
New Insights into YES-Associated Protein Signaling Pathways in Hematological Malignancies: Diagnostic and Therapeutic Challenges

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
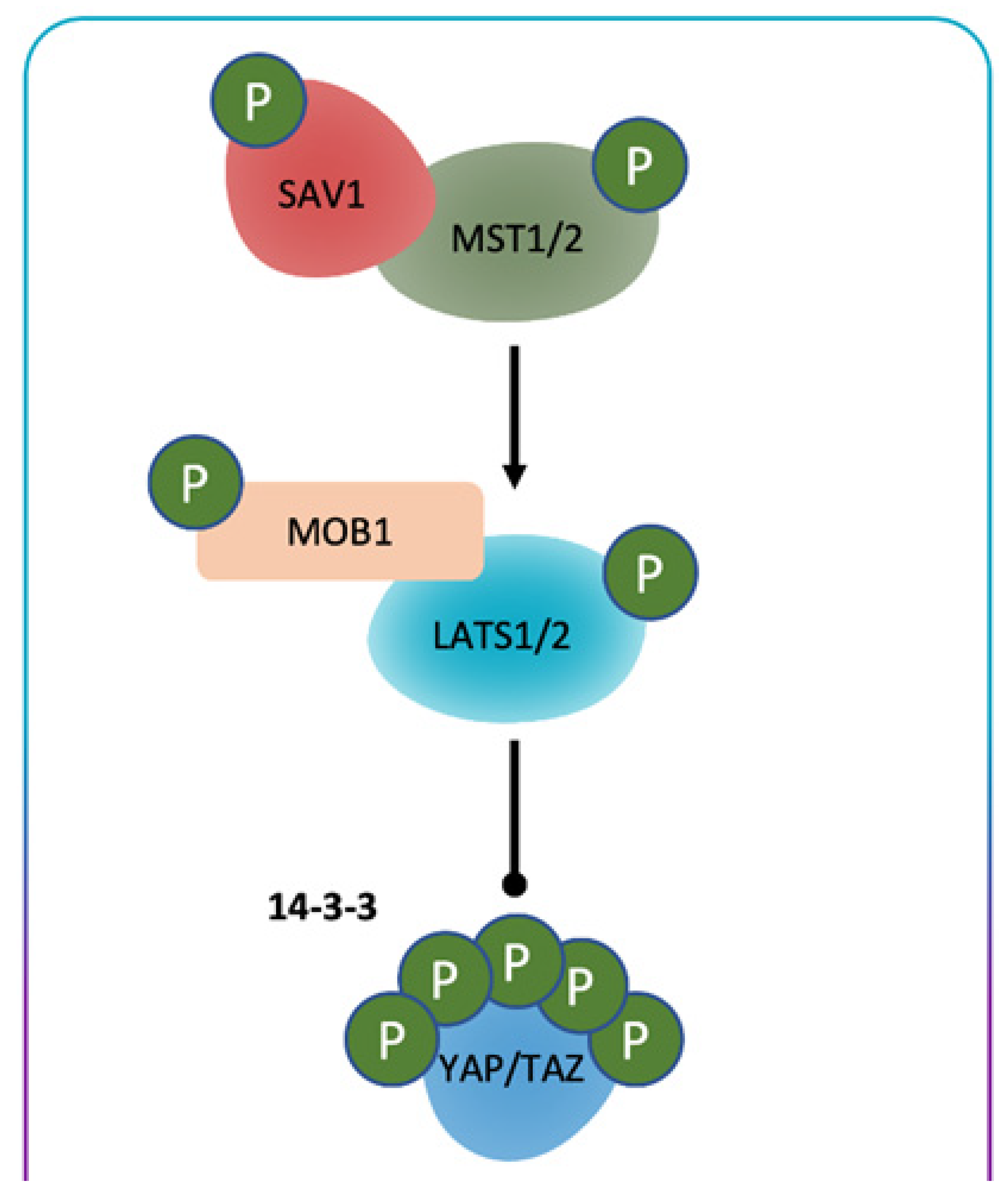
General Consideration of Hippo/YES-Associated Protein Signaling Pathway
2. YAP System and Human Cancer
3. YAP and Hematopoiesis
4. YAP and Hematological Malignancies
4.1. YAP and Lymphoproliferative Diseases
4.2. YAP and Multiple Myeloma
4.3. YAP and Acute Myeloid and Lymphoblastic Leukemias
4.4. YAP and Chronic Myeloid Leukemia
5. Possible Therapeutic Use of the Modulation of the YAP System
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Moya, I.M.; Halder, G. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat. Rev. Mol. Cell Biol. 2019, 20, 211–226. [Google Scholar] [CrossRef]
- Misra, J.R.; Irvine, K.D. The hippo signaling network and its biological functions. Annu. Rev. Genet. 2018, 52, 65–87. [Google Scholar] [CrossRef]
- Sebio, A.; Lenz, H.J. Molecular pathways: Hippo signaling, a critical tumor suppressor. Clin. Cancer Res. 2015, 21, 5002–5007. [Google Scholar] [CrossRef] [Green Version]
- Hoa, L.; Kulaberoglu, Y.; Gundogdu, R.; Cook, D.; Mavis, M.; Gomez, M.; Gomez, V.; Hergovich, A. The characterisation of LATS2 kinase regulation in Hippo-YAP signalling. Cell Signal. 2016, 28, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, S.; Mana-Capelli, S.; Flach, R.J.R.; Danai, L.V.; Amcheslavsky, A.; Nie, Y.; Kaneko, S.; Yao, X.; Chen, X. The conserved misshapen-warts-Yorkie pathway acts in enteroblasts to regulate intestinal stem cells in Drosophila. Dev. Cell 2014, 31, 291–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Wang, W.; Liu, B.; Deng, H.; Uster, E.; Pan, D. Identification of appyhour/MAP4K as alternative Hpo/Mst-like kinases in the Hippo kinase cascade. Dev. Cell 2015, 34, 642–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Z.; Moroishi, T.; Mottier-Pavie, V.; Plou_e, S.W.; Hansen, C.G.; Hong, A.W.; Park, H.W.; Mo, J.S.; Lu, W.; Lu, S.; et al. MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat. Commun. 2015, 6, 8357. [Google Scholar] [CrossRef] [PubMed]
- Maugeri-Sacca, M.; De Maria, R. The hippo pathway in normal development and cancer. Pharmacol. Ther. 2018, 186, 60–72. [Google Scholar] [CrossRef]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.; Chun, A.; Cheung, K.; Rashidi, B.; Yang, X. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J. Biol. Chem. 2008, 283, 5496–5509. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Zhou, Z.; Shah, A.A.; Hong, Y.; Chen, Q.; Wan, Y. New insights into posttranslational modifications of Hippo pathway in carcinogenesis and therapeutics. Cell Div. 2016, 11, 4. [Google Scholar] [CrossRef] [Green Version]
- Lei, Q.-Y.; Zhang, H.; Zhao, B.; Zha, Z.-Y.; Bai, F.; Pei, X.-H.; Zhao, S.; Xiong, Y.; Guan, K.-L. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol. Cell Biol. 2008, 28, 2426–2436. [Google Scholar] [CrossRef] [Green Version]
- Sudol, M.; Harvey, K.F. Modularity in the Hippo signaling pathway. Trends Biochem. Sci. 2010, 35, 627–633. [Google Scholar] [CrossRef]
- Irvine, K.D. Integration of intercellular signaling through the Hippo pathway. Semin. Cell Dev. Biol. 2012, 23, 812–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Li, L.; Lei, Q.; Guan, K.-L. The Hippo–YAP pathway in organ size control and tumorigenesis: An updated version. Genes Dev. 2010, 24, 862–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.-Y.; Zha, Z.-Y.; Zhou, X.; Zhang, H.; Huang, W.; Zhao, D.; Li, T.; Chan, S.W.; Lim, C.J.; Hong, W. The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCF_-TrCP E3 ligase. J. Biol. Chem. 2010, 285, 37159–37169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holden, J.K.; Cunningham, C.N. Targeting the Hippo Pathway and Cancer through the TEAD Family of Transcription Factors. Cancers 2018, 10, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.-K.; Jang, J.-W.; Bae, S.-C. DNA binding partners of YAP/TAZ. BMB Rep. 2018, 51, 126. [Google Scholar] [CrossRef] [Green Version]
- Lai, D.; Ho, K.C.; Hao, Y.; Yang, X. Taxol resistance in breast cancer cells is mediated by the hippo pathway component TAZ and its downstream transcriptional targets Cyr61 and CTGF. Cancer Res. 2011, 71, 2728–2738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, K.C.; Park, H.W.; Guan, K.-L. Regulation of the Hippo pathway transcription factor TEAD. Trends Biochem. Sci. 2017, 42, 862–872. [Google Scholar] [CrossRef]
- Lai, D.; Yang, X. BMP4 is a novel transcriptional target and mediator of mammary cell migration downstream of the Hippo pathway component TAZ. Cell. Signal. 2013, 25, 1720–1728. [Google Scholar] [CrossRef]
- Janse van Rensburg, H.J.J.; Azad, T.; Ling, M.; Hao, Y.; Snetsinger, B.; Khanal, P.; Minassian, L.M.; Graham, C.H.; Rauh, M.J.; Yang, X. The Hippo pathway component TAZ promotes immune evasion in human cancer through PD-L1. Cancer Res. 2018, 78, 1457–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alarcón, C.; Zaromytidou, A.-I.; Xi, Q.; Gao, S.; Yu, J.; Fujisawa, S.; Barlas, A.; Miller, A.N.; Manova-Todorova, K.; Macias, M.J. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell 2009, 139, 757–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varelas, X.; Sakuma, R.; Samavarchi-Tehrani, P.; Peerani, R.; Rao, B.M.; Dembowy, J.; Ya_e, M.B.; Zandstra, P.W.; Wrana, J.L. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat. Cell Biol. 2008, 10, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Passaniti, A.; Brusgard, J.L.; Qiao, Y.; Sudol, M.; Finch-Edmondson, M. Roles of RUNX in Hippo Pathway Signaling. Adv. Exp. Med. Biol. 2017, 962, 435–448. [Google Scholar] [PubMed]
- Murakami, M.; Nakagawa, M.; Olson, E.N.; Nakagawa, O. A WW domain protein TAZ is a critical coactivator for TBX5, a transcription factor implicated in Holt–Oram syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 18034–18039. [Google Scholar] [CrossRef] [Green Version]
- Strano, S.; Munarriz, E.; Rossi, M.; Castagnoli, L.; Shaul, Y.; Sacchi, A.; Oren, M.; Sudol, M.; Cesareni, G.; Blandino, G. Physical interaction with Yes-associated protein enhances p73 transcriptional activity. J. Biol. Chem. 2001, 276, 15164–15173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillmer, R.E.; Link, B.A. The Roles of Hippo Signaling Transducers Yap and Taz in Chromatin Remodeling. Cells 2019, 8, 502. [Google Scholar] [CrossRef] [Green Version]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.-S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.-X.; Alexander, C.M. Alternative Wntsignaling activates YAP/TAZ. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef] [Green Version]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef] [Green Version]
- Fan, R.; Kim, N.-G.; Gumbiner, B.M. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc. Natl. Acad. Sci. USA 2013, 110, 2569–2574. [Google Scholar] [CrossRef] [Green Version]
