
Tyrosine Kinase c-MET as Therapeutic Target for Radiosensitization of Head and Neck Squamous Cell Carcinomas

 ,
,  , and
, and
Abstract
:Simple Summary
Abstract
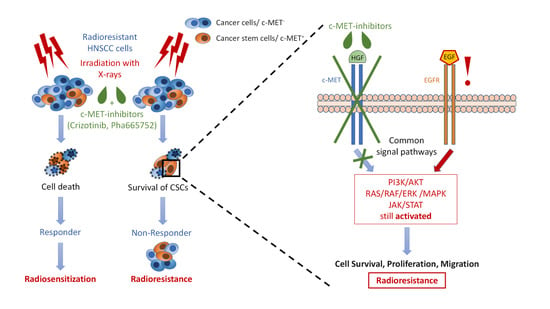
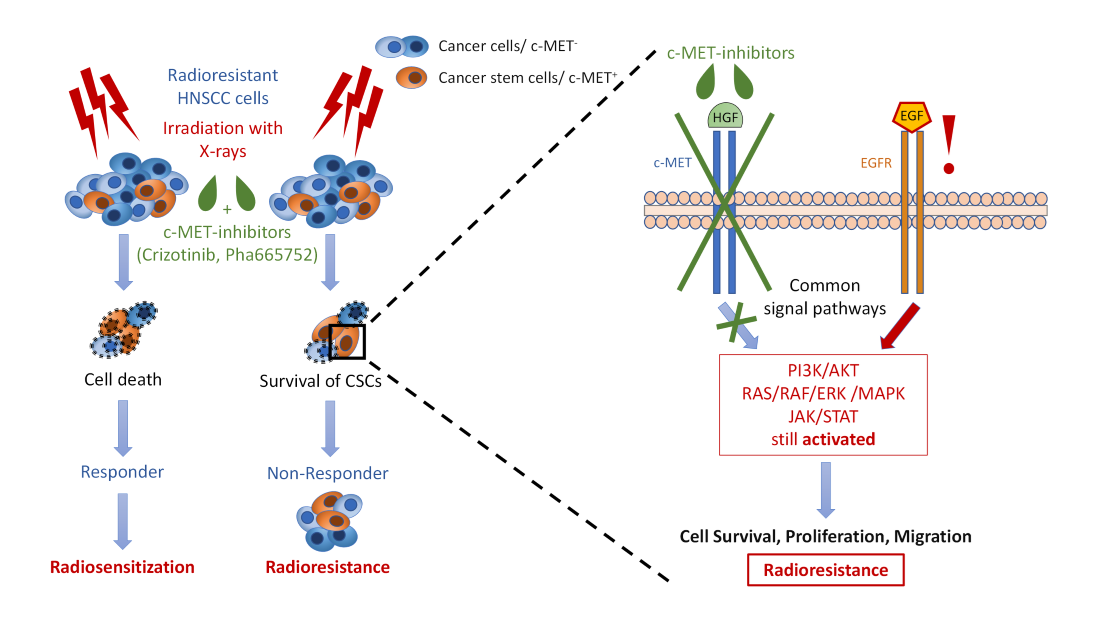{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Colony Formation Assay
2.3. Cell Irradiation
2.4. Viability Assay
2.5. Western Blot Analysis
2.6. Flow Cytometry Analysis and Fluorescence-Activated Cell Sorting (FACS)
2.7. Immunofluorescence Microscopy
2.8. siRNA-Mediated Gene Knock-Down
2.9. Sphere-Formation Assay
2.10. Xenograft Tumor Growth
2.11. Kinome Profiling
2.12. In Silico Gene Expression and Proteome Analysis
2.13. Statistics
3. Results
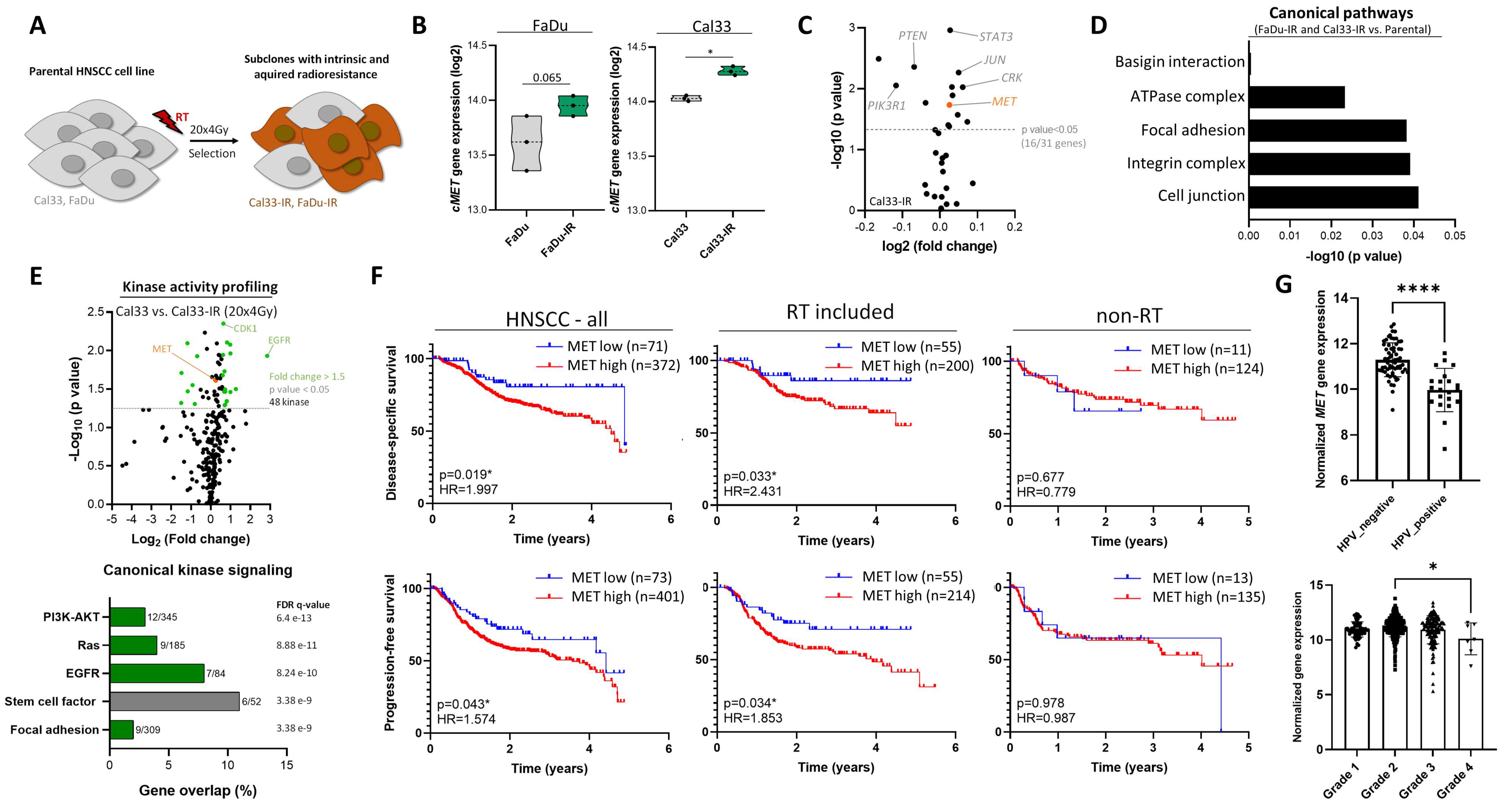
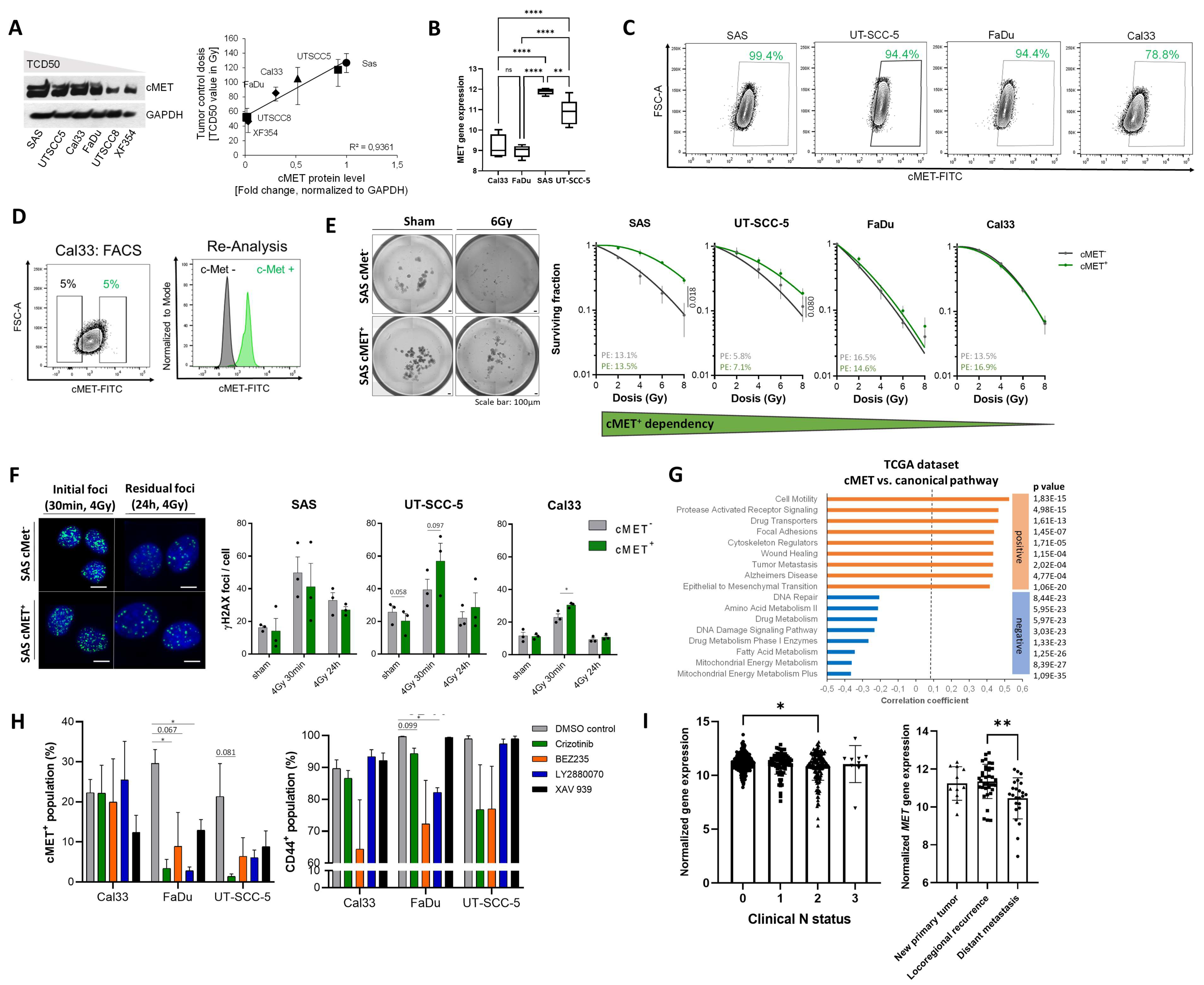
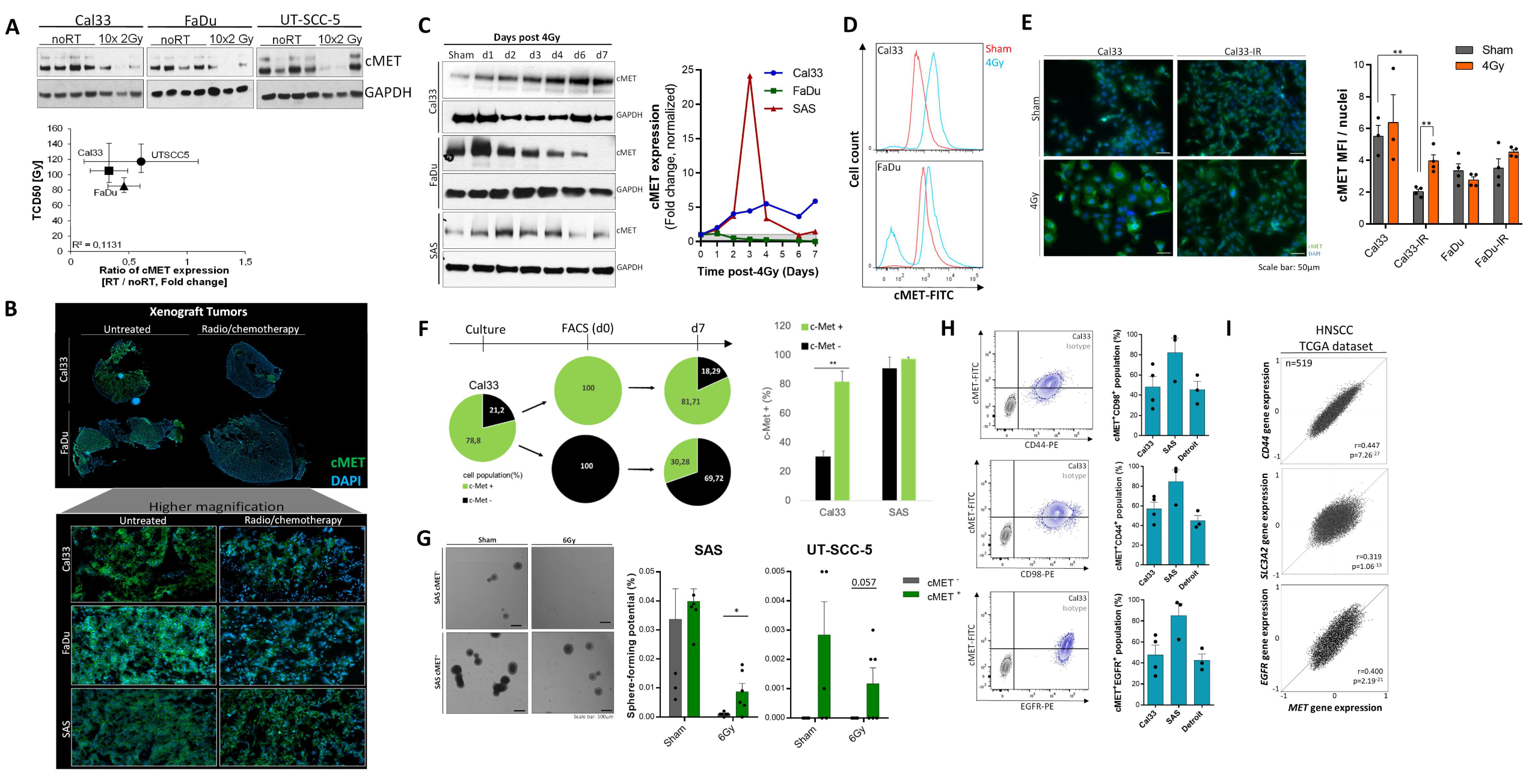
3.1. High c-MET Expression Characterizes a Radioresistant Subpopulation in HNSCC
3.2. c-MET-Expressing HNSCC Cells Are Less Sensitive to Irradiation without Affecting DNA Repair
3.3. The c-MET-Expressing Population in HNSCC Is Dynamically Regulated upon Irradiation and Is Characterized by Stem-Like Features
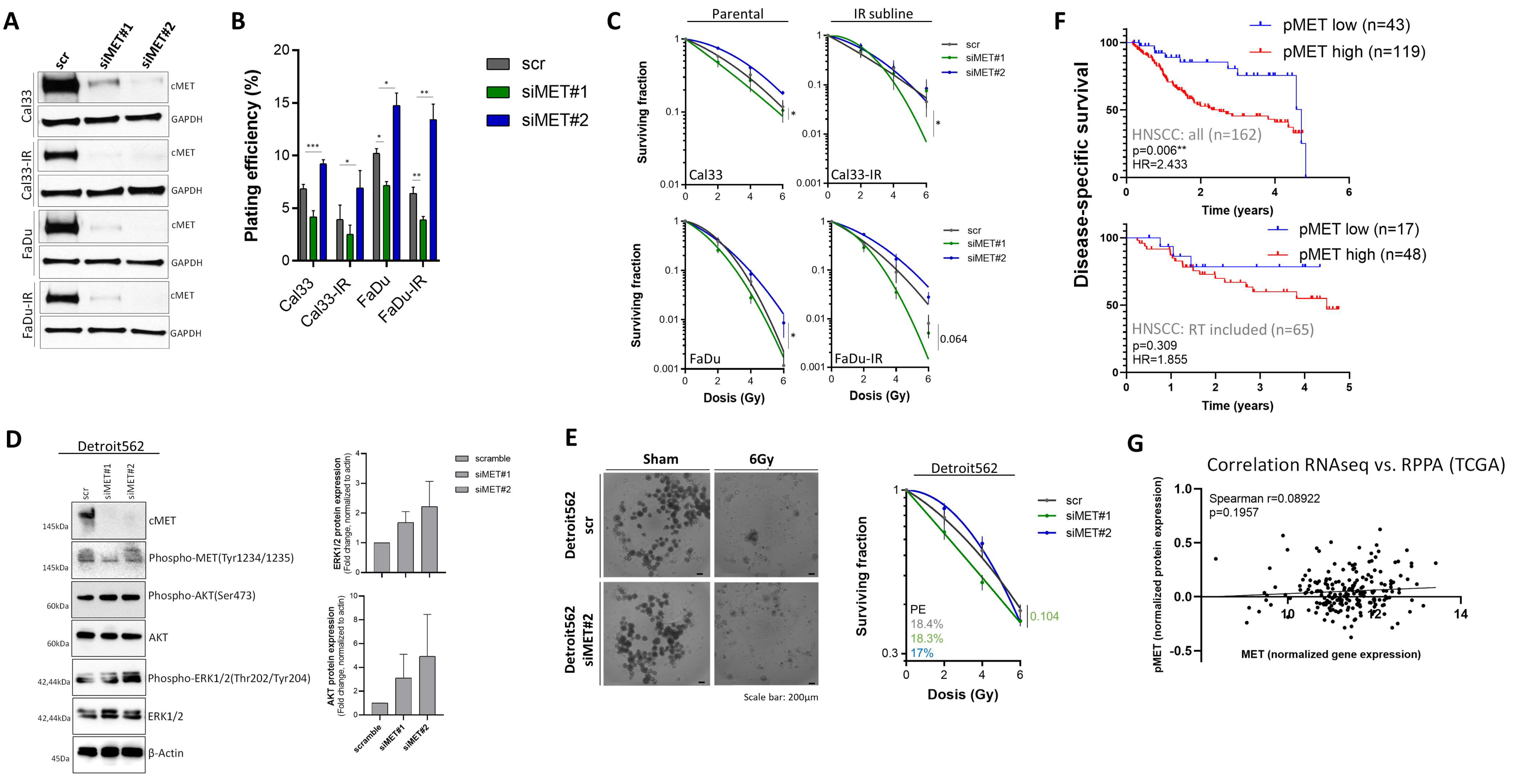
3.4. Compensatory Mechanisms Overcoming c-MET-Mediated Radioresponse Uncovered by MET-Specific Gene Knock-Down
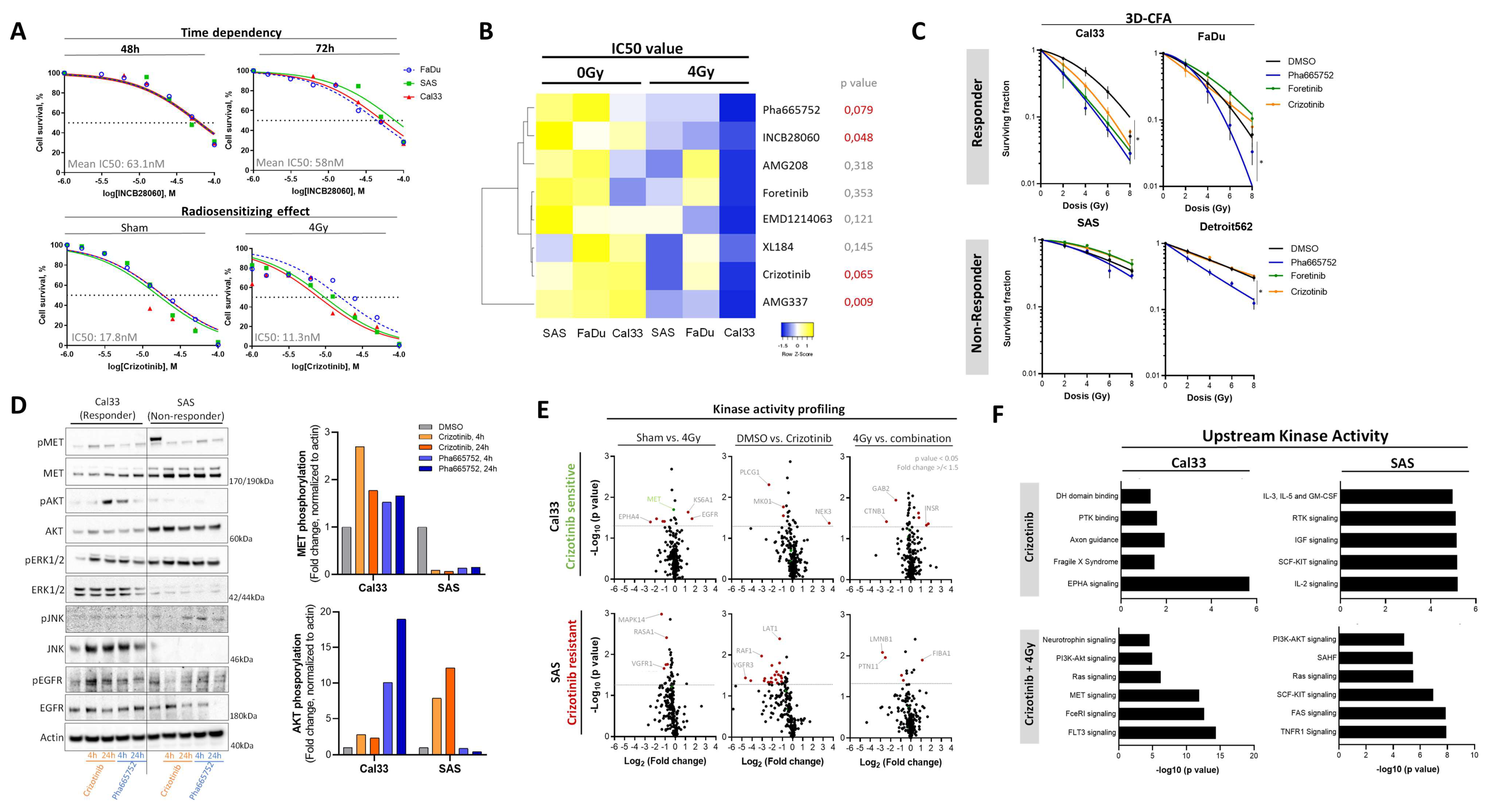
3.5. Chemical c-MET Targeting for HNSCC Radiosensitization
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rettig, E.M.; D’Souza, G. Epidemiology of Head and Neck Cancer. Surg. Oncol. Clin. N. Am. 2015, 24, 379–396. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021. [Google Scholar] [CrossRef] [PubMed]
