
The Tumour Suppressor CYLD Is Required for Clathrin-Mediated Endocytosis of EGFR and Cetuximab-Induced Apoptosis in Head and Neck Squamous Cell Carcinoma

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Cell Culture
2.2. Transfection with siRNA
2.3. Construction of Plasmids
2.4. Cell Viability Assay
2.5. Immunofluorescence
2.6. Measurement of Cell-Surface EGFR Expression
2.7. Apoptosis Assay
2.8. Western Blotting
2.9. Samples from Patients and Patients’ Backgrounds
2.10. Immunohistochemistry
2.11. Statistical Analysis
2.12. Flow Diagram
3. Results
3.1. CME and Degradation of EGFR Are Essential for CTX-Induced Apoptosis
3.2. CYLD Is Required for EGF- and CTX-Induced CME of EGFR
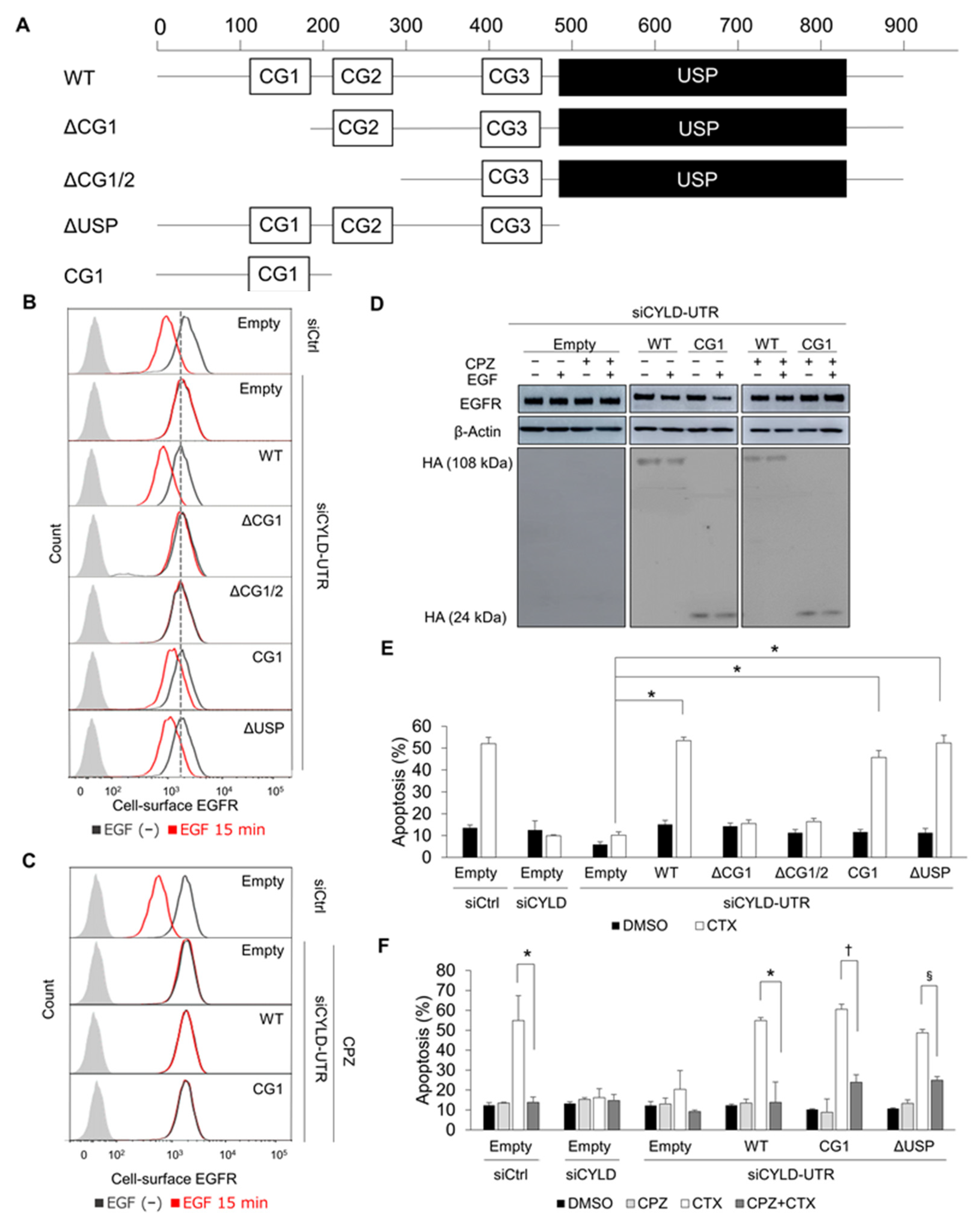
3.3. The N-Terminal Part of CYLD Is Responsible for EGFR CME and CTX-Induced Apoptosis
3.4. Relationship between CYLD Expression and Subcellular EGFR Localization in Human HNSCC Tissues
3.5. Cholesterol Sequestration Restores CYLD Knockdown-Induced Defective EGFR Trafficking and Overcomes CTX Resistance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Joseph, A.W.; D’Souza, G. Epidemiology of human papillomavirus-related head and neck cancer. Otolaryngol. Clin. N. Am. 2012, 45, 739–764. [Google Scholar] [CrossRef]
- van der Heijden, M.; Essers, P.; de Jong, M.C.; de Roest, R.H.; Sanduleanu, S.; Verhagen, C.V.; Vens, C. Biological determinants of chemo-radiotherapy response in HPV-negative head and neck cancer: A multicentric external validation. Front. Oncol. 2020, 9, 1470. [Google Scholar] [CrossRef] [PubMed]
- Machiels, J.P.; Leemans, C.R.; Golusinski, W.; Grau, C.; Licitra, L.; Gregoire, V. Squamous cell carcinoma of the oral cavity, larynx, oropharynx and hypopharynx: EHNS-ESMO-ESTRO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020, 31, 1462–1475. [Google Scholar] [CrossRef] [PubMed]
- Lerch, S.; Berthold, S.; Ziemann, F.; Dreffke, K.; Subtil, F.S.; Senger, Y.; Jensen, A.; Engenhart-Cabillic, R.; Dikomey, E.; Wittig, A.; et al. HPV-positive HNSCC cell lines show strongly enhanced radiosensitivity after photon but not after carbon ion irradiation. Radiother. Oncol. 2020, 151, 134–140. [Google Scholar] [CrossRef]
- Meccariello, G.; Maniaci, A.; Bianchi, G.; Cammaroto, G.; Iannella, G.; Catalano, A.; Vicini, C. Neck dissection and trans oral robotic surgery for oropharyngeal squamous cell carcinoma. Auris Nasus Larynx 2021, S0385-8146, 00163-2. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Sun, M.; Ge, X.; Sun, Y. EGFR tyrosine kinase inhibitors differentially affect autophagy in head and neck squamous cell carcinoma. Biochem. Biophys. Res. Commun. 2017, 486, 1027–1033. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, K.M. Structure-based view of epidermal growth factor receptor regulation. Annu. Rev. Biophys. 2008, 37, 353–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Schmitz, K.R.; Jeffrey, P.D.; Wiltzius, J.J.W.; Kussie, P.; Ferguson, K.M. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005, 7, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, R.; Hua, Y.J.; Liu, Y.P.; Qi, Y.; Zhang, Y.N.; Li, J.B.; Li, C.F.; Zou, X.; Yu, T.; Cao, J.Y.; et al. Concurrent chemoradiotherapy with or without anti-EGFR-targeted treatment for stage II-IVb nasopharyngeal carcinoma: Retrospective analysis with a large cohort and long follow-up. Theranostics 2017, 7, 2314–2324. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Cohen, R.B.; Jones, C.U.; Sur, R.K.; Raben, D.; Baselga, J.; Spencer, S.A.; Zhu, J.; et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010, 11, 21–28. [Google Scholar] [CrossRef]
- Vermorken, J.B.; Trigo, J.; Hitt, R.; Koralewski, P.; Diaz-Rubio, E.; Rolland, F.; Knecht, R.; Amellal, N.; Schueler, A.; Baselga, J. Open-Label, Uncontrolled, Multicenter Phase II Study to Evaluate the Efficacy and Toxicity of Cetuximab As a Single Agent in Patients With Recurrent and/or Metastatic Squamous Cell Carcinoma of the Head and Neck Who Failed to Respond to Platinum-Based Therapy. J. Clin. Oncol. 2007, 25, 2171–2177. [Google Scholar]
- Allegra, C.J.; Rumble, R.B.; Hamilton, S.R.; Mangu, P.B.; Roach, N.; Hantel, A.; Schilsky, R.L. Extended RAS gene mutation testing in metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy: American Society of Clinical Oncology Provisional Clinical Opinion Update 2015. J. Clin. Oncol. 2016, 34, 179–185. [Google Scholar] [CrossRef]
- Loeffler-Ragg, J.; Witsch-Baumgartner, M.; Tzankov, A.; Hilbe, W.; Schwentner, I.; Sprinzl, G.M.; Utermann, G.; Zwierzina, H. Low incidence of mutations in EGFR kinase domain in Caucasian patients with head and neck squamous cell carcinoma. Eur. J. Cancer 2006, 42, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Hwang, C.Y.; Cho, S.; Lee, D.; Gong, J.; Lee, S.; Nam, S.; Cho, K. Systems analysis identifies potential target genes to overcome cetuximab resistance in colorectal cancer cells. FEBS J. 2019, 286, 1305–1318. [Google Scholar] [CrossRef]
- Ortiz-Cuaran, S.; Bouaoud, J.; Karabajakian, A.; Fayette, J.; Saintigny, P. Precision Medicine Approaches to Overcome Resistance to Therapy in Head and Neck Cancers. Front. Oncol. 2021, 11, 614332. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.F.; Cohen, E.E.; Grandis, J.R. New Strategies in Head and Neck Cancer: Understanding Resistance to Epidermal Growth Factor Receptor Inhibitors. Clin. Cancer Res. 2010, 16, 2489–2495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, K.A.; Cohen, E.E. Mechanisms of and therapeutic approaches for overcoming resistance to epidermal growth factor receptor (EGFR)-targeted therapy in squamous cell carcinoma of the head and neck (SCCHN). Oral Oncol. 2015, 51, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Tomas, A.; Futter, C.; Eden, E.R. EGF receptor trafficking: Consequences for signaling and cancer. Trends Cell Biol. 2014, 24, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An Allosteric Mechanism for Activation of the Kinase Domain of Epidermal Growth Factor Receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsahafi, E.; Begg, K.; Amelio, I.; Raulf, N.; Lucarelli, P.; Sauter, T.; Tavassoli, M. Clinical update on head and neck cancer: Molecular biology and ongoing challenges. Cell Death Dis. 2019, 10, 540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.