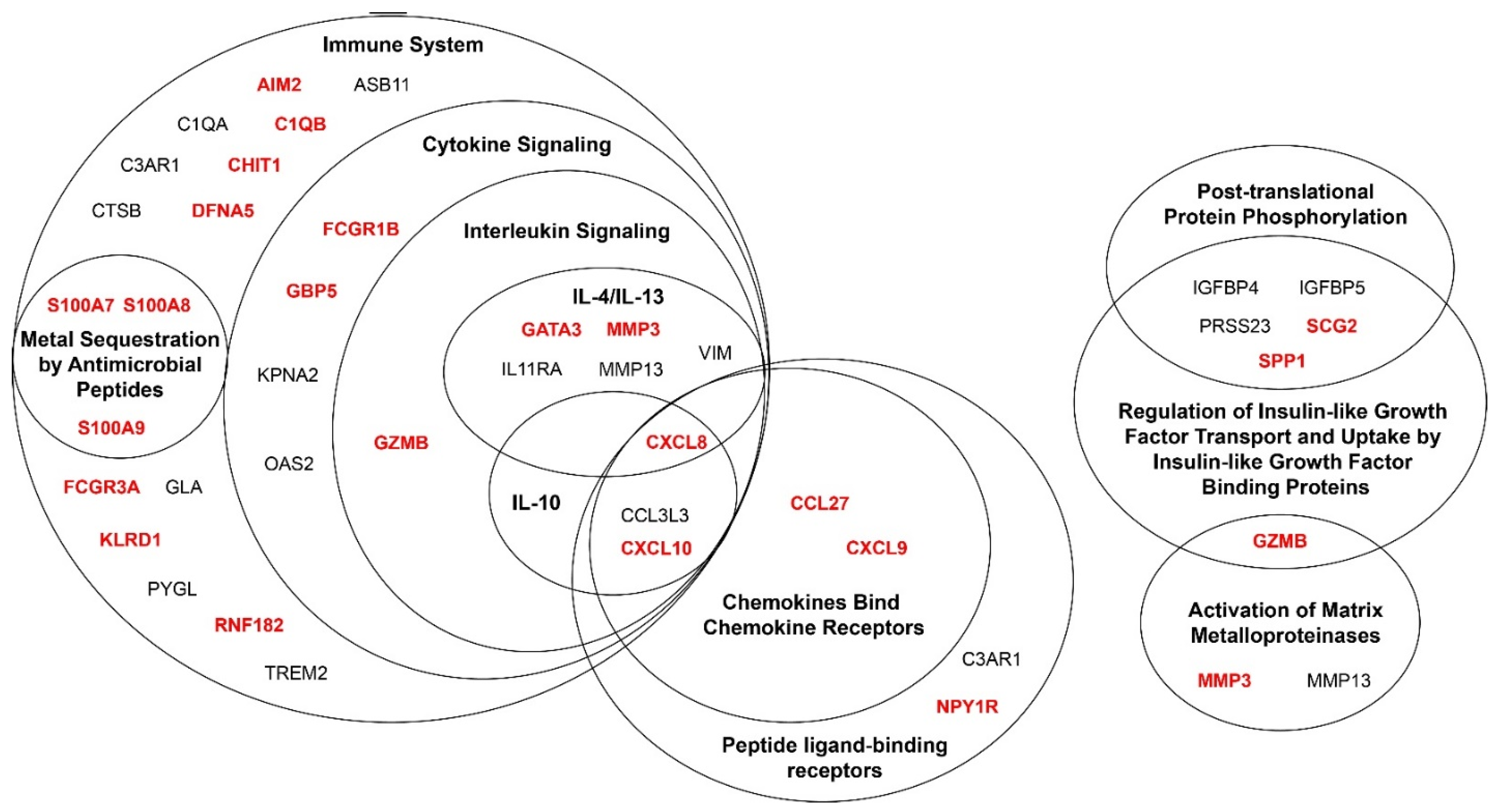{kind=link}
| Gene | Datasets * | Direction | Reported Expression and Function | ||
|---|---|---|---|---|---|
| K | S | B | |||
| Chemokines Bind Chemokine Receptors | |||||
| CXCL8 | ↑CMM | Protein expression increased in CMM vs. nevi by IHC [28,29] | |||
| Promotes melanoma progression and metastasis [33,34,35] | |||||
| CXCL9 CXCL10 | ↑CMM | CXCL9 and CXCL10 increased gene expression in CMM vs. BN by qRT-PCR [30] | |||
| CXCL9 and CXCL10 in gene signature that differentiates BN and CMM [55,56,57] | |||||
| CXCL9 increased gene expression in melanoma metastases vs. BN [58] | |||||
| CXCL9 and CXCL10 recruits CXCR3-expressing effector T cells and natural killer cells into melanoma [59] | |||||
| CXCL10 binds CXCR3 on tumor cells to promote metastases [36] | |||||
| CCL27 | ↓CMM | Recruits T cells in melanoma mouse model [32] | |||
| Peptide Ligand-Binding Receptors | |||||
| C3AR1 | ↑CMM | Increases melanoma tumor growth by inhibiting neutrophil and CD4+ T cell responses [31] | |||
| IL-4 and IL-13 Signaling Pathways | |||||
| GATA3 | ↓CMM | Stabilizes HIF-1α to enhance cancer invasiveness under hypoxic conditions [60] | |||
| MMP3 | ↑CMM | Protein expressed in CMM lesions, but not normal skin, by IHC and IF [61] | |||
| Promotes melanoma tumor growth and metastasis [37] | |||||
| MMP13 | ↑CMM | Protein expression in some CMM cases, absent in BN, by IHC [62] | |||
| VIM | ↓CMM | Protein uniformly expressed in BN and CMM melanocytes by IF [63] | |||
| Cytokine Signaling | |||||
| GZMB | ↑CMM | Increased GZMB-expressing cells in DN (severe atypia) and CMM vs. BN and DN (mild atypia) by IHC [42] | |||
| FCGR1B | ↑CMM | Induced by interferon-γ [64,65] | |||
| GBP5 | ↑CMM | Induced by interferon-γ and type 1 interferons | |||
| Stimulates assembly of NLRP3 inflammasome [39], which expands myeloid-derived suppressor cells in melanoma leading to immunosuppression and increased tumor growth [40] | |||||
| KPNA2 | ↑CMM | Promotes proliferation, migration, and invasion in melanoma cells [38,41] | |||
| OAS2 | ↑CMM | Induced by type 1 interferons and important in anti-viral immune response | |||
| Gene expression induced by UVB in human primary melanocytes (qRT-PCR) [66] | |||||
| Metal Sequestration by Antimicrobial Peptides | |||||
| S100A7 S100A8 S100A9 | ↑CMM | S100A7, S100A8, and S100A9 increased gene expression in CMM vs. BN by qRT-PCR [55,56,57] | |||
| S100A7, S100A8, and S100A9 in gene signature that differentiates BN and CMM [55,56,57] | |||||
| S100A7, S100A8, and S100A9 linked to tumor growth and metastasis in multiple cancers [43] | |||||
| S100A9 promotes migration and metastasis of EMMPRIN-expressing melanoma cells [44] | |||||
| Immune System | |||||
| AIM2 | ↑CMM | Expression in dendritic cells promotes immunosuppressive tumor microenvironment and increased melanoma tumor growth [45] | |||
| ASB11 | ↓CMM | Downregulated by DNA damage, stabilizes pro-apoptotic protein BIK, which increases apoptosis [67] | |||
| C1QA | ↑CMM | C1q protein (composed of C1QA, C1QB, and C1QC) expressed by mesenchymal cells and immune cells in melanoma by IHC [46] C1qa-/- mice have slower melanoma tumor growth, prolonged survival, decreased tumor angiogenesis, and decreased lung metastasis [46] C1q from non-bone marrow-derived cells promotes accelerated melanoma tumor growth [46] C1q promotes cell adhesion, migration, and proliferation of melanoma cells [46] | |||
| C1QB | ↑CMM | ||||
| DFNA5/ GSDME | ↑CMM | Gene expression increased in CMM vs. normal skin by RNAseq [68] | |||
| Included in pyroptosis-related gene signature for BN vs. CMM [68] | |||||
| Triggers pyroptosis and apoptosis [47] | |||||
| Deficiency in melanoma cells increases in vitro and in vivo tumor growth [47] | |||||
| CHIT1 | ↑CMM | Produced by macrophages stimulated by interferon-γ and tumor necrosis factor-α [69] | |||
| Increases transforming growth factor-β SMAD signaling [69], which plays role in melanoma metastasis [70] and angiogenesis in various cancers [71] | |||||