- Gatta, G.; Botta, L.; Sánchez, M.J.; Anderson, L.A.; Pierannunzio, D.; Licitra, L.; EUROCARE Working Group. Prognoses and Improvement for Head and Neck Cancers Diagnosed in Europe in Early 2000s: The EUROCARE-5 Population-Based Study. Eur. J. Cancer 2015, 51, 2130–2143. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Puri, D.R.; Blanco, A.I.; Chao, K.S.C. Intensity-Modulated Radiation Therapy in Head and Neck Cancers: An Update. Head Neck 2007, 29, 387–400. [Google Scholar] [CrossRef]
- Argiris, A.; Li, S.; Savvides, P.; Ohr, J.P.; Gilbert, J.; Levine, M.A.; Chakravarti, A.; Haigentz, M., Jr.; Saba, N.F.; Ikpeazu, C.V.; et al. Phase III Randomized Trial of Chemotherapy with or without Bevacizumab in Patients with Recurrent or Metastatic Head and Neck Cancer. J. Clin. Oncol. 2019, 37, 3266–3274. [Google Scholar] [CrossRef]
- Muzaffar, J.; Bari, S.; Kirtane, K.; Chung, C.H. Recent Advances and Future Directions in Clinical Management of Head and Neck Squamous Cell Carcinoma. Cancers 2021, 13, 338. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Sougnez, C.; Lichtenstein, L.; Cibulskis, K.; Lander, E.; Gabriel, S.B.; Getz, G.; Ally, A.; Balasundaram, M.; Birol, I.; et al. Comprehensive Genomic Characterization of Head and Neck Squamous Cell Carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.H.; Guthrie, V.B.; Masica, D.L.; Tokheim, C.; Kang, H.; Richmon, J.; Agrawal, N.; Fakhry, C.; Quon, H.; Subramaniam, R.M.; et al. Genomic Alterations in Head and Neck Squamous Cell Carcinoma Determined by Cancer Gene-Targeted Sequencing. Ann. Oncol. 2015, 26, 1216–1223. [Google Scholar] [CrossRef]
