
Discovering the Mutational Profile of Early Colorectal Lesions: A Translational Impact

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Hereditary Settings
2.1. Familial Adenomatous Polyposis and MUTYH-Associated Polyposis
Somatic Mutational Profile in FAP and MAP
2.2. Lynch Syndrome
Somatic Mutational Profile in LS Tumors
3. Sporadic Colorectal Adenomas
3.1. Conventional Colorectal Adenomas
3.2. Colorectal Serrated Lesions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Smits, A.M.M.; Bos, J.L.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal—Tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Church, J.M. Clinical significance of small colorectal polyps. Dis. Colon Rectum 2004, 47, 481–485. [Google Scholar] [CrossRef]
- Leggett, B.; Whitehall, V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology 2010, 138, 2088–2100. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.S.; Farraye, F.A.; Yang, S.; O’Brien, M.J. The clinical significance of serrated polyps. Am. J. Gastroenterol. 2011, 106, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.-I.; van Niekerk, D.W.; Owen, D.; Tha, S.P.L.; Turbin, D.A.; Webber, D.L. Longitudinal outcome study of sessile serrated adenomas of the colorectum: An increased risk for subsequent right-sided colorectal carcinoma. Am. J. Surg. Pathol. 2010, 34, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Snover, D.C. Update on the serrated pathway to colorectal carcinoma. Hum. Pathol. 2011, 42, 1–10. [Google Scholar] [CrossRef]
- Pai, R.K.; Mäkinen, M.J.; Rosty, C. Colorectal serrated lesions and polyps. In WHO Classification of Tumours of the Digestive System; Nagtegaal, I.D., Arends, M.J., Odze, R.D., Lam, A.K., Eds.; Lyon IARC Press: Lyon, France, 2019; pp. 163–169. [Google Scholar]
- Snover, D.C.; Ahnen, D.J.; Burt, R.W. Serrated polyps of the colon and rectum and serrated polyposis. In WHO Classification of Tumours of the Digestive System; Nagtegaal, I.D., Arends, M.J., Odze, R.D., Lam, A.K., Eds.; Lyon IARC Press: Lyon, France, 2019. [Google Scholar]
- Løberg, M.; Kalager, M.; Holme, Ø.; Hoff, G.; Adami, H.-O.; Bretthauer, M. Long-term colorectal-cancer mortality after adenoma removal. N. Engl. J. Med. 2014, 371, 799–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Click, B.; Pinsky, P.; Hickey, T.; Doroudi, M.; Schoen, R. Association of colonoscopy adenoma findings with long-term colorectal cancer incidence. JAMA 2018, 319, 2021–2031. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Hang, D.; Wu, K.; Nayor, J.; Drew, D.; Giovannucci, E.; Ogino, S.; Chan, A.; Song, M. Long-term risk of colorectal cancer after removal of conventional adenomas and serrated polyps. Gastroenterology 2020, 158, 852–861. [Google Scholar] [CrossRef]
- Lee, J.; Jensen, C.; Levin, T.; Doubeni, C.; Zauber, A.; Chubak, J.; Kamineni, A.; Schottinger, J.; Ghai, N.; Udaltsova, N.; et al. Long-term risk of colorectal cancer and related death after adenoma removal in a large, community-based population. Gastroenterology 2020, 158, 884–894. [Google Scholar] [CrossRef] [PubMed]
- Holme, Ø.; Bretthauer, M.; Eide, T.J.; Løberg, E.M.; Grzyb, K.; Løberg, M.; Kalager, M.; Adami, H.O.; Kjellevold, Ø.; Hoff, G. Long-term risk of colorectal cancer in individuals with serrated polyps. Gut 2015, 64, 929–936. [Google Scholar] [CrossRef]
- Erichsen, R.; Baron, J.A.; Hamilton-Dutoit, S.J.; Snover, D.C.; Torlakovic, E.E.; Pedersen, L.; Frøslev, T.; Vyberg, M.; Hamilton, S.R.; Sørensen, H.T. Increased risk of colorectal cancer development among patients with serrated polyps. Gastroenterology 2016, 150, 895–902. [Google Scholar] [CrossRef]
- Song, M.; Emilsson, L.; Bozorg, S.R.; Nguyen, L.H.; Joshi, A.D.; Staller, K.; Nayor, J.; Chan, A.T.; Ludvigsson, J.F. Risk of colorectal cancer incidence and mortality after polypectomy: A Swedish record-linkage study. Lancet Gastroenterol. Hepatol. 2020, 5, 537–547. [Google Scholar] [CrossRef]