-Y.; Makino, K.; Xia, W.; Matin, A.; Wen, Y.; Kwong, K.Y.; Bourguignon, L.; Hung, M.-C. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat. Cell Biol. 2001, 3, 802–808. [Google Scholar] [CrossRef]
- Liao, H.-J.; Carpenter, G. Role of the Sec61 Translocon in EGF Receptor Trafficking to the Nucleus and Gene Expression. Mol. Biol. Cell 2007, 18, 1064–1072. [Google Scholar] [CrossRef] [Green Version]
- Jaramillo, M.L.; Leon, Z.; Grothe, S.; Paul-Roc, B.; Abulrob, A.; McCourt, M.O. Effect of the anti-receptor ligand-blocking 225 monoclonal antibody on EGF receptor endocytosis and sorting. Exp. Cell Res. 2006, 312, 2778–2790. [Google Scholar] [CrossRef]
- Liao, H.-J.; Carpenter, G. Cetuximab/C225-Induced Intracellular Trafficking of Epidermal Growth Factor Receptor. Cancer Res. 2009, 69, 6179–6183. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Kimura, T.; Nakagawa, T.; Fukuya, A.; Goji, T.; Fujimoto, S.; Muguruma, N.; Tsuji, Y.; Okahisa, T.; Takayama, T.; et al. EGFR Downregulation after Anti-EGFR Therapy Predicts the Antitumor Effect in Colorectal Cancer. Mol. Cancer Res. 2017, 15, 1445–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bignell, G.R.; Warren, W.; Seal, S.; Takahashi, M.; Rapley, E.; Barfoot, R.; Green, H.; Brown, C.; Biggs, P.J.; Lakhani, S.R.; et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat. Genet. 2000, 25, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Massoumi, R. Ubiquitin chain cleavage: CYLD at work. Trends Biochem. Sci. 2010, 35, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, J. CYLD—A deubiquitylase that acts to fine-tune microtubule properties and functions. J. Cell Sci. 2016, 129, 2289–2295. [Google Scholar] [CrossRef] [Green Version]
- Cui, Z.; Kang, H.; Grandis, J.R.; Johnson, D.E. CYLD alterations in the tumorigenesis and progression of human papillomavirus-associated head and neck cancers. Mol. Cancer Res. 2021, 19, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Jono, H.; Shinriki, S.; Nakamura, T.; Guo, J.; Sueta, A.; Tomiguchi, M.; Fujiwara, S.; Yamamoto-Ibusuki, M.; Murakami, K.-I.; et al. Clinical significance of CYLD downregulation in breast cancer. Breast Cancer Res. Treat. 2014, 143, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Shinriki, S.; Su, Y.; Nakamura, T.; Hayashi, M.; Tsuda, Y.; Murakami, Y.; Tasaki, M.; Hide, T.; Takezaki, T.; et al. Hypoxia suppresses cylindromatosis (CYLD) expression to promote inflammation in glioblastoma: Possible link to acquired resistance to anti-VEGF therapy. Oncotarget 2014, 5, 6353–6364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinoshita, H.; Okabe, H.; Beppu, T.; Chikamoto, A.; Hayashi, H.; Imai, K.; Mima, K.; Nakagawa, S.; Yokoyama, N.; Ishiko, T.; et al. CYLD downregulation is correlated with tumor development in patients with hepatocellular carcinoma. Mol. Clin. Oncol. 