- Kalyankrishna, S.; Grandis, J.R. Epidermal Growth Factor Receptor Biology in Head and Neck Cancer. J. Clin. Oncol. 2006, 24, 2666–2672. [Google Scholar] [CrossRef] [PubMed]
- Ganci, F.; Sacconi, A.; Manciocco, V.; Spriano, G.; Fontemaggi, G.; Carlini, P.; Blandino, G. Radioresistance in Head and Neck Squamous Cell Carcinoma—Possible Molecular Markers for Local Recurrence and New Putative Therapeutic Strategies. Contemp. Issues Head Neck Cancer Manag. 2015, 3–34. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Fisher, G.J. Role of Met Axis in Head and Neck Cancer. Cancers 2013, 5, 1601. [Google Scholar] [CrossRef] [Green Version]
- Alsahafi, E.; Begg, K.; Amelio, I.; Raulf, N.; Lucarelli, P.; Sauter, T.; Tavassoli, M. Clinical Update on Head and Neck Cancer: Molecular Biology and Ongoing Challenges. Cell Death Dis. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linge, A.; Löck, S.; Gudziol, V.; Nowak, A.; Lohaus, F.; von Neubeck, C.; Jütz, M.; Abdollahi, A.; Debus, J.; Tinhofer, I.; et al. Low Cancer Stem Cell Marker Expression and Low Hypoxia Identify Good Prognosis Subgroups in HPV(-) HNSCC after Postoperative Radiochemotherapy: A Multicenter Study of the DKTK-ROG. Clin. Cancer Res. 2016, 22, 2639–2649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothenberger, N.J.; Stabile, L.P. Hepatocyte Growth Factor/c-Met Signaling in Head and Neck Cancer and Implications for Treatment. Cancers 2017, 9, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comoglio, P.M.; Giordano, S.; Trusolino, L. Drug Development of MET Inhibitors: Targeting Oncogene Addiction and Expedience. Nat. Rev. Drug Discov. 2008, 7, 504–516. [Google Scholar] [CrossRef]