| CTSB | ↑CMM | Involved in metastasis [72,73], angiogenesis, and invasion of tumor cells, including melanoma [74,75,76] | |||
| FCGR3A | ↑CMM | Expressed on subset of natural killer cells [77] Mediates antibody-dependent cell-mediated cytotoxicity [77] | |||
| GLA | ↓CMM | Patients with GLA deficiency possibly have increased rate of melanoma [78] | |||
| KLRD1 | ↑CMM | Protein expressed on tumor infiltrating lymphocytes in CMM by IHC [79] | |||
| Regulates natural killer cell cytotoxicity [79] | |||||
| PYGL | ↓CMM | Differentially expressed in BN with and without V600E BRAF mutation by microarray [80] | |||
| Involved in glycogen metabolism, which regulates inflammatory responses and tumorigenesis [81,82,83] | |||||
| RNF182 | ↑CMM | Suppresses Toll-like receptor-triggered immune response and decreases production of proinflammatory cytokines [84] | |||
| TREM2 | ↑CMM | Encodes for innate immune receptor on tumor infiltrating myeloid cells [85] | |||
| TREM2 deletion decreases immunosuppressive regulatory myeloid cells within tumors, which decreases exhausted CD8+ T cell subsets and tumor growth [86] | |||||
| Regulation of Insulin-like Growth Factor Transport and Uptake by Insulin-like Growth Factor Binding Proteins | |||||
| IGFBP4 | ↑CMM | Protein expressed in CMM by IHC [87] | |||
| IGFBP5 | ↓CMM | Gene and protein expression increased in CMM vs. nevi by qRT-PCR and IHC [48] | |||
| Gene expression increased in metastatic melanoma vs. BN by RNAseq [58] | |||||
| Overexpression inhibits in vitro proliferation, migration, invasion, epithelial to mesenchymal transition, and in vivo tumor growth and metastasis of melanoma cell lines [48] | |||||
| PRSS23 | ↑CMM | Knockdown decreases cancer cell proliferation in breast and gastric cancer [49,50] | |||
| SCG2 | ↑CMM | Gene expression increased in VGM and metastatic melanoma vs. normal skin, BN, and melanoma in situ by microarray [27] | |||
| Increases migration of melanoma cells [52] | |||||
| Plays role in chemoattraction and migration [51] | |||||
| SPP1 | ↑CMM | Increased expression in CMM vs. BN across 5 studies (RNAseq, microarray, IHC) [58,88,89,90,91] | |||
| Included in 5 protein assay that distinguishes BN vs. CMM by IHC [89] | |||||
| SPP1 treatment increases melanoma cell migration, invasion, and proliferation [54] | |||||
| SPP1 knockdown decreased in vitro and in vivo proliferation, migration, and invasion of melanoma cells [53] | |||||
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lashway, S.G.; Harris, R.B.; Farland, L.V.; O’Rourke, M.K.; Dennis, L.K. Age and Cohort Trends of Malignant Melanoma in the United States. Cancers 2021, 13, 3866. [Google Scholar] [CrossRef]
- Sacchetto, L.; Zanetti, R.; Comber, H.; Bouchardy, C.; Brewster, D.H.; Broganelli, P.; Chirlaque, M.D.; Coza, D.; Galceran, J.; Gavin, A.; et al. Trends in incidence of thick, thin and in situ melanoma in Europe. Eur. J. Cancer 2018, 92, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Jeter, J.M.; Bowles, T.L.; Curiel-Lewandrowski, C.; Swetter, S.M.; Filipp, F.V.; Abdel-Malek, Z.A.; Geskin, L.J.; Brewer, J.D.; Arbiser, J.L.; Gershenwald, J.E.; et al. Chemoprevention agents for melanoma: A path forward into phase 3 clinical trials. Cancer 2019, 125, 18–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, C.M.; Carroll, H.J.; Whiteman, D.C. Estimating the attributable fraction for cancer: A meta-analysis of nevi and melanoma. Cancer Prev. Res. 2010, 3, 233–245. [Google Scholar] [CrossRef] [Green Version]
- Tsao, H.; Bevona, C.; Goggins, W.; Quinn, T. The transformation rate of moles (melanocytic nevi) into cutaneous melanoma: A population-based estimate. Arch. Dermatol. 2003, 139, 282–288. [Google Scholar] [CrossRef] [Green Version]
- Curiel-Lewandrowski, C.; Swetter, S.M.; Einspahr, J.G.; Hsu, C.H.; Nagle, R.; Sagerman, P.; Tangrea, J.; Parnes, H.; Alberts, D.S.; Chow, H.H. Randomized, double-blind, placebo-controlled trial of sulindac in individuals at risk for melanoma: Evaluation of potential chemopreventive activity. Cancer 2012, 118, 5848–5856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, L.; Meyskens, F.; Levine, N. Effect of oral isotretinoin on dysplastic nevi. J. Am. Acad. Dermatol. 1989, 20, 257–260. [Google Scholar] [CrossRef] [Green Version]