2013, 1, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinriki, S.; Jono, H.; Maeshiro, M.; Nakamura, T.; Guo, J.; Li, J.-D.; Ueda, M.; Yoshida, R.; Shinohara, M.; Nakayama, H.; et al. Loss of CYLD promotes cell invasion via ALK5 stabilization in oral squamous cell carcinoma. J. Pathol. 2017, 244, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Azimifar, S.B.; Böttcher, R.T.; Zanivan, S.; Grashoff, C.; Krüger, M.; Legate, K.R.; Mann, M.; Fässler, R. Induction of membrane circular dorsal ruffles requires co-signalling of integrin-ILK-complex and EGF receptor. J. Cell Sci. 2012, 125, 435–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suenaga, N.; Kuramitsu, M.; Komure, K.; Kanemaru, A.; Takano, K.; Ozeki, K.; Nishimura, Y.; Yoshida, R.; Nakayama, H.; Shinriki, S.; et al. Loss of Tumor Suppressor CYLD Expression Triggers Cisplatin Resistance in Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 5194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanlandingham, P.A.; Ceresa, B.P. Rab7 Regulates Late Endocytic Trafficking Downstream of Multivesicular Body Biogenesis and Cargo Sequestration. J. Biol. Chem. 2009, 284, 12110–12124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roepstorff, K.; Grøvdal, L.; Grandal, M.; Lerdrup, M.; van Deurs, B. Endocytic downregulation of ErbB receptors: Mechanisms and relevance in cancer. Histochem. Cell Biol. 2008, 129, 563–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lajoie, P.; Nabi, I.R. Lipid Rafts, Caveolae, and Their Endocytosis. Int. Rev. Cell Mol. Biol. 2010, 282, 135–163. [Google Scholar] [PubMed]
- LE Roy, C.; Wrana, J.L. Clathrin- and non-clathrin-mediated endocytic regulation of cell signalling. Nat. Rev. Mol. Cell Biol. 2005, 6, 112–126. [Google Scholar] [CrossRef] [PubMed]
- Thuille, N.; Wachowicz, K.; Kleiter, N.; Kaminski, S.; Fresser, F.; Lutz-Nicoladoni, C.; Leitges, M.; Thome, M.; Massoumi, R.; Baier, G. PKCθ/β and CYLD Are Antagonistic Partners in the NFκB and NFAT Transactivation Pathways in Primary Mouse CD3+ T Lymphocytes. PLoS ONE 2013, 8, e53709. [Google Scholar]
- Chen, Y.; Liu, G.; Guo, L.; Wang, H.; Fu, Y.; Luo, Y. Enhancement of tumor uptake and therapeutic efficacy of EGFR-targeted antibody cetuximab and antibody-drug conjugates by cholesterol sequestration. Int. J. Cancer 2015, 136, 182–194. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, S.; Lu, X.; Zhang, H.; Fu, Y.; Luo, Y. Cholesterol sequestration by nystatin enhances the uptake and activity of endostatin in endothelium via regulating distinct endocytic pathways. Blood 2011, 117, 6392–6403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, S.Y.; Brown, A.J.; Chua, N.K.; Yoon, J.-Y.; Lee, J.-J.; Yang, J.O.; Jang, I.; Jeon, S.-J.; Choi, T.-I.; Kim, C.-H.; et al. Reduction of Squalene Epoxidase by Cholesterol Accumulation Accelerates Colorectal Cancer Progression and Metastasis. Gastroenterology 2021, 160, 1194–1207.e28. [Google Scholar] [CrossRef] [PubMed]