- Vasen, H.F.A.; Tomlinson, I.; Castells, A. Clinical management of hereditary colorectal cancer syndromes. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Edelstein, D.L.; Axilbund, J.E.; Hylind, L.M.; Romans, K.; Griffin, C.A.; Cruz-Correa, M.; Giardiello, F.M. Serrated polyposis: Rapid and relentless development of colorectal neoplasia. Gut 2013, 62, 404–408. [Google Scholar] [CrossRef]
- Colussi, D.; Brandi, G.; Bazzoli, F.; Ricciardiello, L. Molecular pathways involved in colorectal cancer: Implications for disease behavior and prevention. Int. J. Mol. Sci. 2013, 14, 16365–16385. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network, T.C.G.A. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Shiovitz, S.; Copeland, W.K.; Passarelli, M.N.; Burnett-Hartman, A.N.; Grady, W.M.; Potter, J.D.; Gallinger, S.; Buchanan, D.D.; Rosty, C.; Win, A.K.; et al. Characterisation of familial colorectal cancer Type X, Lynch syndrome, and non-familial colorectal cancer. Br. J. Cancer 2014, 111. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Kulke, M.H.; Fuchs, C.S.; Allen, B.A.; Uno, H.; Hornick, J.L.; Ukeagbu, C.I.; Brais, L.K.; McNamara, P.G.; Mayer, R.J.; et al. Cancer susceptibility gene mutations in individuals with colorectal cancer. J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wu, W.K.K.; Li, X.; He, J.; Li, X.X.; Ng, S.S.M.; Yu, C.; Gao, Z.; Yang, J.; Li, M.; et al. Novel recurrently mutated genes and a prognostic mutation signature in colorectal cancer. Gut 2015, 64, 636–645. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Grolleman, J.E.; Díaz-Gay, M.; Franch-Expósito, S.; Castellví-Bel, S.; de Voer, R.M. Somatic mutational signatures in polyposis and colorectal cancer. Mol. Asp. Med. 2019, 69, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Doxey, B.W.; Kuwada, S.K.; Burt, R.W. Inherited polyposis syndromes: Molecular mechanisms, clinicopathology, and genetic testing. Clin. Gastroenterol. Hepatol. 2005, 3. [Google Scholar] [CrossRef]
- Aretz, S.; Stienen, D.; Friedrichs, N.; Stemmler, S.; Uhlhaas, S.; Rahner, N.; Propping, P.; Friedl, W. Somatic APC mosaicism: A frequent cause of familial adenomatous polyposis (FAP). Hum. Mutat. 2007, 28, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Ciavarella, M.; Miccoli, S.; Prossomariti, A.; Pippucci, T.; Bonora, E.; Buscherini, F.; Palombo, F.; Zuntini, R.; Balbi, T.; Ceccarelli, C.; et al. Somatic APC mosaicism and oligogenic inheritance in genetically unsolved colorectal adenomatous polyposis patients. Eur. J. Hum. Genet. 2018, 26, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Joerink-van de Beld, M.C.; Jones, N.; Vogt, S.; Tops, C.M.; Vasen, H.F.A.; Sampson, J.R.; Aretz, S.; Hes, F.J. Analysis of MUTYH genotypes and colorectal phenotypes in patients With MUTYH-associated polyposis. Gastroenterology 2009, 136, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Brosens, L.A.A.; Offerhaus, G.J.A.; Giardiello, F.M.; de Leng, W.W.J.; Montgomery, E.A. Pathology and genetics of hereditary colorectal cancer. Pathology 2018, 50, 49–59. [Google Scholar] [CrossRef]
- Borras, E.; San Lucas, F.A.; Chang, K.; Zhou, R.; Masand, G.; Fowler, J.; Mork, M.E.; You, Y.N.; Taggart, M.W.; McAllister, F.; et al. Genomic Landscape of Colorectal Mucosa and Adenomas. Cancer Prev. Res. 2016, 9, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Gausachs, M.; Borras, E.; Chang, K.; Gonzalez, S.; Azuara, D.; Delgado Amador, A.; Lopez-Doriga, A.; San Lucas, F.A.; Sanjuan, X.; Paules, M.J.; et al. Mutational Heterogeneity in APC and KRAS Arises at the Crypt Level and Leads to Polyclonality in Early Colorectal Tumorigenesis. Clin. Cancer Res. 2017, 23, 5936–5947. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, R.; Zhou, X.; Wang, W.; Gao, S.; Mao, Y.; Wu, X.; Guo, L.; Liu, H.; Wen, L.; et al. Genomic and transcriptomic profiling of carcinogenesis in patients with familial adenomatous polyposis. Gut 2020, 69, 1283–1293. [Google Scholar] [CrossRef] [Green Version]