- Linden, K.G.; Leachman, S.A.; Zager, J.S.; Jakowatz, J.G.; Viner, J.L.; McLaren, C.E.; Barr, R.J.; Carpenter, P.M.; Chen, W.P.; Elmets, C.A.; et al. A randomized, double-blind, placebo-controlled phase II clinical trial of lovastatin for various endpoints of melanoma pathobiology. Cancer Prev. Res. 2014, 7, 496–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsui, H.; Kiecker, F.; Shemer, A.; Cannizzaro, M.V.; Wang, C.Q.F.; Gulati, N.; Ohmatsu, H.; Shah, K.R.; Gilleaudeau, P.; Sullivan-Whalen, M.; et al. Discrimination of Dysplastic Nevi from Common Melanocytic Nevi by Cellular and Molecular Criteria. J. Investig. Dermatol. 2016, 136, 2030–2040. [Google Scholar] [CrossRef] [Green Version]
- Yan, B.Y.; Garcet, S.; Gulati, N.; Kiecker, F.; Fuentes-Duculan, J.; Gilleaudeau, P.; Sullivan-Whalen, M.; Shemer, A.; Mitsui, H.; Krueger, J.G. Novel immune signatures associated with dysplastic naevi and primary cutaneous melanoma in human skin. Exp. Dermatol. 2019, 28, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Scatolini, M.; Grand, M.M.; Grosso, E.; Venesio, T.; Pisacane, A.; Balsamo, A.; Sirovich, R.; Risio, M.; Chiorino, G. Altered molecular pathways in melanocytic lesions. Int. J. Cancer 2010, 126, 1869–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Joseph, N.M.; Yu, R.; Benhamida, J.; Liu, S.; Prow, T.; Ruben, B.; North, J.; Pincus, L.; Yeh, I.; et al. Genomic and Transcriptomic Analysis Reveals Incremental Disruption of Key Signaling Pathways during Melanoma Evolution. Cancer Cell 2018, 34, 45–55.e4. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Harr, B.; Schlotterer, C. Comparison of algorithms for the analysis of Affymetrix microarray data as evaluated by co-expression of genes in known operons. Nucleic Acids Res. 2006, 34, e8. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Olney, K.C.; Brotman, S.M.; Andrews, J.P.; Valverde-Vesling, V.A.; Wilson, M.A. Reference genome and transcriptome informed by the sex chromosome complement of the sample increase ability to detect sex differences in gene expression from RNA-Seq data. Biol. Sex. Differ. 2020, 11, 42. [Google Scholar] [CrossRef]
- Webster, T.H.; Couse, M.; Grande, B.M.; Karlins, E.; Phung, T.N.; Richmond, P.A.; Whitford, W.; Wilson, M.A. Identifying, understanding, and correcting technical artifacts on the sex chromosomes in next-generation sequencing data. Gigascience 2019, 8, giz074. [Google Scholar] [CrossRef] [Green Version]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B (Methodol.) 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Engebretsen, S.; Bohlin, J. Statistical predictions with glmnet. Clin. Epigenet. 2019, 11, 123. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.P.; Hoek, K.; Becker, D. Whole-genome expression profiling of the melanoma progression pathway reveals marked molecular differences between nevi/melanoma in situ and advanced-stage melanomas. Cancer Biol. Ther. 2005, 4, 1018–1029. [Google Scholar] [CrossRef] [Green Version]
- Hensley, C.; Spitzler, S.; McAlpine, B.E.; Lynn, M.; Ansel, J.C.; Solomon, A.R.; Armstrong, C.A. In vivo human melanoma cytokine production: Inverse correlation of GM-CSF production with tumor depth. Exp. Dermatol. 1998, 7, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Moretti, S.; Pinzi, C.; Spallanzani, A.; Berti, E.; Chiarugi, A.; Mazzoli, S.; Fabiani, M.; Vallecchi, C.; Herlyn, M. Immunohistochemical evidence of cytokine networks during progression of human melanocytic lesions. Int. J. Cancer 1999, 84, 160–168. [Google Scholar] [CrossRef]