- Toschi, L.; Jänne, P.A. Single-Agent and Combination Therapeutic Strategies to Inhibit Hepatocyte Growth Factor/MET Signaling in Cancer. Clin. Cancer Res. 2008, 14, 5941–5946. [Google Scholar] [CrossRef] [Green Version]
- Ponzetto, C.; Bardelli, A.; Zhen, Z.; Maina, F.; dalla Zonca, P.; Giordano, S.; Graziani, A.; Panayotou, G.; Comoglio, P.M. A Multifunctional Docking Site Mediates Signaling and Transformation by the Hepatocyte Growth Factor/Scatter Factor Receptor Family. Cell 1994, 77, 261–271. [Google Scholar] [CrossRef]
- Sachs, M.; Brohmann, H.; Zechner, D.; Müller, T.; Hülsken, J.; Walther, I.; Schaeper, U.; Birchmeier, C.; Birchmeier, W. Essential Role of Gab1 for Signaling by the C-Met Receptor in Vivo. J. Cell Biol. 2000, 150, 1375–1384. [Google Scholar] [CrossRef] [Green Version]
- Baldanzi, G.; Graziani, A. Physiological Signaling and Structure of the HGF Receptor MET. Biomedicines 2014, 3, 1–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bladt, F.; Riethmacher, D.; Isenmann, S.; Aguzzi, A.; Birchmeier, C. Essential Role for the C- Met Receptor in the Migration of Myogenic Precursor Cells into the Limb Bud. Nature 1995, 376, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Nishizawa, T.; Hagiya, M.; Seki, T.; Shimonishi, M.; Sugimura, A.; Tashiro, K.; Shimizu, S. Molecular Cloning and Expression of Human Hepatocyte Growth Factor. Nature 1989, 342, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.; Giralt, J.; Harari, P.; Spencer, S.; Schulten, J.; Hossain, A.; Chang, S.