- Rashid, M.; Fischer, A.; Wilson, C.H.; Tiffen, J.; Rust, A.G.; Stevens, P.; Idziaszczyk, S.; Maynard, J.; Williams, G.T.; Mustonen, V.; et al. Adenoma development in familial adenomatous polyposis and MUTYH -associated polyposis: Somatic landscape and driver genes. J. Pathol. 2016, 238, 98–108. [Google Scholar] [CrossRef] [Green Version]
- Sekine, S.; Mori, T.; Ogawa, R.; Tanaka, M.; Yoshida, H.; Taniguchi, H.; Nakajima, T.; Sugano, K.; Yoshida, T.; Kato, M.; et al. Mismatch repair deficiency commonly precedes adenoma formation in Lynch syndrome—Associated colorectal tumorigenesis. Mod. Pathol. 2017, 30, 1144–1151. [Google Scholar] [CrossRef]
- Mäki-Nevala, S.; Valo, S.; Ristimäki, A.; Sarhadi, V.; Knuutila, S.; Nyström, M.; Renkonen-Sinisalo, L.; Lepistö, A.; Mecklin, J.-P.; Peltomäki, P. DNA methylation changes and somatic mutations as tumorigenic events in Lynch syndrome-associated adenomas retaining mismatch repair protein expression. EBioMedicine 2019, 39, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Binder, H.; Hopp, L.; Schweiger, M.R.; Hoffmann, S.; Jühling, F.; Kerick, M.; Timmermann, B.; Siebert, S.; Grimm, C.; Nersisyan, L.; et al. Genomic and transcriptomic heterogeneity of colorectal tumours arising in Lynch syndrome. J. Pathol. 2017, 243, 242–254. [Google Scholar] [CrossRef]
- Major, M.B.; Camp, N.D.; Berndt, J.D.; Yi, X.; Goldenberg, S.J.; Hubbert, C.; Biechele, T.L.; Gingras, A.-C.; Zheng, N.; MacCoss, M.J.; et al. Wilms tumor suppressor WTX negatively regulates WNT/ -catenin signaling. Science 2007, 316, 1043–1046. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Pamplona, R.; Lopez-Doriga, A.; Pare-Brunet, L.; Lazaro, K.; Bellido, F.; Alonso, M.H.; Ausso, S.; Guino, E.; Beltran, S.; Castro-Giner, F.; et al. Exome sequencing reveals AMER1 as a frequently mutated gene in colorectal cancer. Clin. Cancer Res. 2015, 21, 4709–4718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, L.E.; Hurley, J.J.; Meuser, E.; Jose, S.; Ashelford, K.E.; Mort, M.; Idziaszczyk, S.; Maynard, J.; Brito, H.L.; Harry, M.; et al. Burden and profile of somatic mutation in duodenal adenomas from patients with familial adenomatous- and MUTYH -associated polyposis. Clin. Cancer Res. 2017, 23, 6721–6732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch syndrome: 1895-2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef]
- De Jong, A.E.; Morreau, H.; Van Puijenbroek, M.; Eilers, P.H.C.; Wijnen, J.; Nagengast, F.M.; Griffioen, G.; Cats, A.; Menko, F.H.; Kleibeuker, J.H.; et al. The role of mismatch repair gene defects in the development of adenomas in patients with HNPCC. Gastroenterology 2004, 126, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Nakajima, T.; Sugano, K.; Yoshida, T.; Taniguchi, H.; Kanemitsu, Y.; Nagino, M.; Sekine, S. Mismatch repair deficiency in Lynch syndrome-associated colorectal adenomas is more prevalent in older patients. Histopathology 2016, 69, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Valo, S.; Kaur, S.; Ristimäki, A.; Renkonen-Sinisalo, L.; Järvinen, H.; Mecklin, J.P.; Nyström, M.; Peltomäki, P. DNA hypermethylation appears early and shows increased frequency with dysplasia in Lynch syndrome-associated colorectal adenomas and carcinomas. Clin. Epigenetics 2015, 7, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahadova, A.; Seppälä, T.T.; Engel, C.; Gallon, R.; Burn, J.; Holinski-Feder, E.; Steinke-Lange, V.; Möslein, G.; Nielsen, M.; ten Broeke, S.W.; et al. The “unnatural” history of colorectal cancer in Lynch syndrome: Lessons from