- Clarke, L.E.; Warf, M.B.; Flake, D.D., 2nd; Hartman, A.R.; Tahan, S.; Shea, C.R.; Gerami, P.; Messina, J.; Florell, S.R.; Wenstrup, R.J.; et al. Clinical validation of a gene expression signature that differentiates benign nevi from malignant melanoma. J. Cutan. Pathol. 2015, 42, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Nabizadeh, J.A.; Manthey, H.D.; Steyn, F.J.; Chen, W.; Widiapradja, A.; Md Akhir, F.N.; Boyle, G.M.; Taylor, S.M.; Woodruff, T.M.; Rolfe, B.E. The Complement C3a Receptor Contributes to Melanoma Tumorigenesis by Inhibiting Neutrophil and CD4+ T Cell Responses. J. Immunol. 2016, 196, 4783–4792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, N.; Sasaki, A.; Niwa, M.; Okada, Y.; Hatanaka, Y.; Tani, Y.; Mizuguchi, H.; Nakagawa, S.; Fujita, T.; Yamamoto, A. Tumor suppressive efficacy through augmentation of tumor-infiltrating immune cells by intratumoral injection of chemokine-expressing adenoviral vector. Cancer Gene Ther. 2006, 13, 393–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schadendorf, D.; Moller, A.; Algermissen, B.; Worm, M.; Sticherling, M.; Czarnetzki, B.M. IL-8 produced by human malignant melanoma cells in vitro is an essential autocrine growth factor. J. Immunol. 1993, 151, 2667–2675. [Google Scholar]
- Singh, R.K.; Gutman, M.; Radinsky, R.; Bucana, C.D.; Fidler, I.J. Expression of interleukin 8 correlates with the metastatic potential of human melanoma cells in nude mice. Cancer Res. 1994, 54, 3242–3247. [Google Scholar] [PubMed]
- Singh, S.; Singh, A.P.; Sharma, B.; Owen, L.B.; Singh, R.K. CXCL8 and its cognate receptors in melanoma progression and metastasis. Future Oncol. 2010, 6, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Wightman, S.C.; Uppal, A.; Pitroda, S.P.; Ganai, S.; Burnette, B.; Stack, M.; Oshima, G.; Khan, S.; Huang, X.; Posner, M.C.; et al. Oncogenic CXCL10 signalling drives metastasis development and poor clinical outcome. Br. J. Cancer 2015, 113, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Shoshan, E.; Braeuer, R.R.; Kamiya, T.; Mobley, A.K.; Huang, L.; Vasquez, M.E.; Velazquez-Torres, G.; Chakravarti, N.; Ivan, C.; Prieto, V.; et al. NFAT1 Directly Regulates IL8 and MMP3 to Promote Melanoma Tumor Growth and Metastasis. Cancer Res. 2016, 76, 3145–3155. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Wang, X. The emerging roles of KPNA2 in cancer. Life Sci. 2020, 241, 117140. [Google Scholar] [CrossRef]
- Shenoy, A.R.; Wellington, D.A.; Kumar, P.; Kassa, H.; Booth, C.J.; Cresswell, P.; MacMicking, J.D. GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science 2012, 336, 481–485. [Google Scholar] [CrossRef]
- Tengesdal, I.W.; Menon, D.R.; Osborne, D.G.; Neff, C.P.; Powers, N.E.; Gamboni, F.; Mauro, A.G.; D’Alessandro, A.; Stefanoni, D.; Henen, M.A.; et al. Targeting tumor-derived NLRP3 reduces melanoma progression by limiting MDSCs expansion. Proc. Natl. Acad. Sci. USA 2021, 118, e2000915118. [Google Scholar] [CrossRef]
- Yang, F.; Li, S.; Cheng, Y.; Li, J.; Han, X. Karyopherin alpha 2 promotes proliferation, migration and invasion through activating NF-kappaB/p65 signaling pathways in melanoma cells. Life Sci. 2020, 252, 117611. [Google Scholar] [CrossRef]
- Martin, J.M.; Pinazo, I.; Jorda, E.; Monteagudo, C. Differential Clinicopathological Features in Spontaneous Regression of Melanomas and Melanocytic Naevi. Acta Derm. Venereol. 2017, 97, 692–697. [Google Scholar] [CrossRef] [Green Version]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef] [Green Version]
- Hibino, T.; Sakaguchi, M.; Miyamoto, S.; Yamamoto, M.; Motoyama, A.; Hosoi, J.; Shimokata, T.; Ito, T.; Tsuboi, R.; Huh, N.H. S100A9 is a novel ligand of EMMPRIN that promotes melanoma metastasis. Cancer Res. 2013, 73, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, K.; Okamura, K.; Riding, R.L.; Fan, X.; Afshari, K.; Haddadi, N.S.; McCauley, S.M.; Guney, M.H.; Luban, J.; Funakoshi, T.; et al. AIM2 regulates anti-tumor immunity and is a viable therapeutic target for melanoma. J. Exp. Med. 2021, 218, e20200962. [Google Scholar] [CrossRef] [PubMed]
- Bulla, R.; Tripodo, C.; Rami, D.; Ling, G.S.; Agostinis, C.; Guarnotta, C.; Zorzet, S.; Durigutto, P.; Botto, M.; Tedesco, F. C1q acts in the tumour microenvironment as a cancer-promoting factor independently of complement activation. Nat. Commun. 2016, 7, 10346. [Google Scholar] [CrossRef] [Green Version]