- Collak, F.K.; Yagiz, K.; Luthringer, D.J.; Erkaya, B.; Cinar, B. Threonine-120 phosphorylation regulated by phosphoinositide-3-kinase/Akt and mammalian target of rapamycin pathway signaling limits the antitumor activity of mammalian sterile 20-like kinase 1. J. Biol. Chem. 2012, 287, 23698–23709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.-X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Chen, Z.; Wang, Y.; Chang, D.; Su, L.; Guo, Y.; Liu, C. WWTR1 promotes cell proliferation and inhibits apoptosis through cyclin A and CTGF regulation in non-small cell lung cancer. Tumor Biol. 2014, 35, 463–468. [Google Scholar] [CrossRef]
- Koontz, L.M.; Liu-Chittenden, Y.; Yin, F.; Zheng, Y.; Yu, J.; Huang, B.; Chen, Q.; Wu, S.; Pan, D. The Hippo effector Yorkie controls normal tissue growth by antagonizing scalloped-mediated default repression. Dev. Cell 2013, 25, 388–401. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Huang, T.; Cheng, A.S.; Yu, J.; Kang, W.; To, K.F. The TEAD family and its oncogenic role in promoting tumorigenesis. Int. J. Mol. Sci. 2016, 17, 138. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Yang, X. Targeting the hippo pathway for breast cancer therapy. Cancers 2018, 10, 422. [Google Scholar] [CrossRef] [Green Version]
- Yeung, B.; Khanal, P.; Mehta, V.; Trinkle-Mulcahy, L.; Yang, X. Identification of Cdk1–LATS–Pin1 as a novel signaling axis in anti-tubulin drug response of cancer cells. Mol. Cancer Res. 2018, 16, 1035–1045. [Google Scholar] [CrossRef] [Green Version]
- Hong, A.W.; Meng, Z.; Yuan, H.X.; Plouffe, S.W.; Moon, S.; Kim, W.; Jho, E.H.; Guan, K.L. Osmotic stress-induced phosphorylation by NLK at Ser128 activates YAP. EMBO Rep. 2017, 18, 72–86. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.; Yamaguchi, H.; Xia, W.; Lim, S.; Khotskaya, Y.; Wu, Y.; Chang, W.; Liu, Q.; Hung, M. Aurora A kinase activates YAP signaling in triple-negative breast cancer. Oncogene 2017, 36, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Qian, M.; He, Q.; Zhu, H.; Yang, B. The posttranslational modifications of Hippo-YAP pathway in cancer. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129397. [Google Scholar] [CrossRef]
- Zhao, Y.; Montminy, T.; Azad, T.; Lightbody, E.; Hao, Y.; SenGupta, S.; Asselin, E.; Nicol, C.; Yang, X. PI3K positively regulates YAP and TAZ in mammary tumorigenesis through multiple signaling pathways. Mol. Cancer Res. 2018, 16, 1046–1058. [Google Scholar] [CrossRef] [Green Version]
- Lamar, J.M.; Xiao, Y.; Norton, E.; Jiang, Z.-G.; Gerhard, G.M.; Kooner, S.; Warren, J.S.; Hynes, R.O. SRC tyrosine kinase activates the YAP/TAZ axis and thereby drives tumor growth and metastasis. J. Biol. Chem. 2019, 294, 2302–2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si, Y.; Ji, X.; Cao, X.; Dai, X.; Xu, L.; Zhao, H.; Guo, X.; Yan, H.; Zhang, H.; Zhu, C. Src inhibits the Hippo tumor suppressor pathway through tyrosine phosphorylation of Lats1. Cancer Res. 2017, 77, 4868–4880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azad, T.; Nouri, K.; Janse van Rensburg, H.J.J.; Maritan, S.M.; Wu, L.; Hao, Y.; Montminy, T.; Yu, J.; Khanal, P.; Mulligan, L.M.; et al. A gain-of-functional screen identifies the Hippo pathway as a central mediator of receptor tyrosine kinases during tumorigenesis. Oncogene 2020, 39, 334–355. [Google Scholar] [CrossRef] [PubMed]
- Azad, T.; Janse Van Rensburg, H.J.; Lightbody, E.; Neveu, B.; Champagne, A.; Gha_ari, A.; Kay, V.; Hao, Y.; Shen, H.; Yeung, B.; et al. A LATS biosensor screen identifies VEGFR as a regulator of the Hippo pathway in angiogenesis. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Nouri, K.; Azad, T.; Lightbody, E.; Khanal, P.; Nicol, C.J.; Yang, X. A kinome-wide screen using a NanoLucLATS luminescent biosensor identifies ALK as a novel regulator of the Hippo pathway in tumorigenesis and immune evasion. FASEB J. 2019, 33, 12487–12499. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef]
- Nicolay, B.N.; Bayarmagnai, B.; Islam, A.B.M.M.K.; Lopez-Bigas, N.; Frolov, M.V. Cooperation between dE2F1 and Yki/Sd defines a distinct transcriptional program necessary to bypass cell cycle exit. Genes Dev. 2011, 25, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, T.; Murakami, H.; Fujii, M.; Ishiguro, F.; Tanaka, I.; Kondo, Y.; Akatsuka, S.; Toyokuni, S.; Yokoi, K.; Osada, H.; et al. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene 2012, 31, 5117–5122. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.J.; Sadanandam, A.; Hu, B.; et al. Yap1 activation enables bypass of oncogenic KRAS addiction in pancreatic cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Croci, O.; De Fazio, S.; Biagioni, F.; Donato, E.; Caganova, M.; Curti, L.; Doni, M.; Sberna, S.; Aldeghi, D.; Biancotto, C.; et al. Transcriptional integration of mitogenic and mechanical signals by Myc and YAP. Genes Dev. 2017, 31, 2017–2022. [Google Scholar] [CrossRef] [Green Version]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer. 2013, 13, 246–257. [Google Scholar] [CrossRef]
- Moon, S.; Yeon Park, S.; Woo Park, H. Regulation of the Hippo pathway in cancer biology. Cell. Mol. Life Sci. 2018, 75, 2303–2319. [Google Scholar] [CrossRef] [PubMed]
- Calses, P.C.; Crawford, J.J.; Lill, J.R.; Dey, A. Hippo Pathway in Cancer: Aberrant Regulation and Therapeutic Opportunities. Trends Cancer 2019, 5, 297–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.X.; Guan, K.L. The Hippo pathway: Regulators and regulations. Genes Dev. 2013, 27, 355–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, M.A.S.; Tao, W.; Fei, X.; Fukumoto, R.; Carcangiu, M.L.; Brownstein, D.G.; Parlow, A.F.; McGrath, J.; Xu, T. Mice deficient of Lats1 develop soft-tissue sarcomas, ovarian tumours and pituitary dysfunction. Nat. Genet. 1999, 21, 182–186. [Google Scholar] [CrossRef]
- Tapon, N.; Harvey, K.F.; Bell, D.W.; Wahrer, D.C.; Schiripo, T.A.; Haber, D.; Hariharan, I.K. salvador Promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell 2002, 110, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Miyoshi, Y.; Takahata, C.; Irahara, N.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Down-regulation of LATS1 and LATS2 mRNA expression by promoter hypermethylation and its association with biologically aggressive phenotype in human breast cancers. Clin. Cancer Res. 2005, 11, 1380–1385. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Gao, Y.; Li, F.; Tong, X.; Ren, Y.; Han, X.; Yao, S.; Long, F.; Yang, Z.; Fan, H.; et al. YAP promotes malignant progression of Lkb1-deficient lung adenocarcinoma through downstream regulation of survivin. Cancer Res. 2015, 75, 4450–4457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, K.; Xu, Q.; Wang, S.; Zhang, W.; Liu, M.; Liang, S.; Zhu, H.; Xu, N. Nuclear accumulation of yes-associated protein (YAP) maintains the survival of doxorubicin-induced senescent cells by promoting survivin expression. Cancer Lett. 2016, 375, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Pobbati, A.V.; Hong, W. Emerging roles of TEAD transcription factors and its coactivators in cancers. Cancer Biol. 2013, 14, 390–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, D.; Guo, J.; Wu, Y.; Du, J.; Yang, L.; Wang, X.; Di, W.; Hu, B.; An, J.; Kong, L.; et al. m6A mRNA methylation initiated by METTL3 directly promotes YAP translation and increases YAP activity by regulating the MALAT1-miR-1914-3p-YAP axis to induce NSCLC drug resistance and metastasis. J. Hematol. Oncol. 