colonoscopy surveillance. Int. J. Cancer 2021, 148, 800–811. [Google Scholar] [CrossRef]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Goel, A.; Hornick, J.L.; Sen, A.; Turgeon, D.K.; Ruffin IV, M.T.; Marcon, N.E.; Baron, J.A.; Bresalier, R.S.; Syngal, S.; et al. Microsatellite instability and DNA mismatch repair protein deficiency in lynch syndrome colorectal polyps. Cancer Prev. Res. 2012, 5, 574–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, C.; Ahadova, A.; Seppälä, T.T.; Aretz, S.; Bigirwamungu-Bargeman, M.; Bläker, H.; Bucksch, K.; Büttner, R.; de Vos Tot Nederveen Cappel, W.T.; Endris, V.; et al. Associations of pathogenic variants in MLH1, MSH2, and MSH6 with risk of colorectal adenomas and tumors and with somatic mutations in patients with Lynch syndrome. Gastroenterology 2020, 158, 1326–1333. [Google Scholar] [CrossRef]
- Lamba, M.; Wakeman, C.; Ebel, R.; Hamilton, S.; Frampton, C.; Kiesanowski, M.; Griffiths, B.; Keating, J.; Parry, S.; Chalmers-Watson, T. Associations between mutations in MSH6 and PMS2 and risk of surveillance-detected colorectal cancer. Clin. Gastroenterol. Hepatol. 2020, 18, 2768–2774. [Google Scholar] [CrossRef]
- Møller, P.; Seppälä, T.T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Evans, D.G.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.H.; et al. Cancer risk and survival in path-MMR carriers by gene and gender up to 75 years of age: A report from the prospective Lynch syndrome database. Gut 2018, 67, 1306–1316. [Google Scholar] [CrossRef] [Green Version]
- ten Broeke, S.W.; van Bavel, T.C.; Jansen, A.M.L.; Gómez-García, E.; Hes, F.J.; van Hest, L.P.; Letteboer, T.G.W.; Olderode-Berends, M.J.W.; Ruano, D.; Spruijt, L.; et al. Molecular background of colorectal tumors from patients with Lynch syndrome associated with germline variants in PMS2. Gastroenterology 2018, 155, 844–851. [Google Scholar] [CrossRef]
- Ahadova, A.; von Knebel Doeberitz, M.; Bläker, H.; Kloor, M. CTNNB1-mutant colorectal carcinomas with immediate invasive growth: A model of interval cancers in Lynch syndrome. Fam. Cancer 2016, 15, 579–586. [Google Scholar] [CrossRef]
- Ahadova, A.; Gallon, R.; Gebert, J.; Ballhausen, A.; Endris, V.; Kirchner, M.; Stenzinger, A.; Burn, J.; von Knebel Doeberitz, M.; Bläker, H.; et al. Three molecular pathways model colorectal carcinogenesis in Lynch syndrome. Int. J. Cancer 2018, 143, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef] [Green Version]
- Lee-Six, H.; Olafsson, S.; Ellis, P.; Osborne, R.J.; Sanders, M.A.; Moore, L.; Georgakopoulos, N.; Torrente, F.; Noorani, A.; Goddard, M.; et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 2019, 574, 532–537. [Google Scholar] [CrossRef]
- Sievers, C.K.; Zou, L.S.; Pickhardt, P.J.; Matkowskyj, K.A.; Albrecht, D.M.; Clipson, L.; Bacher, J.W.; Pooler, B.D.; Moawad, F.J.; Cash, B.D.; et al. Subclonal diversity arises early even in small colorectal tumours and contributes to differential growth fates. Gut 2017, 66, 2132–2140. [Google Scholar] [CrossRef] [Green Version]
- Karczmarski, J.; Goryca, K.; Pachlewski, J.; Dabrowska, M.; Pysniak, K.; Paziewska, A.; Kulecka, M.; Lenarcik, M.; Mroz, A.; Mikula, M.; et al. Mutation profiling of premalignant colorectal neoplasia. Gastroenterol. Res. Pract. 2019, 2019, 2542640. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Yoo, J.; Song, Y.S.; Lim, C.H.; Kim, T.M. Mutation analysis of colorectal and gastric carcinomas originating from adenomas: Insights into genomic evolution associated with malignant progression. Cancers 2020, 12, 325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.-H.; Raju, G.S.; Huff, C.; Ye, Y.; Gu, J.; Chen, J.-S.; Hildebrandt, M.A.T.; Liang, H.; Menter, D.G.; Morris, J.; et al. The somatic mutation landscape of premalignant colorectal adenoma. Gut 2018, 67, 1299–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Niida, A.; Uchi, R.; Hirata, H.; Komatsu, H.; Sakimura, S.; Hayashi, S.; Nambara, S.; Kuroda, Y.; Ito, S.; et al. A temporal shift of the evolutionary principle shaping intratumor heterogeneity in colorectal cancer. Nat. Commun. 2018, 9, 2884. [Google Scholar] [CrossRef]