- Rogers, C.; Erkes, D.A.; Nardone, A.; Aplin, A.E.; Fernandes-Alnemri, T.; Alnemri, E.S. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat. Commun. 2019, 10, 1689. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ding, N.; Li, Y.; Cheng, H.; Wang, D.; Yang, Q.; Deng, Y.; Yang, Y.; Li, Y.; Ruan, X.; et al. Insulin-like growth factor binding protein 5 (IGFBP5) functions as a tumor suppressor in human melanoma cells. Oncotarget 2015, 6, 20636–20649. [Google Scholar] [CrossRef] [Green Version]
- Chan, H.S.; Chang, S.J.; Wang, T.Y.; Ko, H.J.; Lin, Y.C.; Lin, K.T.; Chang, K.M.; Chuang, Y.J. Serine protease PRSS23 is upregulated by estrogen receptor alpha and associated with proliferation of breast cancer cells. PLoS ONE 2012, 7, e30397. [Google Scholar] [CrossRef] [Green Version]
- Han, B.; Yang, Y.; Chen, J.; He, X.; Lv, N.; Yan, R. PRSS23 knockdown inhibits gastric tumorigenesis through EIF2 signaling. Pharmacol. Res. 2019, 142, 50–57. [Google Scholar] [CrossRef]
- Kahler, C.M.; Kirchmair, R.; Kaufmann, G.; Kahler, S.T.; Reinisch, N.; Fischer-Colbrie, R.; Hogue-Angeletti, R.; Winkler, H.; Wiedermann, C.J. Inhibition of proliferation and stimulation of migration of endothelial cells by secretoneurin in vitro. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 932–939. [Google Scholar] [CrossRef]
- Peitsch, W.K.; Doerflinger, Y.; Fischer-Colbrie, R.; Huck, V.; Bauer, A.T.; Utikal, J.; Goerdt, S.; Schneider, S.W. Desmoglein 2 depletion leads to increased migration and upregulation of the chemoattractant secretoneurin in melanoma cells. PLoS ONE 2014, 9, e89491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, G.; Zeng, F.; Su, J.; Zhao, S.; Hu, R.; Zhu, W.; Hu, S.; Chen, X.; Yin, M. BET inhibitor suppresses melanoma progression via the noncanonical NF-kappaB/SPP1 pathway. Theranostics 2020, 10, 11428–11443. [Google Scholar] [CrossRef]
- Philip, S.; Bulbule, A.; Kundu, G.C. Osteopontin stimulates tumor growth and activation of promatrix metalloproteinase-2 through nuclear factor-kappa B-mediated induction of membrane type 1 matrix metalloproteinase in murine melanoma cells. J. Biol. Chem. 2001, 276, 44926–44935. [Google Scholar] [CrossRef] [Green Version]
- Clarke, L.E.; Flake, D.D., 2nd; Busam, K.; Cockerell, C.; Helm, K.; McNiff, J.; Reed, J.; Tschen, J.; Kim, J.; Barnhill, R.; et al. An independent validation of a gene expression signature to differentiate malignant melanoma from benign melanocytic nevi. Cancer 2017, 123, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.E.; Mabey, B.; Flake Ii, D.D.; Meek, S.; Cassarino, D.S.; Duncan, L.M.; High, W.A.; Napekoski, K.M.; Prieto, V.G.; Tetzlaff, M.T.; et al. Clinical validity of a gene expression signature in diagnostically uncertain neoplasms. Per. Med. 2020, 17, 361–371. [Google Scholar] [CrossRef]
- Ko, J.S.; Matharoo-Ball, B.; Billings, S.D.; Thomson, B.J.; Tang, J.Y.; Sarin, K.Y.; Cai, E.; Kim, J.; Rock, C.; Kimbrell, H.Z.; et al. Diagnostic Distinction of Malignant Melanoma and Benign Nevi by a Gene Expression Signature and Correlation to Clinical Outcomes. Cancer Epidemiol. Biomark. Prev. 2017, 26, 1107–1113. [Google Scholar] [CrossRef] [Green Version]
- Meves, A.; Nikolova, E.; Heim, J.B.; Squirewell, E.J.; Cappel, M.A.; Pittelkow, M.R.; Otley, C.C.; Behrendt, N.; Saunte, D.M.; Lock-Andersen, J.; et al. Tumor Cell Adhesion As a Risk Factor for Sentinel Lymph Node Metastasis in Primary Cutaneous Melanoma. J. Clin. Oncol. 2015, 33, 2509–2515. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.C.; Lin, J.J.; Hsu, C.L.; Juan, H.F.; Lou, P.J.; Huang, M.C. GATA3 interacts with and stabilizes HIF-1alpha to enhance cancer cell invasiveness. Oncogene 2017, 36, 4243–4252. [Google Scholar] [CrossRef]