- Colin, D.; Limagne, E.; Jeanningros, S.; Jacquel, A.; Lizard, G.; Athias, A.; Gambert, P.; Hichami, A.; Latruffe, N.; Solary, E.; et al. Endocytosis of Resveratrol via Lipid Rafts and Activation of Downstream Signaling Pathways in Cancer Cells. Cancer Prev. Res. 2011, 4, 1095–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Quiles, V.; Akimov, V.; Osinalde, N.; Francavilla, C.; Puglia, M.; Barrio-Hernandez, I.; Kratchmarova, I.; Olsen, J.V.; Blagoev, B. Cylindromatosis tumor suppressor protein (CYLD) deubiquitinase is necessary for proper ubiquitination and degradation of the epidermal growth factor receptor. Mol. Cell. Proteom. 2017, 16, 1433–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, K.; Kigawa, T.; Koshiba, S.; Sato, K.; Matsuo, Y.; Sakamoto, A.; Takagi, T.; Shirouzu, M.; Yabuki, T.; Nunokawa, E.; et al. The CAP-Gly domain of CYLD associates with the proline-rich sequence in NEMO/IKK. Structure 2004, 12, 1719–1728. [Google Scholar] [CrossRef] [Green Version]
- Wickström, S.A.; Masoumi, K.C.; Khochbin, S.; Fässler, R.; Massoumi, R. CYLD negatively regulates cell-cycle progression by inactivating HDAC6 and increasing the levels of acetylated tubulin. EMBO J. 2010, 29, 131–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, J.A.; Halverson-Tamboli, R.A.; Rasenick, M.M. Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci. 2006, 8, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Montagnac, G.; Meas-Yedid, V.; Irondelle, M.; Castro-Castro, A.; Franco, M.; Shida, T.; Nachury, M.; Benmerah, A.; Olivo-Marin, J.-C.; Chavrier, P. αTAT1 catalyses microtubule acetylation at clathrin-coated pits. Nat. Cell Biol. 2013, 502, 567–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muraro, E.; Fanetti, G.; Lupato, V.; Giacomarra, V.; Steffan, A.; Gobitti, C.; Vaccher, E.; Franchin, G. Cetuximab in locally advanced head and neck squamous cell carcinoma: Biological mechanisms involved in efficacy, toxicity and resistance. Crit. Rev. Oncol. 2021, 164, 103424. [Google Scholar] [CrossRef]
- Lansiaux, A.; Rebucci, M.; Peixoto, P.; Dewitte, A.; Wattez, N.; De Nuncques, M.-A.; Rezvoy, N.; Vautravers-Dewas, C.; Buisine, M.-P.; Guerin, E.; et al. Mechanisms underlying resistance to cetuximab in the HNSCC cell line: Role of AKT inhibition in bypassing this resistance. Int. J. Oncol. 2010, 38, 189–200. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Huang, S.; Kruser, T.; Nechrebecki, M.M.; Armstrong, E.A.; Benavente, S.; Gondi, V.; Hsu, K.-T.; Harari, P.M. Mechanisms of acquired resistance to cetuximab: Role of HER (ErbB) family members. Oncogene 2008, 27, 3944–3956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, A.W.K.; Wasan, K.M.; Lopez-Berestein, G. Development of liposomal polyene antibiotics: An historical perspective. J. Pharm. Pharm. Sci. 2003, 6, 67–83. [Google Scholar]