- Testa, U.; Pelosi, E.; Castelli, G. Colorectal cancer: Genetic abnormalities, tumor progression, tumor heterogeneity, clonal evolution and tumor-initiating cells. Med. Sci. 2018, 6, 31. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Zhang, X.Y.; Hu, Z.; Hou, Q.; Zhang, H.; Li, Y.; Li, S.; Yue, J.; Jiang, Z.; Weissman, S.M.; et al. Evolution and heterogeneity of non-hereditary colorectal cancer revealed by single-cell exome sequencing. Oncogene 2017, 36, 2857–2867. [Google Scholar] [CrossRef]
- Irahara, N.; Baba, Y.; Nosho, K.; Shima, K.; Yan, L.; Dias-Santagata, D.; Iafrate, A.J.; Fuchs, C.S.; Haigis, K.M.; Ogino, S. NRAS mutations are rare in colorectal cancer. Diagn. Mol. Pathol. 2010, 19, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Jung, S.H.; Kim, T.M.; Rhee, J.K.; Park, H.C.; Kim, M.S.; Kim, S.S.; An, C.H.; Lee, S.H.; Chung, Y.J. Whole-exome sequencing identified mutational profiles of high-grade colon adenomas. Oncotarget 2017, 8, 6579–6588. [Google Scholar] [CrossRef] [Green Version]
- Druliner, B.R.; Wang, P.; Bae, T.; Baheti, S.; Slettedahl, S.; Mahoney, D.; Vasmatzis, N.; Xu, H.; Kim, M.; Bockol, M.; et al. Molecular characterization of colorectal adenomas with and without malignancy reveals distinguishing genome, transcriptome and methylome alterations. Sci. Rep. 2018, 8, 3161. [Google Scholar] [CrossRef] [PubMed]
- Gala, M.K.; Mizukami, Y.; Le, L.P.; Moriichi, K.; Austin, T.; Yamamoto, M.; Lauwers, G.Y.; Bardeesy, N.; Chung, D.C. Germline mutations in oncogene-induced senescence pathways are associated with multiple sessile serrated adenomas. Gastroenterology 2014, 146, 520–529.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.H.N.; Lai, J.C.W.; Ho, S.L.; Leung, W.K.; Law, W.L.; Lee, J.F.Y.; Chan, A.K.W.; Tsui, W.Y.; Chan, A.S.Y.; Lee, B.C.H.; et al. RNF43 germline and somatic mutation in serrated neoplasia pathway and its association with BRAF mutation. Gut 2017, 66, 1645–1656. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.-H.; Liau, J.-Y.; Yuan, C.-T.; Lin, Y.-L.; Tseng, L.-H.; Cheng, M.-L.; Jeng, Y.-M. RNF43 Is an Early and Specific Mutated Gene in the Serrated Pathway, With Increased Frequency in Traditional Serrated Adenoma and Its Associated Malignancy. Am. J. Surg. Pathol. 2016, 40, 1352–1359. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Yamashita, S.; Tanabe, T.; Hashimoto, T.; Yoshida, H.; Taniguchi, H.; Kojima, M.; Shinmura, K.; Saito, Y.; Hiraoka, N.; et al. Frequent PTPRK-RSPO3 fusions and RNF43 mutations in colorectal traditional serrated adenoma. J. Pathol. 2016, 239, 133–138. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, T.; Yamashita, S.; Yoshida, H.; Taniguchi, H.; Ushijima, T.; Yamada, T.; Saito, Y.; Ochiai, A.; Sekine, S.; Hiraoka, N. WNT pathway gene mutations are associated with the presence of dysplasia in colorectal sessile serrated adenoma/polyps. Am. J. Surg. Pathol. 2017, 41, 1188–1197. [Google Scholar] [CrossRef]
- Bettington, M.L.; Walker, N.I.; Rosty, C.; Brown, I.S.; Clouston, A.D.; McKeone, D.M.; Pearson, S.-A.; Klein, K.; Leggett, B.A.; Whitehall, V.L. A clinicopathological and molecular analysis of 200 traditional serrated adenomas. Mod. Pathol. 2015, 28, 414–427. [Google Scholar] [CrossRef]
- Murakami, T.; Akazawa, Y.; Yatagai, N.; Hiromoto, T.; Sasahara, N.; Saito, T.; Sakamoto, N.; Nagahara, A.; Yao, T. Molecular characterization of sessile serrated adenoma/polyps with dysplasia/carcinoma based on immunohistochemistry, next-generation sequencing, and microsatellite instability testing: A case series study. Diagn. Pathol. 2018, 13, 88. [Google Scholar] [CrossRef]