- Walker, R.A.; Woolley, D.E. Immunolocalisation studies of matrix metalloproteinases-1, -2 and -3 in human melanoma. Virchows Arch. 1999, 435, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Corte, M.D.; Gonzalez, L.O.; Corte, M.G.; Quintela, I.; Pidal, I.; Bongera, M.; Vizoso, F. Collagenase-3 (MMP-13) expression in cutaneous malignant melanoma. Int. J. Biol. Mark. 2005, 20, 242–248. [Google Scholar] [CrossRef]
- Miettinen, M.; Lehto, V.P.; Virtanen, I. Presence of fibroblast-type intermediate filaments (vimentin) and absence of neurofilaments in pigmented nevi and malignant melanomas. J. Cutan. Pathol. 1983, 10, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Ernst, L.K.; Duchemin, A.M.; Miller, K.L.; Anderson, C.L. Molecular characterization of six variant Fcgamma receptor class I (CD64) transcripts. Mol. Immunol. 1998, 35, 943–954. [Google Scholar] [CrossRef]
- Porges, A.J.; Redecha, P.B.; Doebele, R.; Pan, L.C.; Salmon, J.E.; Kimberly, R.P. Novel Fc gamma receptor I family gene products in human mononuclear cells. J. Clin. Investig. 1992, 90, 2102–2109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Anumanthan, G.; Scheaffer, S.; Cornelius, L.A. HMGB1/RAGE Mediates UVB-Induced Secretory Inflammatory Response and Resistance to Apoptosis in Human Melanocytes. J. Investig. Dermatol. 2019, 139, 202–212. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.Y.; Huang, M.Y.; Lin, Y.M.; Ho, C.H.; Lin, S.Y.; Chen, H.Y.; Hung, M.C.; Chen, R.H. BIK ubiquitination by the E3 ligase Cul5-ASB11 determines cell fate during cellular stress. J. Cell Biol. 2019, 218, 3002–3018. [Google Scholar] [CrossRef] [Green Version]
- Ju, A.; Tang, J.; Chen, S.; Fu, Y.; Luo, Y. Pyroptosis-Related Gene Signatures Can Robustly Diagnose Skin Cutaneous Melanoma and Predict the Prognosis. Front. Oncol. 2021, 11, 709077. [Google Scholar] [CrossRef]
- Lee, C.G.; Herzog, E.L.; Ahangari, F.; Zhou, Y.; Gulati, M.; Lee, C.M.; Peng, X.; Feghali-Bostwick, C.; Jimenez, S.A.; Varga, J.; et al. Chitinase 1 is a biomarker for and therapeutic target in scleroderma-associated interstitial lung disease that augments TGF-beta1 signaling. J. Immunol. 2012, 189, 2635–2644. [Google Scholar] [CrossRef] [Green Version]
- Janji, B.; Melchior, C.; Gouon, V.; Vallar, L.; Kieffer, N. Autocrine TGF-beta-regulated expression of adhesion receptors and integrin-linked kinase in HT-144 melanoma cells correlates with their metastatic phenotype. Int. J. Cancer 1999, 83, 255–262. [Google Scholar] [CrossRef]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef]
- Sloane, B.F.; Dunn, J.R.; Honn, K.V. Lysosomal cathepsin B: Correlation with metastatic potential. Science 1981, 212, 1151–1153. [Google Scholar] [CrossRef] [PubMed]
- Sloane, B.F.; Honn, K.V.; Sadler, J.G.; Turner, W.A.; Kimpson, J.J.; Taylor, J.D. Cathepsin B activity in B16 melanoma cells: A possible marker for metastatic potential. Cancer Res. 1982, 42, 980–986. [Google Scholar]
- Aggarwal, N.; Sloane, B.F. Cathepsin B: Multiple roles in cancer. Proteom. Clin. Appl. 2014, 8, 427–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengsch, F.; Buck, A.; Gunther, S.C.; Seiz, J.R.; Tacke, M.; Pfeifer, D.; von Elverfeldt, D.; Sevenich, L.; Hillebrand, L.E.; Kern, U.; et al. Cell type-dependent pathogenic functions of overexpressed human cathepsin B in murine breast cancer progression. Oncogene 2014, 33, 4474–4484. [Google Scholar] [CrossRef] [Green Version]
- Tummalapalli, P.; Spomar, D.; Gondi, C.S.; Olivero, W.C.; Gujrati, M.; Dinh, D.H.; Rao, J.S. RNAi-mediated abrogation of cathepsin B and MMP-9 gene expression in a malignant meningioma cell line leads to decreased tumor growth, invasion and angiogenesis. Int. J. Oncol. 2007, 31, 1039–1050. [Google Scholar]
- Seidel, U.J.; Schlegel, P.; Lang, P. Natural killer cell mediated antibody-dependent cellular cytotoxicity in tumor immunotherapy with therapeutic antibodies. Front. Immunol. 2013, 4, 76. [Google Scholar] [CrossRef] [Green Version]
- Bird, S.; Hadjimichael, E.; Mehta, A.; Ramaswami, U.; Hughes, D. Fabry disease and incidence of cancer. Orphanet J. Rare Dis. 2017, 12, 150. [Google Scholar] [CrossRef] [Green Version]