-C.; Chin, S.; Baselga, J. Cetuximab and Radiotherapy in Laryngeal Preservation for Cancers of the Larynx and Hypopharynx: A Secondary Analysis of a Randomized Clinical Trial. JAMA Otolaryngol. Head Neck Surg. 2016, 142, 842–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takes, R.P.; Baatenburg de Jong, R.J.; Schuuring, E.; Litvinov, S.V.; Hermans, J.; Van Krieken, J.H. Differences in Expression of Oncogenes and Tumor Suppressor Genes in Different Sites of Head and Neck Squamous Cell. Anticancer Res. 1998, 18, 4793–4800. [Google Scholar] [PubMed]
- Mehanna, H.; Robinson, M.; Hartley, A.; Kong, A.; Foran, B.; Fulton-Lieuw, T.; Dalby, M.; Mistry, P.; Sen, M.; O’Toole, L.; et al. Radiotherapy plus Cisplatin or Cetuximab in Low-Risk Human Papillomavirus-Positive Oropharyngeal Cancer (De-ESCALaTE HPV): An Open-Label Randomised Controlled Phase 3 Trial. Lancet 2019, 393, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Gebre-Medhin, M.; Brun, E.; Engström, P.; Haugen Cange, H.; Hammarstedt-Nordenvall, L.; Reizenstein, J.; Nyman, J.; Abel, E.; Friesland, S.; Sjödin, H.; et al. ARTSCAN III: A Randomized Phase III Study Comparing Chemoradiotherapy With Cisplatin Versus Cetuximab in Patients With Locoregionally Advanced Head and Neck Squamous Cell Cancer. J. Clin. Oncol. 2021, 39, 38–47. [Google Scholar] [CrossRef]