- Borowsky, J.; Dumenil, T.; Bettington, M.; Pearson, S.A.; Bond, C.; Fennell, L.; Liu, C.; McKeone, D.; Rosty, C.; Brown, I.; et al. The role of APC in WNT pathway activation in serrated neoplasia. Mod. Pathol. 2018, 31, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, T.; Ogawa, R.; Yoshida, H.; Taniguchi, H.; Kojima, M.; Saito, Y.; Sekine, S. Acquisition of WNT pathway gene alterations coincides with the transition from precursor polyps to traditional serrated adenomas. Am. J. Surg. Pathol. 2019, 43, 132–139. [Google Scholar] [CrossRef]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef]
- Koo, B.-K.; Spit, M.; Jordens, I.; Low, T.Y.; Stange, D.E.; van de Wetering, M.; van Es, J.H.; Mohammed, S.; Heck, A.J.R.; Maurice, M.M.; et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012, 488, 665–669. [Google Scholar] [CrossRef]
- de Lau, W.; Peng, W.C.; Gros, P.; Clevers, H. The R-spondin/Lgr5/Rnf43 module: Regulator of Wnt signal strength. Genes Dev. 2014, 28, 305–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, N.; Yamaguchi, K.; Ikenoue, T.; Fujii, T.; Furukawa, Y. Identification of Two Wnt-Responsive Elements in the Intron of RING Finger Protein 43 (RNF43) Gene. PLoS ONE 2014, 9, e86582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jen, J.; Powell, S.M.; Papadopoulos, N.; Smith, K.J.; Hamilton, S.R.; Vogelstein, B.; Kinzler, K.W. Molecular determinants of dysplasia in colorectal lesions. Cancer Res. 1994, 54, 5523–5526. [Google Scholar] [PubMed]
- Carmon, K.; Gong, X.; Lin, Q.; Thomas, A.; Liu, Q. R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/beta-catenin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 11452–11457. [Google Scholar] [CrossRef] [Green Version]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Ogawa, R.; Hashimoto, T.; Motohiro, K.; Yoshida, H.; Taniguchi, H.; Saito, Y.; Yasuhiro, O.; Ochiai, A.; Hiraoka, N. Comprehensive characterization of RSPO fusions in colorectal traditional serrated adenomas. Histopathology 2017, 71, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Ogawa, R.; Yoshida, H.; Taniguchi, H.; Kojima, M.; Saito, Y.; Sekine, S. EIF3E-RSPO2 and PIEZO1-RSPO2 fusions in colorectal traditional serrated adenoma. Histopathology 2019, 75, 266–273. [Google Scholar] [CrossRef] [PubMed]


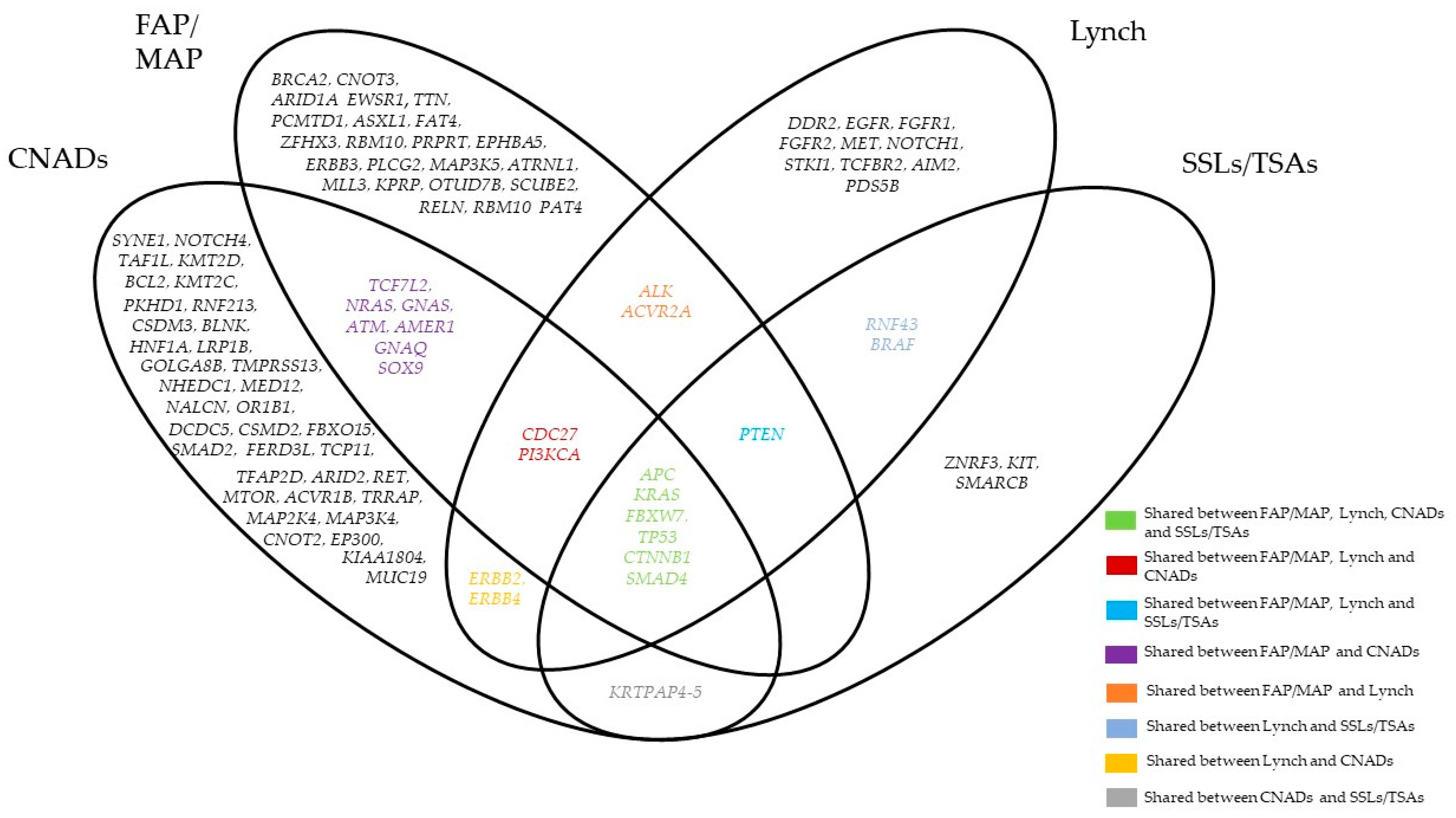{kind=link}
| Syndrome | Study Population | Samples Analyzed | Study Methods | Frequent Mutated Genes and Candidate Driver Genes | Notes | References |
|---|---|---|---|---|---|---|
| FAP | 12 FAP patients | 25 adenomas, 10 adjacent NM | WES | APC, KRAS, FBXW7, TCF7L2, BRCA2, ALK, CNOT3, ARID1A, CDC27, EWSR1, GNAQ, TTN, PCMTD1 | [32] | |
| FAP | 14 FAP patients | 37 adenomas and matched NM | Targeted Ampliseq sequencing | APC, KRAS | [33] | |
| FAP and MAP | 5 FAP patients 1 MAP patient | 20 LGD adenomas, 4 HGD adenomas, 7 carcinomas and 8 adjacent NM | WES, WGS | APC (69%), TTN (37%), SMAD4 (35%), GNAS (33%), ASXL1 (33%), KRAS (23%), FAT4 (22%), ZFHX3 (22%), FBXW7 (22%), PTPRT (20%), SOX9 (16%) | Additional potential driver events with lower frequency: ERBB3 (8%), ARID1A (8%), TP53 (8%), ACVR2A (6%), EPHA5 (6%), TCF7L2 (6%), PIK3CA (6%), RBM10 (5%), CTNNB1 (5%), ATM (5%), AMER1 (5%). | [34] |
| FAP and MAP | 2 FAP patients and 2 MAP patients | 6 adenomas 8 adenomas | WES | APC, KRAS, WTX/FAM123B, SCUBE2, RELN, FBXW7, MLL3, OTUD7B, KPRP, ATRNL1, MAP3K5, NRAS, PLCG2, PTEN, TP53 | Except for APC, WTX and KRAS, few adenomas shared the same set of mutated driver genes | [35] |
| 3 FAP patients and 4 MAP patients | 22 adenomas 33 adenomas | Targeted exome sequencing | ||||
| 7 FAP patients and 3 MAP patients | 41 adenomas 22 adenomas | WTX/KRAS capillary sequencing | ||||