- Vetter, C.S.; Straten, P.T.; Terheyden, P.; Zeuthen, J.; Brocker, E.B.; Becker, J.C. Expression of CD94/NKG2 subtypes on tumor-infiltrating lymphocytes in primary and metastatic melanoma. J. Investig. Dermatol. 2000, 114, 941–947. [Google Scholar] [CrossRef] [Green Version]
- Bloethner, S.; Snellman, E.; Bermejo, J.L.; Hiripi, E.; Gast, A.; Thirumaran, R.K.; Wellenreuther, R.; Hemminki, K.; Kumar, R. Differential gene expression in melanocytic nevi with the V600E BRAF mutation. Genes Chromosomes Cancer 2007, 46, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Favaro, E.; Bensaad, K.; Chong, M.G.; Tennant, D.A.; Ferguson, D.J.; Snell, C.; Steers, G.; Turley, H.; Li, J.L.; Gunther, U.L.; et al. Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell Metab. 2012, 16, 751–764. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Wei, K.; Liu, J.; Tang, K.; Zhang, H.; Zhu, L.; Chen, J.; Li, F.; Xu, P.; Chen, J.; et al. Glycogen metabolism regulates macrophage-mediated acute inflammatory responses. Nat. Commun. 2020, 11, 1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Navarro, F.J.; Martinez-Morcillo, F.J.; Lopez-Munoz, A.; Pardo-Sanchez, I.; Martinez-Menchon, T.; Corbalan-Velez, R.; Cayuela, M.L.; Perez-Oliva, A.B.; Garcia-Moreno, D.; Mulero, V. The vitamin B6-regulated enzymes PYGL and G6PD fuel NADPH oxidases to promote skin inflammation. Dev. Comp. Immunol. 2020, 108, 103666. [Google Scholar] [CrossRef]
- Cao, Y.; Sun, Y.; Chang, H.; Sun, X.; Yang, S. The E3 ubiquitin ligase RNF182 inhibits TLR-triggered cytokine production through promoting p65 ubiquitination and degradation. FEBS Lett. 2019, 593, 3210–3219. [Google Scholar] [CrossRef]
- Molgora, M.; Esaulova, E.; Vermi, W.; Hou, J.; Chen, Y.; Luo, J.; Brioschi, S.; Bugatti, M.; Omodei, A.S.; Ricci, B.; et al. TREM2 Modulation Remodels the Tumor Myeloid Landscape Enhancing Anti-PD-1 Immunotherapy. Cell 2020, 182, 886–900.e17. [Google Scholar] [CrossRef] [PubMed]
- Katzenelenbogen, Y.; Sheban, F.; Yalin, A.; Yofe, I.; Svetlichnyy, D.; Jaitin, D.A.; Bornstein, C.; Moshe, A.; Keren-Shaul, H.; Cohen, M.; et al. Coupled scRNA-Seq and Intracellular Protein Activity Reveal an Immunosuppressive Role of TREM2 in Cancer. Cell 2020, 182, 872–885.e19. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.Z.; Warycha, M.A.; Christos, P.J.; Darvishian, F.; Yee, H.; Kaminio, H.; Berman, R.S.; Shapiro, R.L.; Buckley, M.T.; Liebes, L.F.; et al. Assessing the clinical utility of measuring Insulin-like Growth Factor Binding Proteins in tissues and sera of melanoma patients. J. Transl. Med. 2008, 6, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haqq, C.; Nosrati, M.; Sudilovsky, D.; Crothers, J.; Khodabakhsh, D.; Pulliam, B.L.; Federman, S.; Miller, J.R., 3rd; Allen, R.E.; Singer, M.I.; et al. The gene expression signatures of melanoma progression. Proc. Natl. Acad. Sci. USA 2005, 102, 6092–6097. [Google Scholar] [CrossRef] [Green Version]
- Kashani-Sabet, M.; Rangel, J.; Torabian, S.; Nosrati, M.; Simko, J.; Jablons, D.M.; Moore, D.H.; Haqq, C.; Miller, J.R., 3rd; Sagebiel, R.W. A multi-marker assay to distinguish malignant melanomas from benign nevi. Proc. Natl. Acad. Sci. USA 2009, 106, 6268–6272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talantov, D.; Mazumder, A.; Yu, J.X.; Briggs, T.; Jiang, Y.; Backus, J.; Atkins, D.; Wang, Y. Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin. Cancer Res. 2005, 11, 7234–7242. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Dai, D.L.; Martinka, M.; Su, M.; Zhang, Y.; Campos, E.I.; Dorocicz, I.; Tang, L.; Huntsman, D.; Nelson, C.; et al. Osteopontin expression correlates with melanoma invasion. J. Investig. Dermatol. 2005, 124, 1044–1052. [Google Scholar] [CrossRef]
- Lezcano, C.; Jungbluth, A.A.; Nehal, K.S.; Hollmann, T.J.; Busam, K.J. PRAME Expression in Melanocytic Tumors. Am. J. Surg. Pathol. 2018, 42, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Ferris, L.K.; Jansen, B.; Ho, J.; Busam, K.J.; Gross, K.; Hansen, D.D.; Alsobrook, J.P., 2nd; Yao, Z.; Peck, G.L.; Gerami, P. Utility of a Noninvasive 2-Gene Molecular Assay for Cutaneous Melanoma and Effect on the Decision to Biopsy. JAMA Dermatol. 2017, 153, 675–680. [Google Scholar] [CrossRef]