2019, 12, 135. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP and TAZ: A signalling hub of the tumour microenvironment. Nat. Rev. Cancer 2019, 19, 454–464. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the roots of cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.W.; Lee, S.S.; Kim, S.B.; Sohn, B.H.; Lee, H.S.; Jang, H.J.; Park, Y.Y.; Kopetz, S.; Kim, S.S.; Oh, S.C.; et al. Significant association of oncogene YAP1 with poor prognosis and cetuximab resistance in colorectal cancer patients. Clin. Cancer Res. 2015, 21, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.Z.; Yao, T.J.; Lee, N.P.; Ng, I.O.; Chan, Y.T.; Zender, L.; Lowe, S.W.; Poon, R.T.; Luk, J.M. Yes-associated protein is an independent prognostic marker in hepatocellular carcinoma. Cancer 2009, 115, 4576–4585. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, L.A.; Northcott, P.A.; Dalton, J.; Fraga, C.; Ellison, D.; Angers, S.; Taylor, M.D.; Kenney, A.M. YAP1 is amplified and up-regulated in hedgehog-associated medulloblastomas and mediates Sonic hedgehog-driven neural precursor proliferation. Genes Dev. 2009, 23, 2729–2741. [Google Scholar] [CrossRef] [Green Version]
- Li, S.Y.; Hu, J.A.; Wang, H.M. Expression of Yes-associated protein 1 gene and protein in oral squamous cell carcinoma. Chin. Med. J. 2013, 126, 655–658. [Google Scholar]
- Liu, J.Y.; Li, Y.H.; Lin, H.X.; Liao, Y.J.; Mai, S.J.; Liu, Z.W.; Zhang, Z.L.; Jiang, L.J.; Zhang, J.X.; Kung, H.F.; et al. Overexpression of YAP 1 contributes to progressive features and poor prognosis of human urothelial carcinoma of the bladder. BMC Cancer 2013, 13, 349. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Wang, J.; Ou, C.; Zhang, Y.; Ma, L.; Weng, W.; Pan, Q.; Sun, F. Mutual interaction between YAP and c-Myc is critical for carcinogenesis in liver cancer. Biochem. Biophys. Res. Commun. 2013, 439, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Xu, R.Z.; Zhang, L.; Zhao, X.Y. Berbamine, a novel nuclear factor kappaB inhibitor, inhibits growth and induces apoptosis in human myeloma cells. Acta Pharm. Sin. 2009, 30, 1659–1665. [Google Scholar] [CrossRef] [PubMed]
- Schoumacher, M.; Burbridge, M. Key Roles of AXL and MER Receptor Tyrosine Kinases in Resistance to Multiple Anticancer Therapies. Curr. Oncol. Rep. 2017, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.H.; Pelissier, F.A.; Zhang, H.; Lakins, J.; Weaver, V.M.; Park, C.; LaBarge, M.A. Microenvironment rigidity modulates responses to the HER2 receptor tyrosine kinase inhibitor lapatinib via YAP and TAZ transcription factors. Mol. Biol. Cell 2015, 26, 3946–3953. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.; Halder, G. The two faces of Hippo: Targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Han, H.; Liu, G.-P.; Ma, Y.-X.; Pan, R.-L.; Sang, L.-J.; Li, R.-H.; Yang, L.-J.; Marks, J.R.; Wang, W.; et al. LncRNA wires up Hippo and Hedgehog signaling to reprogramme glucose metabolism. EMBO J. 2017, 36, 3325–3335. [Google Scholar] [CrossRef]
- Cox, A.G.; Hwang, K.L.; Brown, K.K.; Evason, K.J.; Beltz, S.; Tsomides, A.; O’Connor, K.; Galli, G.G.; Yimlamai, D.; Chhangawala, S.; et al. Yap reprograms glutamine metabolism to increase nucleotide biosynthesis and enable liver growth. Nat. Cell Biol. 2016, 18, 886–896. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.K.H.; Du, W.; Shelton, S.J.; Oldham, M.C.; DiPersio, C.M.; Klein, O.D. An FAKYAP-mTOR Signaling Axis Regulates Stem Cell-Based Tissue Renewal in Mice. Cell Stem Cell 2017, 21, 91–106. [Google Scholar] [CrossRef] [Green Version]
- Hansen, C.G.; Ng, Y.L.D.; Lam, W.L.M.; Plouffe, S.W.; Guan, K.L. The Hippo pathway effectors YAP and TAZ promote cell growth by modulating amino acid signaling to mTORC1. Cell Res. 2015, 25, 1299–1313. [Google Scholar] [CrossRef]
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol. Cell 2008, 29, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Strano, S.; Monti, O.; Pediconi, N.; Baccarini, A.; Fontemaggi, G.; Lapi, E.; Mantovani, F.; Damalas, A.; Citro, G.; Sacchi, A.; et al. The transcriptional coactivator yes-associated protein drives p73 gene-target specificity in response to DNA damage. Mol. Cell. 2005, 18, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Cottini, F.; Hideshima, T.; Xu, C.; Sattler, M.; Dori, M.; Agnelli, L.; ten Hacken, E.; Bertilaccio, M.T.; Antonini, E.; Neri, A.; et al. Rescue of Hippo coactivator YAP1 triggers DNA damage-induced apoptosis in hematological cancers. Nat. Med. 2014, 20, 599–606. [Google Scholar] [CrossRef] [Green Version]
- Shtivelman, E.; Lifshitz, B.; Gale, R.P.; Roe, B.A.; Canaani, E. Alternative splicing of RNAs transcribed from the human abl gene and from the bcr-abl fused gene. Cell 1986, 47, 277–284. [Google Scholar] [CrossRef]
- Baskaran, R.; Wood, L.D.; Whitaker, L.L.; Canman, C.E.; Morgan, S.E.; Xu, Y.; Barlow, C.; Baltimore, D.; Wynshaw-Boris, A.; Kastan, M.B.; et al. Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature 1997, 387, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Agami, R.; Blandino, G.; Oren, M.; Shaul, Y. Interaction of c-Abl and p73alpha and their collaboration to induce apoptosis. Nature 1999, 399, 809–813. [Google Scholar] [CrossRef]
- Goldberg, Z.; Vogt Sionov, R.; Berger, M.; Zwang, Y.; Perets, R.; Van Etten, R.A.; Oren, M.; Taya, Y.; Haupt, Y. Tyrosine phosphorylation of Mdm2 by c-Abl: Implications for p53 regulation. EMBO J. 2002, 21, 3715–3727. [Google Scholar] [CrossRef]
- Allington, T.M.; Galliher-Beckley, A.J.; Schiemann, W.P. Activated Abl kinase inhibits oncogenic transforming growth factor-beta signaling and tumorigenesis in mammary tumors. FASEB J. 2009, 23, 4231–4243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keshet, R.; Adler, J.; Ricardo Lax, I.; Shanzer, M.; Porat, Z.; Reuven, N.; Shaul, Y. c-Abl antagonizes the YAP oncogenic function. Cell Death Differ. 2015, 22, 935–945. [Google Scholar] [CrossRef] [Green Version]
- Huh, H.D.; Kim, D.H.; Jeong, H.S.; Park, H.W. Regulation of TEAD Transcription Factors in Cancer Biology. Cells 2019, 8, 600. [Google Scholar] [CrossRef] [Green Version]
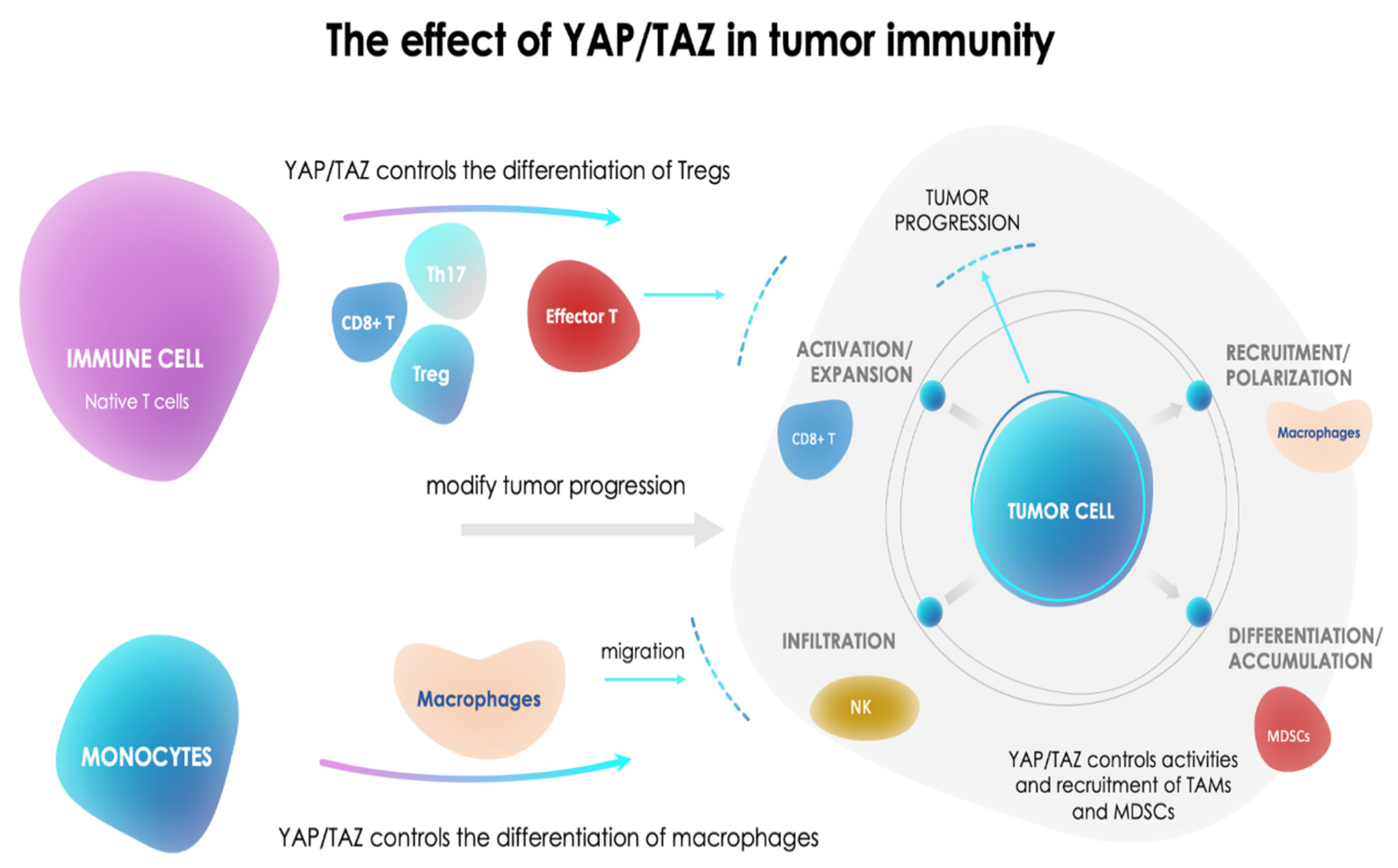
- Pan, Z.; Tian, Y.; Cao, C.; Niu, G. The Emerging Role of YAP/TAZ in Tumor Immunity. Mol. Cancer Res. 2019, 17, 1777–1786. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Peng, Z.; Qin, M.; Liu, Y.; Wang, J.; Zhang, C.; Lin, J.; Dong, T.; Wang, L.; Li, S.; et al. Interferon-γ induces tumor resistance to anti-PD-1 immunotherapy by promoting YAP phase separation. Mol. Cell 2021, 81, 1216–1230. [Google Scholar] [CrossRef]