| Lynch | 44 patients | 86 adenomas, 36 adenocarcinomas | Target NGS | APC (40% of LS-associated adenomas, 28% of LS-adenocarcinomas and 60% sporadic adenomas); CTNNB1 (5% LS-adenomas and 10% sporadic adenomas); RNF43 (52% LS-associated adenomas; 56% LS-adenocarcinomas). | KRAS, BRAF and NRAS mutations uncommon in both LS- and sporadic adenomas | [36] |
| 84 sporadic adenomas | ||||||
| Lynch | 57 patients | 59 adenomas: 16 MMR-P; 43 MMR-D (41 LGD and 18 HGD) | Amplicon-based NGS | TP53 (24%), KRAS (22%), SMAD4 (19%), CTNNB1 (15%) | Additional potential driver events with lower frequency: ALK (2%), BRAF (9%), DDR2 (2%), EGFR (10%), ERBB2 (3%), ERBB4 (5%), FBXW7 (9%), FGFR1 (2%), FGFR2 (3%), MET (2%), NOTCH1 (5%), PTEN (10%), PIK3CA (5%), STKI1 (3%) | |
| [37] | ||||||
| Lynch | 11 patients | Paired tumor (adenoma and cancer) and tumor-distant NM | Whole- genome DNA- sequencing | ACVR2A, TGFBR2, CDC27, AIM2, PDS5B, TP53, KRAS (Frequent in the G1 LS-CRC subgroup) | Paired patient-matched specimens of tumors were stratified into two subgroups based on their genomic characteristics (G1 with higher amount of mutation and MS slippage than G2) | [38] |
| Study Population | Samples Analyzed | Study Methods | Frequent Mutated Genes and Candidate Driver Genes | Notes | Reference |
|---|---|---|---|---|---|
| 36 patients | 48 colorectal polyps: 33 tubular adenomas, 5 tubulovillous adenomas, 4 SSLs, 6 HPs | Target NGS | APC (67%), KRAS (15%), NRAS (2%), TP53 (8%), FBXW7 (10%), BRAFV600E (17%, mainly in SSLs and HPs) | [57] | |
| 58 patients with colorectal adenomas (retrospective study) | 85 samples from 58 adenomas ≥ 2 cm: 19 LGD adenomas (10 tubular and 9 tubulovillous adenomas); 21 premalignant adenomas; 28 HGD adenomas; and 17 invasive adenocarcinomas | Target NGS | APC (76.5%), KRAS (62.4%), SYNE1 (35.3%), NOTCH4 (23.5%), TCF7L2 (18.8%), GNAS (17.6%), FBXW7 (15.3%), TAF1L (15.3%), KMT2D (15.33%) BCL2 (12.9%), KMT2C (11.8%), PKHD1 (11.8%), RNF213 (10.6%), CSDM3 (10.6%), TP53 (20%), BLNK (17.6%), HNF1A (12.9%), LRP1B (10.6%) | The percentages refer to all the analyzed adenomatous samples. Data on cancer samples are described in Supplementary Table S2 | [58] |
| NA | 149 adenomas from two independent projects: 100 CNADs (first study); 35 CNADs and 14 SSLs (second study) | WES and Target sequencing | CNADs: APC, CTNNB1, KRTAP4-5, GOLGA8B, TMPRSS13, TP53, NHEDC1, PI3KCA, KRAS, FBXW7, SOX9, ATM, CDC27, MED12, NALCN; SSLs: KRTAP4-5, BRAFV600E | [60] | |
| 2 CRC patients | 24 NM single cells; 48 adenoma single cells (tubular adenomatous polyps and inflammatory fibroid polyp normal appearance) | single-cell WES and bulk WES | OR1B1, DCDC5, CSMD1, FBXO15, TCP11, TFAP2D | Small sample size. Data on CRC single cells are described in Supplementary Table S2 | [63] |
| NA | 11 colorectal adenoma-carcinoma pairs | WES | APC, CTNNB1, KRAS, TP53, TMPRSS13, TFC7L2, NRAS, FERD3L | Data on gastric cancer not included. Data on cancer samples are described in Supplementary Table S2 | [59] |
| 2 large prospective cohort studies: Nurses’ Health Study (N = 121,700 women followed since 1976) and Health Professionals Follow-up Study (N = 51,500 men followed since 1986) | 225 colorectal cancers LGD (=50% gland formation) vs. HGD (<50% gland formation) | Pyrosequencing and Sanger sequencing | NRAS activating mutations (5/225 CNADs c.34G>A, c.35G>A and c.35G>T in codon 12 and c.181C>A in codon 61) | Only NRAS mutations were analyzed in this study | [64] |
| 12 Korean patients | 12 high-grade colon adenoma samples (11 non-hypermutated and 1 hypermutated (POLE-mutated) tubulovillous adenomas) and matched NM | WES | APC, KRAS, SMAD4, ERBB4, TCF7L2, AMER1, TP53, GNAS, ARID2, RET, MTOR, NRAS, ACVR1B, GNAQ, ATM, PIK3CA, ERBB2, TRRAP, MAP2K4, MAP3K4, CNOT2, EP300 | [65] | |