- Montagut, C.; Dalmases, A.; Bellosillo, B.; Crespo, M.; Pairet, S.; Iglesias, M.; Salido, M.; Gallen, M.; Marsters, S.; Tsai, S.P.; et al. Identification of a Mutation in the Extracellular Domain of the Epidermal Growth Factor Receptor Conferring Cetuximab Resistance in Colorectal Cancer. Nat. Med. 2012, 18, 221–223. [Google Scholar] [CrossRef]
- Da Costa, A.A.B.A.; Costa, F.D.; Araújo, D.V.; Camandaroba, M.P.G.; de Jesus, V.H.F.; Oliveira, A.; Alves, A.C.F.; Stecca, C.; Machado, L.; de Oliveira, A.C.F.; et al. The Roles of PTEN, CMET, and P16 in Resistance to Cetuximab in Head and Neck Squamous Cell Carcinoma. Med. Oncol. 2018, 36, 8. [Google Scholar] [CrossRef]
- Scagliotti, G.V.; Novello, S.; von Pawel, J. The Emerging Role of MET/HGF Inhibitors in Oncology. Cancer Treat. Rev. 2013, 39, 793–801. [Google Scholar] [CrossRef]
- Huang, X.; Li, E.; Shen, H.; Wang, X.; Tang, T.; Zhang, X.; Xu, J.; Tang, Z.; Guo, C.; Bai, X.; et al. Targeting the HGF/MET Axis in Cancer Therapy: Challenges in Resistance and Opportunities for Improvement. Front. Cell Dev. Biol. 2020, 8, 152. [Google Scholar] [CrossRef]
- Puccini, A.; Marín-Ramos, N.I.; Bergamo, F.; Schirripa, M.; Lonardi, S.; Lenz, H.-J.; Loupakis, F.; Battaglin, F. Safety and Tolerability of C-MET Inhibitors in Cancer. Drug Saf. 2019, 42, 211–233. [Google Scholar] [CrossRef]
- Wilson, G.D.; Thibodeau, B.J.; Fortier, L.E.; Pruetz, B.L.; Galoforo, S.; Marples, B.; Baschnagel, A.M.; Akervall, J.; Huang, J. Cancer Stem Cell Signaling during Repopulation in Head and Neck Cancer. Available online: https://www.hindawi.com/journals/sci/2016/1894782/ (accessed on 7 January 2021).