- Quartarone, E.; Alonci, A.; Allegra, A.; Bellomo, G.; Calabrò, L.; D’Angelo, A.; Del Fabro, V.; Grasso, A.; Cincotta, M.; Musolino, C. Differential levels of soluble angiopoietin-2 and Tie-2 in patients with haematological malignancies. Eur. J. Haematol. 2006, 77, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Hooglugt, A.; van der Stoel, M.M.; Boon, R.A.; Huveneers, S. Endothelial YAP/TAZ Signaling in Angiogenesis and Tumor Vasculature. Front. Oncol. 2021, 10, 612802. [Google Scholar] [CrossRef] [PubMed]
- Avenoso, A.; Campo, S.; Scuruchi, M.; Mania, M.; Innao, V.; D’Ascola, A.; Mandraffino, G.; Allegra, A.G.; Musolino, C.; Allegra, A. Quantitative polymerase Chain reaction profiling of microRNAs in peripheral lymph-monocytes from MGUS subjects. Pathol. Res. Pract. 2020, 13, 153317. [Google Scholar] [CrossRef]
- Allegra, A.; Musolino, C.; Tonacci, A.; Pioggia, G.; Gangemi, S. Interactions between the MicroRNAs and Microbiota in Cancer Development: Roles and Therapeutic Opportunities. Cancers 2020, 12, 805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Innao, V.; Allegra, A.; Pulvirenti, N.; Allegra, A.G.; Musolino, C. Therapeutic potential of antagomiRs in haematological and oncological neoplasms. Eur. J. Cancer Care 2020, 29, e13208. [Google Scholar] [CrossRef]
- Musolino, C.; Oteri, G.; Allegra, A.; Mania, M.; D’Ascola, A.; Avenoso, A.; Innao, V.; Allegra, A.G.; Campo, S. Altered microRNA expression profile in the peripheral lymphoid compartment of multiple myeloma patients with bisphosphonate-induced osteonecrosis of the jaw. Ann. Hematol. 2018, 97, 1259–1269. [Google Scholar] [CrossRef]
- Campo, S.; Allegra, A.; D’Ascola, A.; Alonci, A.; Scuruchi, M.; Russo, S.; Avenoso, A.; Gerace, D.; Campo, G.M.; Musolino, C. MiRNome expression is deregulated in the peripheral lymphoid compartment of multiple myeloma. Br. J. Haematol. 2014, 165, 801–813. [Google Scholar] [CrossRef]
- Allegra, A.; Alonci, A.; Campo, S.; Penna, G.; Petrungaro, A.; Gerace, D.; Musolino, C. Circulating microRNAs: New biomarkers in diagnosis, prognosis and treatment of cancer (review). Int. J. Oncol. 2012, 41, 1897–1912. [Google Scholar] [CrossRef] [Green Version]
- Mori, M.; Triboulet, R.; Mohseni, M.; Schlegelmilch, K.; Shrestha, K.; Camargo, F.D.; Gregory, R.I. Hippo Signaling Regulates Microprocessor and Links Cell-Density-Dependent miRNA Biogenesis to Cancer. Cell 2014, 156, 893–906. [Google Scholar] [CrossRef] [Green Version]
- Chaulk, S.G.; Lattanzi, V.J.; Hiemer, S.E.; Fahlman, R.P.; Varelas, X. The Hippo pathway effectors TAZ/YAP regulate dicer expression and microRNA biogenesis through Let-7. J. Biol. Chem. 2014, 289, 1886–1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samji, P.; Rajendran, M.K.; Warrier, V.P.; Ganesh, A.; Devarajan, K. Regulation of Hippo signaling pathway in cancer: A MicroRNA perspective. Cell Signal. 2021, 109858. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xu, X.; Maglic, D.; Dill, M.T.; Mojumdar, K.; Ng, P.K.-S.; Jeong, K.J.; Tsang, Y.H.; Moreno, D.; Bhavana, V.H.; et al. Comprehensive Molecular Characterization of the Hippo Signaling Pathway in Cancer. Cell Rep. 2018, 25, 1304–1317.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, T.; Li, Y.; Wang, J.; Wang, H.; Liang, Y.; Shi, H.; Sun, B.; Yin, D.; Sun, J.; Song, R.; et al. YAP is a critical oncogene in human cholangiocarcinoma. Oncotarget 2015, 6, 17206–17220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, C.A.; Wang, R.; Miao, J.; Oliva, E.; Shen, X.; Wheeler, T.; Hilsenbeck, S.G.; Orsulic, S.; Goode, S. Hippo pathway effector Yap is an ovarian cancer oncogene. Cancer Res. 2010, 70, 8517–8525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, W.; Tong, J.H.; Chan, A.W.; Lee, T.L.; Lung, R.W.; Leung, P.P.; So, K.K.; Wu, K.; Fan, D.; Yu, J.; et al. Yes-associated protein 1 exhibits oncogenic property in gastric cancer and its nuclear accumulation associates with poor prognosis. Clin. Cancer Res. 2011, 17, 2130–2139. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Mao, D.; Hua, G.; Lv, X.; Chen, X.; Angeletti, P.C.; Dong, J.; Remmenga, S.W.; Rodabaugh, K.J.; Zhou, J.; et al. The hippo/YAP pathway interacts with EGFR signaling and HPV oncoproteins to regulate cervical cancer progression. EMBO Mol. Med. 2015, 7, 1426–1449. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.H.; Kuo, C.C.; Lin, B.X.; Huang, Y.H.; Lin, C.W. Elevation of YAP promotes the epithelial-mesenchymal transition and tumor aggressiveness in colorectal cancer. Exp. Cell Res. 2017, 350, 218–225. [Google Scholar] [CrossRef]
- Chen, H.; Chen, Q.; Luo, Q. Expression of netrin-1 by hypoxia contributes to the invasion and migration of prostate carcinoma cells by regulating YAP activity. Exp. Cell Res. 2016, 349, 302–309. [Google Scholar] [CrossRef]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [Green Version]
- Panciera, T.; Azzolin, L.; Fujimura, A.; Di Biagio, D.; Frasson, C.; Bresolin, S.; Soligo, S.; Basso, G.; Bicciato, S.; Rosato, A.; et al. Induction of expandable tissue-specific stem/progenitor cells through transient expression of YAP/TAZ. Cell Stem Cell 2016, 19, 725–737. [Google Scholar] [CrossRef] [Green Version]
- Yimlamai, D.; Christodoulou, C.; Galli, G.G.; Yanger, K.; Pepe- Mooney, B.; Gurung, B.; Shrestha, K.; Cahan, P.; Stanger, B.Z.; Camargo, F.D. Hippo pathway activity influences liver cell fate. Cell 2014, 157, 1324–1338. [Google Scholar] [CrossRef] [Green Version]
- Geng, J.; Yu, S.; Zhao, H.; Sun, X.; Li, X.; Wang, P.; Xiong, X.; Hong, L.; Xie, C.; Gao, J.; et al. The transcriptional coactivator TAZ regulates reciprocal differentiation of TH17 cells and Treg cells. Nat. Immunol. 2017, 18, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Shi, H.; Li, J.; Dong, Y.; Liang, J.; Ye, J.; Kong, S.; Zhang, S.; Zhong, T.; Yuan, Z.; et al. Mst1/Mst2 regulate development and function of regulatory T cells through modulation of Foxo1/Foxo3 stability in autoimmune disease. J. Immunol. 2014, 192, 1525–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salojin, K.V.; Hamman, B.D.; Chang, W.C.; Jhaver, K.G.; Al-Shami, A.; Crisostomo, J.; Crisostomo, J.; Wilkins, C.; Digeorge-Foushee, A.M.; Allen, J.; et al. Genetic deletion of Mst1 alters T cell function and protects against autoimmunity. PLoS ONE 2014, 9, e98151. [Google Scholar] [CrossRef] [PubMed]
- Donato, E.; Biagioni, F.; Bisso, A.; Caganova, M.; Amati, B.; Campaner, S. YAP and TAZ are dispensable for physiological and malignant haematopoiesis. Leukemia 2018, 32, 2037–2040. [Google Scholar] [CrossRef] [Green Version]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xie, F.; Chu, F.; Zhang, Z.; Yang, B.; Dai, T.; Gao, L.; Wang, L.; Ling, L.; Jia, J.; et al. YAP antagonizes innate antiviral immunity and is targeted for lysosomal degradation through IKK epsilon-mediated phosphorylation. Nat. Immunol. 2017, 18, 733–743. [Google Scholar] [CrossRef]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef] [Green Version]