- Gerami, P.; Yao, Z.; Polsky, D.; Jansen, B.; Busam, K.; Ho, J.; Martini, M.; Ferris, L.K. Development and validation of a noninvasive 2-gene molecular assay for cutaneous melanoma. J. Am. Acad. Dermatol. 2017, 76, 114–120.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepe, M.S.; Feng, Z.; Janes, H.; Bossuyt, P.M.; Potter, J.D. Pivotal evaluation of the accuracy of a biomarker used for classification or prediction: Standards for study design. J. Natl. Cancer Inst. 2008, 100, 1432–1438. [Google Scholar] [CrossRef] [Green Version]
- Altman, D.G.; McShane, L.M.; Sauerbrei, W.; Taube, S.E. Reporting Recommendations for Tumor Marker Prognostic Studies (REMARK): Explanation and elaboration. PLoS Med. 2012, 9, e1001216. [Google Scholar] [CrossRef] [Green Version]
- Simon, R.M.; Paik, S.; Hayes, D.F. Use of archived specimens in evaluation of prognostic and predictive biomarkers. J. Natl. Cancer Inst. 2009, 101, 1446–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, W.C. Residual dysplastic and other nevi in superficial spreading melanoma. Clinical correlations and association with sun damage. Cancer 1988, 62, 163–173. [Google Scholar] [CrossRef]
- Reddy, K.K.; Farber, M.J.; Bhawan, J.; Geronemus, R.G.; Rogers, G.S. Atypical (dysplastic) nevi: Outcomes of surgical excision and association with melanoma. JAMA Dermatol. 2013, 149, 928–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagebiel, R.W. Melanocytic nevi in histologic association with primary cutaneous melanoma of superficial spreading and nodular types: Effect of tumor thickness. J. Investig. Dermatol. 1993, 100, 322S–325S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Titus-Ernstoff, L.; Barnhill, R.L.; Duray, P.H.; Ernstoff, M.S.; Kirkwood, J.M. Dysplastic nevi in relation to superficial spreading melanoma. Cancer Epidemiol. Biomark. Prev. 1993, 2, 99–101. [Google Scholar]
- Kunz, M.; Loffler-Wirth, H.; Dannemann, M.; Willscher, E.; Doose, G.; Kelso, J.; Kottek, T.; Nickel, B.; Hopp, L.; Landsberg, J.; et al. RNA-seq analysis identifies different transcriptomic types and developmental trajectories of primary melanomas. Oncogene 2018, 37, 6136–6151. [Google Scholar] [CrossRef] [PubMed]





| Lesion Types 1 (n) | Sample Type | Relationship | Data Type | Data Access | References |
|---|---|---|---|---|---|
| BN (5), DN (7), CMM (16) | Frozen sections | Independent samples | 1-channel microarray | GSE114445 | Krueger: |
| Mitsui et al., 2016 [10] | |||||
| Yan et al., 2019 [11] | |||||
| BN (18), DN (11), RGM (8), VGM (15) | Fresh biopsy (VGM limited to dermal portion) | Independent samples | 2-channel microarray | GSE12391 | Scatolini et al. 2010 [12] |
| PN (17), CMM (20) | Manual microdissection of FFPE sections | Paired lesions | RNAseq | phs001550.v2.pl | Bastian: |
| Shain et al., 2015 [13] | |||||
| Shain et al., 2018 [14] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borden, E.S.; Adams, A.C.; Buetow, K.H.; Wilson, M.A.; Bauman, J.E.; Curiel-Lewandrowski, C.; Chow, H.-H.S.; LaFleur, B.J.; Hastings, K.T. Shared Gene Expression and Immune Pathway Changes Associated with Progression from Nevi to Melanoma. Cancers 2022, 14, 3. https://doi.org/10.3390/cancers14010003
Borden ES, Adams AC, Buetow KH, Wilson MA, Bauman JE, Curiel-Lewandrowski C, Chow H-HS, LaFleur BJ, Hastings KT. Shared Gene Expression and Immune Pathway Changes Associated with Progression from Nevi to Melanoma. Cancers. 2022; 14(1):3. https://doi.org/10.3390/cancers14010003
Chicago/Turabian StyleBorden, Elizabeth S., Anngela C. Adams, Kenneth H. Buetow, Melissa A. Wilson, Julie E. Bauman, Clara Curiel-Lewandrowski, H.-H. Sherry Chow, Bonnie J. LaFleur, and Karen Taraszka Hastings. 2022. "Shared Gene Expression and Immune Pathway Changes Associated with Progression from Nevi to Melanoma" Cancers 14, no. 1: 3. https://doi.org/10.3390/cancers14010003