| 31 patients | 90 tissues: 16 CAP cases matched with 15 CFP cases-all polyps are adenomatous polyps with villous features -tubulovillous or villous- and LGD) | WES | TP53, FBXW7, PIK3CA, KIAA1804, SMAD2 and SMAD4 (mutations exclusively in CAP samples); APC (significantly mutated in both polyp groups, 70% CFPs and 80% CAPs); MUC19 (CFPs) | [66] | |
| 20 patients with multiple SSLs (16 fulfill WHO clinical criteria for SPS) | 1 SSL from each individual (19/20) | BRAF/ KRAS SNaPshot genotyping | BRAFV600E (18/19 SSLs) | [67] | |
| 5 patients(carriers of RNF43 c.953-1, G>A germline mutation) from 1 SPS family | 16 serrated lesions (SSLs/TSAs/HPs), 5 tubulovillous/ villous adenomas and 1 cancer (from 5 SPS patients); 90 sporadic lesions (14 HPs, 47 SSLs/TSAs, 29 CNADs); | WES, Target Gene Sanger Sequencing | RNF43 (18.8% serrated lesions from SPS patients; 20% conventional adenomas from SPS patients; 34% sporadic SSLs/TSAs; 0% sporadic HPs; 3.4% sporadic CNADs); BRAFV600E (62.5% SSLs/TSAs from SPS patients; 66% sporadic SSLs/TSAs; 81.2% sporadic SSLs/TSAs RNF43-mutated) | SSLs and TSAs analyzed as a single group. Data on cancer samples are described in Supplementary Table S2 | [68] |
| NA | 20 SSLs; 36 TSAs; 37 TSAs with cytologic dysplasia; 30 tubulovillous/ villous adenomas | Sanger sequencing | RNF43 (10% SSLs; 28% TSAs; 19% TSAs with dysplasia; 0% tubulovillous/villous adenomas), BRAF (82% TSAs RNF43- mutated with/without dysplasia), KRAS (11.7% TSAs RNF43- mutated with/without dysplasia) | Data on cancer samples are reported in Supplementary Table S2 | [69] |
| NA | 130 serrated lesions (26 HPs, 34 SSLs, 70 TSAs) and 58 CNADs (27 tubular adenomas, 31 tubulovillous adenomas) | Target NGS, Sanger sequencing | RNF43 (6% SSLs; 24% TSAs; 0% CNADs); BRAF (73% HPs; 74% SSLs; 60% TSAs; 0% CNADs); APC (0% HPs and SSLs; 13% TSAs; 59% CNADs) | [70] | |
| NA | 46 dysplastic SSLs; 45 SSLs without dysplasia | Target NGS, Sanger sequencing | RNF43 (50% dysplastic SSLs); APC (9% dysplastic SSLs), ZNRF3 (7% dysplastic SSLs); BRAF (87% dysplastic SSLs; 84% SSLs without dysplasia) | [71] | |
| 196 patients | 200 TSAs (162 ordinary, 38 advanced); 50 tubulovillous adenomas | Allele Specific PCR | BRAFV600E (67% TSAs) | [72] | |
| 8 patients | 8 SSLs: 4 SSLs with HGD, 4 SSLs with submucosal carcinoma | Target NGS | BRAFV600E (88% SSLs); FBXW7 (38% SSLs); TP53 (25% SSLs); KIT, PTEN, SMAD4, SMARCB (13% SSLs each) | [73] | |
| NA | 189 samples: 20 SSLs; 20 dysplastic SSLs; 14 TSAs; 6 dysplastic TSAs; 19 tubular and tubulovillous adenomas | Targeted amplicon sequencing | APC (5% SSLs; 20% dysplastic SSLs; 36% TSAs; 33% dysplastic TSAs; 89% tubular and tubulovillous adenomas) | Only APC mutations were analyzed in this study. Data on cancer samples are reported in Supplementary Table S2 | [74] |
| NA | 15 TSAs associated with precursors polyps: 9 associated with HPs and 6 associated with SSLs | Laser microdissection based sequencing | RNF43, APC, CTNNB1; BRAFV600E | [75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alquati, C.; Prossomariti, A.; Piazzi, G.; Buttitta, F.; Bazzoli, F.; Laghi, L.; Ricciardiello, L. Discovering the Mutational Profile of Early Colorectal Lesions: A Translational Impact. Cancers 2021, 13, 2081. https://doi.org/10.3390/cancers13092081
Alquati C, Prossomariti A, Piazzi G, Buttitta F, Bazzoli F, Laghi L, Ricciardiello L. Discovering the Mutational Profile of Early Colorectal Lesions: A Translational Impact. Cancers. 2021; 13(9):2081. https://doi.org/10.3390/cancers13092081
Chicago/Turabian StyleAlquati, Chiara, Anna Prossomariti, Giulia Piazzi, Francesco Buttitta, Franco Bazzoli, Luigi Laghi, and Luigi Ricciardiello. 2021. "Discovering the Mutational Profile of Early Colorectal Lesions: A Translational Impact" Cancers 13, no. 9: 2081. https://doi.org/10.3390/cancers13092081