- Lian, I.; Kim, J.; Okazawa, H.; Zhao, J.; Zhao, B.; Yu, J.; Chinnaiyan, A.; Israel, M.A.; Goldstein, L.S.; Abujarour, R.; et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 2010, 24, 1106–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramalho-Santos, M.; Yoon, S.; Matsuzaki, Y.; Mulligan, R.C.; Melton, D.A. “Stemness”: Transcriptional profiling of embryonic and adult stem cells. Science 2002, 298, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhang, Z.; Kashiwagi, M.; Yoshida, T.; Joshi, I.; Jena, N.; Somasundaram, R.; Emmanuel, A.O.; Sigvardsson, M.; Fitamant, J.; et al. Superenhancer reprogramming drives a B-cell-epithelial transition and high-risk leukemia. Genes Dev. 2016, 30, 1971–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.S.; Lee, D.H.; Kim, S.K.; Shin, S.Y.; Seo, E.J.; Lim, D.S. Mammalian sterile 20-like kinase 1 suppresses lymphoma development by promoting faithful chromosome segregation. Cancer Res. 2012, 72, 5386–5395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, E.M.; Campo, E.; Wright, G.; Lenz, G.; Salaverria, I.; Jares, P.; Xiao, W.; Braziel, R.M.; Rimsza, L.M.; Chan, W.C.; et al. Pathway discovery in mantle cell lymphoma by integrated analysis of high-resolution gene expression and copy number profiling. Blood 2010, 116, 953–961. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, N.; Xu, H.; Zhou, X.; Wang, J.; Fang, X.; Zhang, Y.; Li, Y.; Yang, J.; Wang, X. Regulation of Hippo-YAP signaling by insulin-like growth factor-1 receptor in the tumorigenesis of diffuse large B-cell lymphoma. J. Hematol. Oncol. 2020, 16, 1–15. [Google Scholar] [CrossRef]
- Chang, Y.; Fu, X.R.; Cui, M.; Li, W.M.; Zhang, L.; Li, X.; Li, L.; Sun, Z.C.; Zhang, X.D.; Li, Z.M.; et al. Activated hippo signal pathway inhibits cell proliferation and promotes apoptosis in NK/T cell lymphoma cells. Cancer Med. 2019, 8, 3892–3904. [Google Scholar] [CrossRef] [Green Version]
- Hansen, C.G.; Moroishi, T.; Guan, K.L. YAP and TAZ: A nexus for Hippo signaling and beyond. Trends Cell Biol. 2015, 25, 499–513. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, J.; Inami, K.; Michishita, F.; Jiang, X.; Iwasa, H.; Nakagawa, K.; Ishigami-Yuasa, M.; Kagechika, H.; Miyamura, N.; Hirayama, J.; et al. Novel YAP1 activator, identified by transcription-based functional screen, limits multiple myeloma growth. Mol. Cancer Res. 2018, 16, 197–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, S.; Price, T.; Huang, W.; Plue, M.; Warren, J.; Sundaramoorthy, P.; Paul, B.; Feinberg, D.; MacIver, N.; Chao, N.; et al. PINK1-Dependent Mitophagy Regulates the Migration and Homing of Multiple Myeloma Cells via the MOB1B-Mediated Hippo-YAP/TAZ Pathway. Adv. Sci. 2020, 7, 1900860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, E.; Basu-Roy, U.; Gunaratne, P.H.; Coarfa, C.; Lim, D.S.; Basilico, C.; Mansukhani, A. SOX2 regulates YAP1 to maintain stemness and determine cell fate in the osteo-adipo lineage. Cell Rep. 2013, 3, 2075–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.; Xiao, X.; Yang, S. LncRNA MALAT1 acts as an oncogene in multiple myeloma through sponging miR-509-5p to modulate FOXP1 expression. Oncotarget 2017, 8, 101984–101993. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Jiang, T.; Jia, Y.; Zou, J.; Wang, X.; Gu, W. LncRNA MALAT1/miR-181a-5p affects the proliferation and adhesion of myeloma cells via regulation of Hippo-YAP signaling pathway. Cell Cycle 2019, 18, 2509–2523. [Google Scholar] [CrossRef] [PubMed]
- Kyriazoglou, A.; Ntanasis-Stathopoulos, I.; Terpos, E.; Fotiou, D.; Kastritis, E.; Dimopoulos, M.A.; Gavriatopoulou, M. Emerging Insights into the Role of the Hippo Pathway in Multiple Myeloma and Associated Bone Disease. Clin. Lymphoma Myeloma Leuk. 2020, 20, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Han, W.; Qin, A.; Wang, Z.; Xu, J.; Qian, Y. The emerging role of Hippo signaling pathway in regulating osteoclast formation. J. Cell Physiol. 2018, 233, 4606–4617. [Google Scholar] [CrossRef]
- Pan, J.X.; Xiong, L.; Zhao, K.; Zeng, P.; Wang, B.; Tang, F.L.; Sun, D.; Guo, H.H.; Yang, X.; Cui, S.; et al. YAP promotes osteogenesis and suppresses adipogenic differentiation by regulating beta-catenin signaling. Bone Res. 2018, 6, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.; Yang, H.; Wan, J.; Deng, X.; Chen, L.; Hao, S.; Ma, L. Knockdown of the Hippo transducer YAP reduces proliferation and promotes apoptosis in the Jurkat leukemia cell. Mol. Med. Rep. 2018, 18, 5379–5388. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Wang, J.; Yao, S.F.; Zhao, Y.; Liu, L.; Li, L.W.; Xu, T.; Gan, L.G.; Xiao, C.L.; Shan, Z.L.; et al. Effect of YAP Inhibition on Human Leukemia HL-60 Cells. Int. J. Med. Sci. 2017, 14, 902–910. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Zhong, L.; Yao, S.F.; Zhao, Y.; Liu, L.; Li, L.W.; Xu, T.; Gan, L.G.; Xiao, C.L.; Shan, Z.L.; et al. Verteporfin Inhibits Cell Proliferation and Induces Apoptosis in Human Leukemia NB4 Cells without Light Activation. Int. J. Med. Sci. 2017, 14, 1031–1039. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Pasqualucci, L.; Dalla-Favera, R. Genetics of diffuse large B-cell lymphoma. Blood 2018, 131, 2307–2319. [Google Scholar] [CrossRef]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef]
- Henney, J.E. From the Food and Drug Administration. JAMA 2000, 283, 2779. [Google Scholar] [CrossRef]
- Ziemssen, F.; Heimann, H. Evaluation of verteporfin pharmakokinetics--redefining the need of photosensitizers in ophthalmology. Expert Opin. Drug Metab. Toxicol. 2012, 8, 1023–1041. [Google Scholar] [CrossRef]
- Ma, W.; Ma, J.; Lei, T.; Zhao, M.; Zhang, M. Targeting immunotherapy for bladder cancer by using anti-CD3 x CD155 bispecific antibody. J. Cancer 2019, 10, 5153–5161. [Google Scholar] [CrossRef]
- Ma, Y.W.; Liu, Y.Z.; Pan, J.X. Verteporfin induces apoptosis and eliminates cancer stem-like cells in uveal melanoma in the absence of light activation. Am. J. Cancer Res. 2016, 6, 2816–2830. [Google Scholar] [PubMed]
- Wen, B.; Deutsch, E.; Marangoni, E.; Frascona, V.; Maggiorella, L.; Abdulkarim, B.; Chavaudra, N.; Bourhis, J.; Tyrphostin, A.G. 1024 modulates radiosensitivity in human breast cancer cells. Br. J. Cancer 2001, 85, 2017–2021. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Huang, J.; Ma, L.; Shan, J.; Shen, J.; Yang, Z.; Liu, L.; Luo, Y.; Yao, C.; Qian, C. MicroRNA-122 confers sorafenib resistance to hepatocellular carcinoma cells by targeting IGF-1R to regulate RAS/RAF/ERK signaling pathways. Cancer Lett. 2016, 371, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, C.S.; Mitsiades, N.S.; McMullan, C.J.; Poulaki, V.; Shringarpure, R.; Akiyama, M.; Hideshima, T.; Chauhan, D.; Joseph, M.; Libermann, T.A.; et al. Inhibition of the insulin-like growth factor receptor-1 tyrosine kinase activity as a therapeutic strategy for multiple myeloma, other hematologic malignancies, and solid tumors. Cancer Cell 2004, 5, 221–230. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 10 May 2019).