- Schulz, A.; Meyer, F.; Dubrovska, A.; Borgmann, K. Cancer Stem Cells and Radioresistance: DNA Repair and Beyond. Cancers 2019, 11, 862. [Google Scholar] [CrossRef] [Green Version]
- Digomann, D.; Kurth, I.; Tyutyunnykova, A.; Chen, O.; Löck, S.; Gorodetska, I.; Peitzsch, C.; Skvortsova, I.-I.; Negro, G.; Aschenbrenner, B.; et al. The CD98 Heavy Chain Is a Marker and Regulator of Head and Neck Squamous Cell Carcinoma Radiosensitivity. Clin. Cancer Res. 2019, 25, 3152–3163. [Google Scholar] [CrossRef] [PubMed]
- Linge, A.; Löck, S.; Krenn, C.; Appold, S.; Lohaus, F.; Nowak, A.; Gudziol, V.; Baretton, G.B.; Buchholz, F.; Baumann, M.; et al. Independent Validation of the Prognostic Value of Cancer Stem Cell Marker Expression and Hypoxia-Induced Gene Expression for Patients with Locally Advanced HNSCC after Postoperative Radiotherapy. Clin. Transl. Radiat. Oncol. 2016, 1, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linge, A.; Lohaus, F.; Löck, S.; Nowak, A.; Gudziol, V.; Valentini, C.; von Neubeck, C.; Jütz, M.; Tinhofer, I.; Budach, V.; et al. HPV Status, Cancer Stem Cell Marker Expression, Hypoxia Gene Signatures and Tumour Volume Identify Good Prognosis Subgroups in Patients with HNSCC after Primary Radiochemotherapy: A Multicentre Retrospective Study of the German Cancer Consortium Radiation Oncology Group (DKTK-ROG). Radiother. Oncol. 2016, 121, 364–373. [Google Scholar] [CrossRef]
- Kurth, I.; Hein, L.; Mäbert, K.; Peitzsch, C.; Koi, L.; Cojoc, M.; Kunz-Schughart, L.; Baumann, M.; Dubrovska, A. Cancer Stem Cell Related Markers of Radioresistance in Head and Neck Squamous Cell Carcinoma. Oncotarget 2015, 6, 34494–34509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prince, M.E.P.; Ailles, L.E. Cancer Stem Cells in Head and Neck Squamous Cell Cancer. J. Clin. Oncol. 2008, 26, 2871–2875. [Google Scholar] [CrossRef]
- Zhang, Z.; Filho, M.S.; Nör, J.E. The Biology of Head and Neck Cancer Stem Cells. Oral. Oncol. 2012, 48, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Helbig, L.; Koi, L.; Brüchner, K.; Gurtner, K.; Hess-Stumpp, H.; Unterschemmann, K.; Baumann, M.; Zips, D.; Yaromina, A. BAY 87-2243, a Novel Inhibitor of Hypoxia-Induced Gene Activation, Improves Local Tumor Control after Fractionated Irradiation in a Schedule-Dependent Manner in Head and Neck Human Xenografts. Radiat. Oncol. 2014, 9, 207. [Google Scholar] [CrossRef] [Green Version]
- Koi, L.; Bergmann, R.; Brüchner, K.; Pietzsch, J.; Pietzsch, H.-J.; Krause, M.; Steinbach, J.; Zips, D.; Baumann, M. Radiolabeled Anti-EGFR-Antibody Improves Local Tumor Control after External Beam Radiotherapy and Offers Theragnostic Potential. Radiother. Oncol. 2014, 110, 362–369. [Google Scholar] [CrossRef]
- Koi, L.; Löck, S.; Linge, A.; Thurow, C.; Hering, S.; Baumann, M.; Krause, M.; Gurtner, K. EGFR-Amplification plus Gene Expression Profiling Predicts Response to Combined Radiotherapy with EGFR-Inhibition: A Preclinical Trial in 10 HNSCC-Tumour-Xenograft Models. Radiother. Oncol. 2017, 124, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, K.; Molfenter, B.; Laureano, N.K.; Tawk, B.; Bieg, M.; Hostench, X.P.; Weichenhan, D.; Ullrich, N.D.; Shang, V.; Richter, D.; et al. Somatic Mutations and Promotor Methylation of the Ryanodine Receptor 2 Is a Common Event in the Pathogenesis of Head and Neck Cancer. Int. J. Cancer 2019, 145, 3299–3310. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Shen, Y.; Hostench, X.P.; Bieg, M.; Plath, M.; Ishaque, N.; Eils, R.; Freier, K.; Weichert, W.; Zaoui, K.; et al. Integrative Analysis of Multi-Omics Data Identified EGFR and PTGS2 as Key Nodes in a Gene Regulatory Network Related to Immune Phenotypes in Head and Neck Cancer. Clin. Cancer Res. 2020, 26, 3616–3628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linge, A.; Schmidt, S.; Lohaus, F.; Krenn, C.; Bandurska-Luque, A.; Platzek, I.; von Neubeck, C.; Appold, S.; Nowak, A.; Gudziol, V.; et al. Independent Validation of Tumour Volume, Cancer Stem Cell Markers and Hypoxia-Associated Gene Expressions for HNSCC after Primary Radiochemotherapy. Clin. Transl. Radiat. Oncol. 