- Stoehr, M.; Mozet, C.; Boehm, A.; Aigner, A.; Dietz, A.; Wichmann, G. Simvastatin suppresses head and neck squamous cell carcinoma ex vivo and enhances the cytostatic effects of chemotherapeutics. Cancer Chemother. Pharmacol. 2014, 73, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, I.; Han, B.; Park, J.O.; Jang, J.; Park, C.; Kang, W.K. Effect of Simvastatin on Cetuximab Resistance in Human Colorectal Cancer with KRAS Mutations. J. Natl. Cancer Inst. 2011, 103, 674–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Wang, X.; Song, D.; Liu, X.; Gu, Y.; Xu, Z.; Wang, X.; Zhang, X.; Ye, Q.; Tong, Z.; et al. Cholesterol induces epithelial-to-mesenchymal transition of prostate cancer cells by suppressing degradation of EGFR through APMAP. Cancer Res. 2019, 79, 3063–3075. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Gordon, M.; Schultheis, A.M.; Yang, D.; Nagashima, F.; Azuma, M.; Chang, H.-M.; Borucka, E.; Lurje, G.; Sherrod, A.E.; et al. FCGR2A and FCGR3A Polymorphisms Associated With Clinical Outcome of Epidermal Growth Factor Receptor–Expressing Metastatic Colorectal Cancer Patients Treated With Single-Agent Cetuximab. J. Clin. Oncol. 2007, 25, 3712–3718. [Google Scholar] [CrossRef]
- Chew, H.Y.; De Lima, P.O.; Cruz, J.L.G.; Banushi, B.; Echejoh, G.; Hu, L.; Joseph, S.R.; Lum, B.; Rae, J.; O’Donnell, J.S.; et al. Endocytosis Inhibition in Humans to Improve Responses to ADCC-Mediating Antibodies. Cell 2020, 180, 895–914. [Google Scholar] [CrossRef]
- Sun, S.-C. Deubiquitylation and regulation of the immune response. Nat. Rev. Immunol. 2008, 8, 501–511. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutants | Forward Primers (5′–3′) | Reverse Primers (5′–3′) |
|---|---|---|
| ΔCG1 | ATGCAGGTCGAACTTCCTCCTTTGG | GGGCCGGCCAGCGTAGTCTGGTACA |
| ΔCG1/2 | ATGCTTGCCTTTATGTCAAGAGGTG | GGGCCGGCCAGCGTAGTCTGGTACA |
| CG1 | GGCGCGCCTCTAGAACTATAGTGAG | TTATGCAGTGTCATCATCTTCTAT |
| ΔUSP | GGCGCGCCTCTAGAACTATAGTGAG | TTAAATCATTATCTCCAAGCCTTC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, R.; Shinriki, S.; Maeshiro, M.; Hirayama, M.; Jono, H.; Yoshida, R.; Nakayama, H.; Matsui, H. The Tumour Suppressor CYLD Is Required for Clathrin-Mediated Endocytosis of EGFR and Cetuximab-Induced Apoptosis in Head and Neck Squamous Cell Carcinoma. Cancers 2022, 14, 173. https://doi.org/10.3390/cancers14010173
Liu R, Shinriki S, Maeshiro M, Hirayama M, Jono H, Yoshida R, Nakayama H, Matsui H. The Tumour Suppressor CYLD Is Required for Clathrin-Mediated Endocytosis of EGFR and Cetuximab-Induced Apoptosis in Head and Neck Squamous Cell Carcinoma. Cancers. 2022; 14(1):173. https://doi.org/10.3390/cancers14010173
Chicago/Turabian StyleLiu, Rin, Satoru Shinriki, Manabu Maeshiro, Mayumi Hirayama, Hirofumi Jono, Ryoji Yoshida, Hideki Nakayama, and Hirotaka Matsui. 2022. "The Tumour Suppressor CYLD Is Required for Clathrin-Mediated Endocytosis of EGFR and Cetuximab-Induced Apoptosis in Head and Neck Squamous Cell Carcinoma" Cancers 14, no. 1: 173. https://doi.org/10.3390/cancers14010173