- Tse, E.; Kwong, Y.L. The diagnosis and management of NK/T-cell lymphomas. J. Hematol. Oncol. 2017, 10, 85. [Google Scholar] [CrossRef] [Green Version]
- Segrelles, C.; Paramio, J.M.; Lorz, C. The transcriptional co-activator YAP: A new player in head and neck cancer. Oral Oncol. 2018, 86, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Yi, S.; Zhang, Y.; Li, Z.; Qiu, L. Expression of LATS mRNA in mantle cell lymphoma and its clinical significance. Zhonghua Yi Xue Za Zhi 2015, 95, 3285–3288. [Google Scholar] [PubMed]
- Ettari, R.; Zappalà, M.; Grasso, S.; Musolino, C.; Innao, V.; Allegra, A. Immunoproteasome-selective and non-selective inhibitors: A promising approach for the treatment of multiple myeloma. Pharmacol. Ther. 2018, 182, 176–192. [Google Scholar] [CrossRef]
- Allegra, A.; Alonci, A.; Gerace, D.; Russo, S.; Innao, V.; Calabrò, L.; Musolino, C. New orally active proteasome inhibitors in multiple myeloma. Leuk. Res. 2014, 38, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Penna, G.; Alonci, A.; Russo, S.; Greve, B.; Innao, V.; Minardi, V.; Musolino, C. Monoclonal antibodies: Potential new therapeutic treatment against multiple myeloma. Eur. J. Haematol. 2013, 90, 441–468. [Google Scholar] [CrossRef] [Green Version]
- Caserta, S.; Innao, V.; Musolino, C.; Allegra, A. Immune checkpoint inhibitors in multiple myeloma: A review of the literature. Pathol. Res. Pract. 2020, 216, 153114. [Google Scholar] [CrossRef]
- Allegra, A.; Sant’antonio, E.; Penna, G.; Alonci, A.; D’Angelo, A.; Russo, S.; Cannavò, A.; Gerace, D.; Musolino, C. Novel therapeutic strategies in multiple myeloma: Role of the heat shock protein inhibitors. Eur. J. Haematol. 2011, 86, 93–110. [Google Scholar] [CrossRef]
- Oancea, M.; Mani, A.; Hussein, M.A.; Almasan, A. Apoptosis of multiple myeloma. Int. J. Hematol. 2004, 80, 224–231. [Google Scholar] [CrossRef] [Green Version]
- Yun, Z.; Zhichao, J.; Hao, Y.; Ou, J.; Ran, Y.; Wen, D.; Qun, S. Targeting autophagy in multiple myeloma. Leuk. Res. 2017, 59, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moroishi, T.; Hansen, C.G.; Guan, K.L. The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 2015, 15, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.Y.; Yi, H.S.; Kim, H.W.; Shong, M. Dysregulation of mitophagy in carcinogenesis and tumor progression. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J. Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol. 2014, 2, 749–754. [Google Scholar] [CrossRef] [Green Version]
- Dai, K.; Radin, D.P.; Leonardi, D. Deciphering the dual role and prognostic potential of PINK1 across cancer types. Neural Regen. Res. 2021, 16, 659–665. [Google Scholar] [CrossRef]
- Sureshbabu, A.; Bhandari, V. Targeting mitochondrial dysfunction in lung diseases: Emphasis on mitophagy. Front. Physiol. 2013, 4, 384. [Google Scholar] [CrossRef] [Green Version]
- Ibata, S.; Kobune, M.; Kikuchi, S.; Yoshida, M.; Miura, S.; Horiguchi, H.; Murase, K.; Iyama, S.; Takada, K.; Miyanishi, K.; et al. High expression of nucleoporin 133 mRNA in bone marrow CD138+ cells is a poor prognostic factor in multiple myeloma. Oncotarget 2018, 9, 25127–25135. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Sun, H.; Song, F.; Yu, M.; Wu, Y.; Wang, J. YAP1 negatively regulates chondrocyte differentiation partly by activating the beta-catenin signaling pathway. Int. J. Biochem. Cell Biol. 2017, 87, 104–113. [Google Scholar] [CrossRef]
- Song, J.; Ye, B.; Liu, H.; Zhang, N.; Hu, J.; Luo, E. Fak-Mapk, Hippo and Wnt signalling pathway expression and regulation in distraction osteogenesis. Cell Prolif. 2018, 51, e12453. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Shi, M.; Li, J.; Zhang, H.; Chen, B.; Chen, L.; Gao, W.; Giuliani, N.; Zhao, R.C. Elevated tumor necrosis factor-alpha suppresses TAZ expression and impairs osteogenic potential of Flk-1+ mesenchymal stem cells in patients with multiple myeloma. Stem Cells Dev. 2007, 16, 921–930. [Google Scholar] [CrossRef]
- Matsumoto, Y.; La Rose, J.; Kent, O.A.; Wagner, M.J.; Narimatsu, M.; Levy, A.D.; Omar, M.H.; Tong, J.; Krieger, J.R.; Riggs, E.; et al. Reciprocal stabilization of ABL and TAZ regulates osteoblastogenesis through transcription factor RUNX2. J. Clin. Investig. 2016, 126, 4482–4496. [Google Scholar] [CrossRef] [Green Version]
- Eda, H.; Aoki, K.; Kato, S.; Okawa, Y.; Takada, K.; Tanaka, T.; Marumo, K.; Ohkawa, K. The proteasome inhibitor bortezomib inhibits FGF-2 induced reduction of TAZ levels in osteoblast-like cells. Eur. J. Haematol. 2010, 85, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E. A 360 degrees view of circular RNAs: From biogenesis to functions. Wires RNA 2018, 9, e1478. [Google Scholar] [CrossRef] [Green Version]
- Panda, A.C. Circular RNAs act as miRNA sponges. Adv. Exp. Med. Biol. 2018, 1087, 67–79. [Google Scholar] [PubMed]
- Rong, D.; Sun, H.; Li, Z.; Liu, S.; Dong, C.; Fu, K.; Tang, W.; Cao, H. An emerging function of circRNA-miRNAs-mRNA axis in human diseases. Oncotarget 2017, 8, 73271–73281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Du, Y.; Yang, X.; Mo, Y.; Fan, C.; Xiong, F.; Ren, D.; Ye, X.; Li, C.; Wang, Y.; et al. Circular RNAs function as ceRNAs to regulate and control human cancer progression. Mol. Cancer 2018, 17, 79. [Google Scholar] [CrossRef]
- Su, M.; Xiao, Y.; Ma, J.; Tang, Y.; Tian, B.; Zhang, Y.; Li, X.; Wu, Z.; Yang, D.; Zhou, Y.; et al. Circular RNAs in cancer: Emerging functions in hallmarks, stemness, resistance and roles as potential biomarkers. Mol. Cancer 2019, 18, 90. [Google Scholar] [CrossRef]
- Mei, M.; Wang, Y.; Wang, Q.; Liu, Y.; Song, W.; Zhang, M. CircCDYL serves as a new biomarker in mantle cell lymphoma and promotes cell proliferation. Cancer Manag. Res. 2019, 11, 10215–10221. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Wang, X.; Fu, S.; Wang, S.; Fu, Y.; Zhang, J.; Liu, Z. Circular RNA circ-CDYL sponges miR-1180 to elevate yes-associated protein in multiple myeloma. Exp. Biol. Med. 2020, 245, 925–932. [Google Scholar] [CrossRef]
- Hu, Y.; Lin, J.H.; Fang, H.; Fang, J.; Li, C.; Chen, W.; Liu, S.; Ondrejka, S.; Gong, Z.; Reu, F.; et al. Targeting the MALAT1/PARP1/LIG3 complex induces DNA damage and apoptosis in multiple myeloma. Leukemia 2018, 32, 2250–2262. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.-F.; Chang, Y.C.; Chang, C.-S.; Lin, S.F.; Liu, Y.C.; Hsiao, H.H.; Chang, J.G.; Liu, T.C. MALAT1 long non-coding RNA is overexpressed in multiple myeloma and may serve as a marker to predict disease progression. BMC Cancer 2014, 14, 809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allegra, A.; Mania, M.; D’Ascola, A.; Oteri, G.; Siniscalchi, E.N.; Avenoso, A.; Innao, V.; Scuruchi, M.; Allegra, A.G.; Musolino, C.; et al. Altered Long Noncoding RNA Expression Profile in Multiple Myeloma Patients with Bisphosphonate-Induced Osteonecrosis of the Jaw. Biomed Res. Int. 2020, 2020, 9879876. [Google Scholar] [CrossRef] [PubMed]
- Gholami, M.; Mirfakhraie, R.; Movafagh, A.; Jalaeekhoo, H.; Kalahroodi, R.; Zare-Abdollahi, D. Zare-Karizi, S. The expression analysis of LATS2 gene in de novo AML patients. Med. Oncol. 2014, 31, 961. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Yokoyama, Y.; Suzukawa, K.; Nanmoku, T.; Kurita, N.; Seki, M.; Maie, K.; Suyama, T.; Takaiwa, N.; Sakata-Yanagimoto, M.; et al. Identification of a fusion gene composed of a Hippo pathway gene MST2 and a common translocation partner ETV6 in a recurrent translocation t(8;12) (q22;p13) in acute myeloid leukemia. Ann. Hematol. 2015, 94, 1431–1433. [Google Scholar] [CrossRef]
- Hill, V.K.; Dunwell, T.L.; Catchpoole, D.; Krex, D.; Brini, A.T.; Griffiths, M.; Craddock, C.; Maher, E.R.; Latif, F. Frequent epigenetic inactivation of KIBRA, an upstream member of the Salvador/Warts/Hippo (SWH) tumor suppressor network, is associated with specific genetic event in B-cell acute lymphocytic leukemia. Epigenetics 2011, 6, 326–332. [Google Scholar] [CrossRef] [Green Version]
- Jiménez-Velasco, A.; Román-Gómez, J.; Agirre, X.; Barrios, M.; Navarro, G.; Vázquez, I.; Prósper, F.; Torres, A.; Heiniger, A. Down regulation of the large tumor suppressor 2(LATS2/KPM) gene is associated with poor prognosis in acute lymphoblastic leukemia. Leukemia 2005, 19, 2347–2350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Pan, S.; Liu, J.; Wang, S.; Ni, Y.; Xiao, L.; Wei, Q.; Peng, Y.; Ding, Z.; Zhao, W. HIF-1α forms regulatory loop with YAP to coordinate hypoxia-induced adriamycin resistance in acute myeloid leukemia cells. Cell Biol. Int. 2020, 44, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Badran, A.; Yoshida, A.; Wano, Y.; Imamura, S.; Kawai, Y.; Tsutani, H.; Inuzuka, M.; Ueda, T. Expression of the antiapoptotic gene survivin in chronic myeloid leukemia. Anticancer Res. 2003, 23, 589–592. [Google Scholar]
- Hernandez-Boluda, J.C.; Bellosillo, B.; Vela, M.C.; Colomer, D.; Alvarez-Larran, A.; Cervantes, F. Survivin expression in the progression of chronic myeloid leukemia: A sequential study in 16 patients. Leuk. Lymphoma 2005, 46, 717–722. [Google Scholar] [CrossRef]
- Gomez-Casares, M.T.; Vaque, J.P.; Lemes, A.; Molero, T.; Delgado, M.D.; Leon, J. C-myc expression in cell lines derived from chronic myeloid leukemia. Haematologica 2004, 89, 241–243. [Google Scholar] [PubMed]
- Albajar, M.; Gomez-Casares, M.T.; Llorca, J.; Mauleon, I.; Vaque, J.P.; Acosta, J.C.; Bermudez, A.; Donato, N.; Delgado, M.D.; Leon, J. MYC in chronic myeloid leukemia: Induction of aberrant DNA synthesis and association with poor response to imatinib. Mol. Cancer Res. 2011, 9, 564–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriyama, K.; Hori, T. BCR-ABL induces tyrosine phosphorylation of YAP leading to expression of Survivin and Cyclin D1 in chronic myeloid leukemia cells. Int. J. Hematol. 2019, 110, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Chorzalska, A.; Kim, J.F.; Order, K.; Tepper, A.; Ahsan, N.; Rao, R.S.P.; Olszewski, A.J.; Yu, X.; Terentyev, D.; Morgan, J.; et al. Long-Term Exposure to Imatinib Mesylate Downregulates Hippo Pathway and Activates YAP in a Model of Chronic Myelogenous Leukemia. Stem Cells Dev. 2017, 26, 656–677. [Google Scholar] [CrossRef]
- Moroishi, T.; Park, H.W.; Qin, B.; Chen, Q.; Meng, Z.; Plouffe, S.W.; Taniguchi, K.; Yu, F.-X.; Karin, M.; Pan, D.; et al. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev. 2015, 29, 1271–1284. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ji, J.Y.; Yu, M.; Overholtzer, M.; Smolen, G.A.; Wang, R.; Brugge, J.S.; Dyson, N.J.; Haber, D.A. YAP-dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nat. Cell Biol. 2009, 11, 1444–1450. [Google Scholar] [CrossRef]
- Liu, H.; Jiang, D.; Chi, F.; Zhao, B. The Hippo pathway regulates stem cell proliferation, self-renewal, and differentiation. Protein Cell 2012, 3, 291–304. [Google Scholar] [CrossRef]
- Marsola, A.P.Z.C.; Simões, B.P.; Palma, L.C.; Berzoti-Coelho, M.G.; Burin, S.M.; de Castro, F.A. Expression of Hippo signaling pathway and Aurora kinase genes in chronic myeloid leukemia. Med. Oncol. 2018, 35, 26. [Google Scholar] [CrossRef] [PubMed]
- Reuven, N.; Adler, J.; Meltser, V.; Shaul, Y. The Hippo pathway kinase Lats2 prevents DNA damage-induced apoptosis through inhibition of the tyrosine kinase c-Abl. Cell Death Differ. 2013, 20, 1330–1340. [Google Scholar] [CrossRef] [Green Version]
- Sasi, N.K.; Bhutkar, A.; Lanning, N.J.; MacKeigan, J.P.; Weinreich, M. DDK promotes tumor chemoresistance and survival via multiple pathways. Neoplasia 2017, 19, 439–450. [Google Scholar] [CrossRef]
- Ferreira, A.F.; de Oliveira, G.L.; Tognon, R.; Collassanti, M.D.; Zanichelli, M.A.; Hamerschlak, N.; de Souza, A.M.; Covas, D.T.; Kashima, S.; de Castro, F.A. Apoptosis-related gene expression profile in chronic myeloid leukemia patients after imatinib mesylate and dasatinib therapy. Acta Haematol. 2015, 133, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Huang, Z.; Gao, M.; Huang, N.; Luo, Z.; Shen, H.; Wang, X.; Wang, T.; Hu, J.; Feng, W. Inhibition of YAP suppresses CML cell proliferation and enhances efficacy of imatinib in vitro and in vivo. J. Exp. Clin. Cancer Res. 2016, 35, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pobbati, A.V.; Hong, W. A combat with the YAP/TAZ-TEAD oncoproteins for cancer Therapy. Theranostics 2020, 10, 3622–3635. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, T.; Dou, Y.; Zhang, S.; Liu, H.; Khishignyam, T.; Li, X.; Zuo, D.; Zhang, Z.; Jin, M.; et al. Atorvastatin Exerts Antileukemia Activity via Inhibiting Mevalonate-YAP Axis in K562 and HL60 Cells. Front. Oncol. 2019, 9, 1032. [Google Scholar] [CrossRef] [PubMed]
- Huggett, M.T.; Jermyn, M.; Gillams, A.; Illing, R.; Mosse, S.; Novelli, M.; Kent, E.; Bown, S.G.; Hasan, T.; Pogue, B.W.; et al. Phase I/II study of verteporfin photodynamic therapy in locally advanced pancreatic cancer. Br. J. Cancer 2014, 110, 1698–1704. [Google Scholar] [CrossRef] [Green Version]
- Fenske, T.S.; Shah, N.M.; Kim, K.M.; Saha, S.; Zhang, C.; Baim, A.E.; Farnen, J.P.; Onitilo, A.A.; Blank, J.H.; Ahuja, H.; et al. A phase 2 study of weekly temsirolimus and bortezomib for relapsed or refractory B-cell non-Hodgkin lymphoma: A Wisconsin Oncology Network study. Cancer 2015, 121, 3465–3471. [Google Scholar] [CrossRef] [Green Version]
- Pobbati, A.V.; Rubin, B.P. Protein-Protein Interaction Disruptors of the YAP/TAZ-TEAD Transcriptional Complex. Molecules 2020, 25, 6001. [Google Scholar] [CrossRef] [PubMed]
- Corvaisier, M.; Bauzone, M.; Corfiotti, F.; Renaud, F.; El Amrani, M.; Monté, D.; Truant, S.; Leteurtre, E.; Formstecher, P.; Van Seuningen, I.; et al. Regulation of cellular quiescence by YAP/TAZ and Cyclin E1 in colon cancer cells: Implication in chemoresistance and cancer relapse. Oncotarget 2016, 7, 56699–56712. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Si, X.; Gu, S.; Wang, M.; Shen, J.; Li, H.; Shen, J.; Li, D.; Fang, Y.; Liu, C.; et al. Allosteric Inhibitors of SHP2 with Therapeutic Potential for Cancer Treatment. J. Med. Chem. 2017, 60, 10205–10219. [Google Scholar] [CrossRef]
- Cottini, F.; Anderson, K.C.; Tonon, G. Awakening the Hippo co-activator YAP1, a mercurial cancer gene, in hematologic cancers. Mol. Cell Oncol. 2014, 1, e970055. [Google Scholar] [CrossRef] [Green Version]
- Allegra, A.; Speciale, A.; Molonia, M.S.; Guglielmo, L.; Musolino, C.; Ferlazzo, G.; Costa, G.; Saija, A.; Cimino, F. Curcumin ameliorates the in vitro efficacy of carfilzomib in human multiple myeloma U266 cells targeting p53 and NF-κB pathways. Toxicol. Vitr. 2018, 47, 186–194. [Google Scholar] [CrossRef]
- Allegra, A.; Innao, V.; Russo, S.; Gerace, D.; Alonci, A.; Musolino, C. Anticancer Activity of Curcumin and Its Analogues: Preclinical and Clinical Studies. Cancer Investig. 2017, 35, 1–22. [Google Scholar] [CrossRef]
- Shen, Y.; Han, Z.; Liu, S.; Jiao, Y.; Li, Y.; Yuan, H. Curcumin inhibits the tumorigenesis of breast cancer by blocking tafazzin/yes-associated protein axis. Cancer Manag. Res. 2020, 12, 1493–1502. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Shi, Q.; Xu, S.; Du, C.; Liang, L.; Wu, K.; Wang, K.; Wang, X.; Chang, L.S.; He, D.; et al. Curcumin promotes KLF5 proteasome degradation through downregulating YAP/TAZ in bladder cancer cells. Int. J. Mol. Sci. 2014, 15, 15173–15187. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, H.; Wang, L.; Tan, R.; Zhu, M.; Zhong, X.; Zhang, Y.; Chen, B.; Wang, L. Decursin inhibits the growth of HepG2 hepatocellular carcinoma cells via Hippo/YAP signaling pathway. Phyther. Res. 2018, 32, 2456–2465. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.W.; Xu, J.; Zhu, G.Y.; Huang, Z.J.; Lu, Y.; Li, X.Q.; Wang, N.; Zhang, F.X. Apigenin suppresses the stem cell-like properties of triple-negative breast cancer cells by inhibiting YAP/TAZ activity. Cell Death Discov. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.; Xiang, K.; Wu, Y.; Zhang, T.; Liu, Y.; Liu, X.; Zhen, W.; Si, Y. Cucurbitacin b inhibits the hippo-YAP signaling pathway and exerts anticancer activity in colorectal cancer cells. Med. Sci. Monit. 2018, 24, 9251–9258. [Google Scholar] [CrossRef] [PubMed]
- Thurnher, M.; Nussbaumer, O.; Gruenbacher, G. Novel aspects of mevalonate pathway inhibitors as antitumor agents. Clin. Cancer Res. 2012, 18, 3524–3531. [Google Scholar] [CrossRef] [Green Version]
- Bathaie, S.Z.; Ashrafi, M.; Azizian, M.; Tamanoi, F. Mevalonate pathway and human cancers. Curr. Mol. Pharm. 2017, 10, 77–85. [Google Scholar] [CrossRef]
- Swanson, K.M.; Hohl, R.J. Anti-cancer therapy: Targeting the mevalonate pathway. Curr. Cancer Drug Targets 2006, 6, 15–37. [Google Scholar] [CrossRef] [PubMed]
- Thurnher, M.; Gruenbacher, G.; Nussbaumer, O. Regulation of mevalonate metabolism in cancer and immune cells. Biochim. Biophys. Acta. 2013, 1831, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.; Poklepovic, A.; Dent, P. Not the comfy chair! Cancer drugs that act against multiple active sites. Expert Opin. Ther. Targets 2019, 23, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Dent, P.; Booth, L.; Poklepovic, A.; Hancock, J.F. Signaling alterations caused by drugs and autophagy. Cell. Signal. 2019, 64, 109416. [Google Scholar] [CrossRef] [PubMed]
- Dent, P.; Booth, L.; Poklepovic, A.; Martinez, J.; Hoff, D.V.; Hancock, J.F. Neratinib degrades MST4 via autophagy that reduces membrane stiffness and is essential for the inactivation of PI3K, ERK1/2, and YAP/TAZ signaling. J. Cell Physiol. 2020, 235, 7889–7899. [Google Scholar] [CrossRef]
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat. Genet. 2015, 47, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.C.; Yang, C.T.; Jablons, D.M.; You, L. The role of yes-associated protein (YAP) in regulating programmed death-ligand 1 (PD-L1) in thoracic cancer. Biomedicines 2018, 6, E114. [Google Scholar] [CrossRef] [Green Version]
- Sarmasti Emami, S.; Zhang, D.; Yang, X. Interaction of the Hippo Pathway and Phosphatases in Tumorigenesis. Cancers 2020, 12, 2438. [Google Scholar] [CrossRef]
- Pocaterra, A.; Romani, P.; Dupont, S. YAP/TAZ functions and their regulation at a glance. J. Cell Sci. 2020, 133, jcs230425. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Yamamoto, K.; Agarwala, S.; Terai, K.; Fukui, H.; Fukuhara, S.; Ando, K.; Miyazaki, T.; Yokota, Y.; Schmelzer, E.; et al. Flow-dependent endothelial YAP Regulation contributes to vessel maintenance. Dev. Cell 2017, 40, 523–536.e6. [Google Scholar] [CrossRef] [Green Version]
- Meng, Z.; Qiu, Y.; Lin, K.C.; Kumar, A.; Placone, J.K.; Fang, C.; Wang, K.-C.; Lu, S.; Pan, M.; Hong, A.W.; et al. RAP2 mediates mechanoresponses of the Hippo pathway. Nature 2018, 560, 655–660. [Google Scholar] [CrossRef] [Green Version]
- Pocaterra, A.; Santinon, G.; Romani, P.; Brian, I.; Dimitracopoulos, A.; Ghisleni, A.; Carnicer-Lombarte, A.; Forcato, M.; Braghetta, P.; Montagner, M.; et al. F-actin dynamics regulates mammalian organ growth and cell fate maintenance. J. Hepatol. 2019, 71, 130–142. [Google Scholar] [CrossRef] [Green Version]
- Xin, M.; Kim, Y.; Sutherland, L.B.; Qi, X.; McAnally, J.; Schwartz, R.J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci. Signal. 2011, 4, ra70. [Google Scholar] [CrossRef] [Green Version]
- Mahoney, J.E.; Mori, M.; Szymaniak, A.D.; Varelas, X.; Cardoso, W.V. The hippo pathway effector Yap controls patterning and differentiation of airway epithelial progenitors. Dev. Cell 2014, 30, 137–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reginensi, A.; Scott, R.P.; Gregorieff, A.; Bagherie-Lachidan, M.; Chung, C.; Lim, D.S.; Pawson, T.; Wrana, J.; McNeill, H. Yap- and Cdc42-dependent nephrogenesis and morphogenesis during mouse kidney development. PLoS Genet. 2013, 9, e1003380. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Finegold, M.J.; Johnson, R.L. Hippo pathway coactivators Yap and Taz are required to coordinate mammalian liver regeneration. Exp. Mol. Med. 2018, 50, e43. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, N.; Xie, R.; Wang, W.; Cai, J.; Choi, K.S.; David, K.K.; Huang, B.; Yabuta, N.; Nojima, H.; et al. Homeostatic control of Hippo signaling activity revealed by an endogenous activating mutation in YAP. Genes Dev. 2015, 29, 1285–1297. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Hematological Malignancies | Yap Status | Mechanisms | Therapeutic Approach | Study | Ref. |
|---|---|---|---|---|---|
| Diffuse large B-cell lymphoma | Amplified Expression | Effects on cell proliferation and cell cycle arrest | Verteporfin (VP), CRISPR/Cas9 genome editing system, IGF-1R inhibitors | In vivo and in vitro | [128] |
| Natural Killer T–Cell Lymphoma | Amplified expression (phosphorylation blocked) | Effects on Bcl-2/Bax ratio, CTGF, surviving, c-myc, cyclin D 1, AP-1, SMADs, TBX5, p73, ICD of ERBB4 | Effects on Bcl-2/Bax ratio, CTGF, surviving, c-myc, cyclin D 1, AP-1, SMADs, TBX5, p73, ICD of ERBB4 | In vivo In vitro In vitro | [126] [129] [130] |
| Multiple Myeloma | Reduced levels in MM cells (hyper-stimulated) | Generation of the serine ethreonine kinase STK4, stimulation of MOB1B, effect on mitophagy Effect on circ-CDYL/miRNA-1180 (reduced apoptosis) LncRNA MALAT/MiRNA-181a-5p (reduced apoptosis). Action on bone disease (APC/β -catenin complex, Wnt signal, generation of Runx2, fibroblast growth factor 2 | STK4 block, Sirtuin reduction. miRNA-1180 silencing; LncRNA MALAT1 interference. Bortezomib | In vitro In vitro and in vivo In vitro In vitro and in vivo In vitro | [92,131,132] [133,134,135] [136,137] [138] [137] |
| Acute T cell leukemia | Increased YAP expression | Augmented leukemia cell growth and reduced programmed cell death (effects on protein kinase B, B-cell lymphoma 2 and BCL2 like protein 1) | Lentivirus transduced short hairpin RNA method | In vitro | [139] |
| Acute promyelocytic leukemia | Increased expression | Reduced concentrations of Bax and cleaved PARP, increased levels of Bcl-2, survivin, PARP, and cyclinD1. Possible effects on c-Myc, survivin, p-ERK, p-AKT, cleaved caspase3, and p-p38 MAPK | YAP knockdown or inhibition (shRNA or VP) | In vitro | [140,141] |
| Chronic myeloid leukemia | Constitutively phosphorylated | Effects on Src family kinases, survivin and cyclin D | Imatinib mesylate, RK-20449, block of miRNA-181a, VP | In vitro In vivo and in vitro | [194,195,199] [203] |
| Groups | Targets | Substances | Effects | Ref. |
|---|---|---|---|---|
| Group I | Upstream proteins, SFKs, AMPY, Phosphatases, EGFR, CPCR, Integrins, Adenylyl cyclase families, gamma-secretase, ErbB signaling, ILK | Kinase inhibitors, MEK/MAK inhibitors (trametinib, CAY10561, FR180204), Gamma secretase inhibitors, Epigenetic modulators (Panobinostat, Dacinostat, vorinostat), Actine modulators (Blebbistatin, ML-7, Cytochalasin D, Latrunculin A), Phosphatases inhibitors (okadaic acid, Calyculin A), SK2 inhibitors (Dasatinib, PP2, SV6656, AZ D0530), PI3 K inhibitors (BX795, Wortmannin/LY294002, Temserolimus, MLN0128), Mevalonate pathway inhibitors (statins, zoledronic acid), Cellular stress modulators (metformin, phenformin), AICAR. Erlotinib, AG-1478, Losartan, Dihydrexidine, Gallein, Clengitide, Agrin, RGD peptides, VEGFR inhibitors (Apatinib, SU4312, Axitinib, Pazopanib), Forskoli, Theophylline, IBMX, Odulilast, Rolipran, Dibenzapine, QLT0267, FAK inhibitors (PF-562271, PF-573228, CT-707) | Activate YAP and TAZ, promote TAZ degradation | [204,205,206,207] |
| Group II | Disruptors preventing the formation of the YAP/TAZ-TEAD complex | Verteporfin, YAP cyclic peptide (peptide 17), cystine-dense peptide (TB1 G1), Vgll1–4, substances targeting TEADs’ palmitate-binding pocket (fenamate derivatives, vinylsulfonamide derivatives, K-975) | Target either TEAD family of transcription factors or YAP/TAZ | [146,147,148,204,208,209] |
| Group III | Downstream YAP/TAZ targets: metabolic enzymes (aldehyde dehydrogenase, aspartate transaminase, cyclooxygenase 2), kinases, ligands and proteins (BCL-xL, FOXM1, TG2) | A37, celecoxib, WZ400, CXCL5 neutralizing antibody, SB255002, Jagged-1 neutralizing antibody, deoxybouvardin, CYR61 (093 G9) antibodies, navitoclax, thiostrepton, NC9 | Inhibition of proteins that are expressed under YAP/TAZ influence | [204] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allegra, A.; Pioggia, G.; Innao, V.; Musolino, C.; Gangemi, S. New Insights into YES-Associated Protein Signaling Pathways in Hematological Malignancies: Diagnostic and Therapeutic Challenges. Cancers 2021, 13, 1981. https://doi.org/10.3390/cancers13081981
Allegra A, Pioggia G, Innao V, Musolino C, Gangemi S. New Insights into YES-Associated Protein Signaling Pathways in Hematological Malignancies: Diagnostic and Therapeutic Challenges. Cancers. 2021; 13(8):1981. https://doi.org/10.3390/cancers13081981
Chicago/Turabian StyleAllegra, Alessandro, Giovanni Pioggia, Vanessa Innao, Caterina Musolino, and Sebastiano Gangemi. 2021. "New Insights into YES-Associated Protein Signaling Pathways in Hematological Malignancies: Diagnostic and Therapeutic Challenges" Cancers 13, no. 8: 1981. https://doi.org/10.3390/cancers13081981