2019, 16, 40–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaromina, A.; Krause, M.; Thames, H.; Rosner, A.; Krause, M.; Hessel, F.; Grenman, R.; Zips, D.; Baumann, M. Pre-Treatment Number of Clonogenic Cells and Their Radiosensitivity Are Major Determinants of Local Tumour Control after Fractionated Irradiation. Radiother. Oncol. 2007, 83, 304–310. [Google Scholar] [CrossRef]
- De Bacco, F.; Casanova, E.; Medico, E.; Pellegatta, S.; Orzan, F.; Albano, R.; Luraghi, P.; Reato, G.; D’Ambrosio, A.; Porrati, P.; et al. The MET Oncogene Is a Functional Marker of a Glioblastoma Stem Cell Subtype. Cancer Res. 2012, 72, 4537–4550. [Google Scholar] [CrossRef] [Green Version]
- Gastaldi, S.; Sassi, F.; Accornero, P.; Torti, D.; Galimi, F.; Migliardi, G.; Molyneux, G.; Perera, T.; Comoglio, P.M.; Boccaccio, C.; et al. Met Signaling Regulates Growth, Repopulating Potential and Basal Cell-Fate Commitment of Mammary Luminal Progenitors: Implications for Basal-like Breast Cancer. Oncogene 2013, 32, 1428–1440. [Google Scholar] [CrossRef] [Green Version]
- Saini, M.; Verma, A.; Mathew, S.J. SPRY2 Is a Novel MET Interactor That Regulates Metastatic Potential and Differentiation in Rhabdomyosarcoma. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Corso, S.; Migliore, C.; Ghiso, E.; De Rosa, G.; Comoglio, P.M.; Giordano, S. Silencing the MET Oncogene Leads to Regression of Experimental Tumors and Metastases. Oncogene 2008, 27, 684–693. [Google Scholar] [CrossRef] [Green Version]
- Peschard, P.; Park, M. From Tpr-Met to Met, Tumorigenesis and Tubes. Oncogene 2007, 26, 1276–1285. [Google Scholar] [CrossRef] [Green Version]
- Migliore, C.; Giordano, S. Molecular Cancer Therapy: Can Our Expectation Be MET? Eur. J. Cancer 2008, 44, 641–651. [Google Scholar] [CrossRef]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET Signalling: Principles and Functions in Development, Organ Regeneration and Cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Eder, J.P.; Vande Woude, G.F.; Boerner, S.A.; LoRusso, P.M. Novel Therapeutic Inhibitors of the C-Met Signaling Pathway in Cancer. Clin. Cancer Res. 2009, 15, 2207–2214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baschnagel, A.M.; Williams, L.; Hanna, A.; Chen, P.Y.; Krauss, D.J.; Pruetz, B.L.; Akervall, J.; Wilson, G.D. C-Met Expression Is a Marker of Poor Prognosis in Patients with Locally Advanced Head and Neck Squamous Cell Carcinoma Treated with Chemoradiation. Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 701–707. [Google Scholar] [CrossRef]
- Akervall, J.; Guo, X.; Qian, C.-N.; Schoumans, J.; Leeser, B.; Kort, E.; Cole, A.; Resau, J.; Bradford, C.; Carey, T.; et al. Genetic and Expression Profiles of Squamous Cell Carcinoma of the Head and Neck Correlate with Cisplatin Sensitivity and Resistance in Cell Lines and Patients. Clin. Cancer Res. 2004, 10, 8204–8213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madoz-Gúrpide, J.; Zazo, S.; Chamizo, C.; Casado, V.; Caramés, C.; Gavín, E.; Cristóbal, I.; García-Foncillas, J.; Rojo, F. Activation of MET Pathway Predicts Poor Outcome to Cetuximab in Patients with Recurrent or Metastatic Head and Neck Cancer. J. Transl. Med. 2015, 13, 282. [Google Scholar] [CrossRef]
- Wong, S.J.; Heron, D.E.; Stenson, K.; Ling, D.C.; Vargo, J.A. Locoregional Recurrent or Second Primary Head and Neck Cancer: Management Strategies and Challenges. Am. Soc. Clin. Oncol. Educ. Book 2016, 36, e284–e292. [Google Scholar] [CrossRef] [PubMed]
- Bensimon, A.; Koch, J.P.; Francica, P.; Roth, S.M.; Riedo, R.; Glück, A.A.; Orlando, E.; Blaukat, A.; Aebersold, D.M.; Zimmer, Y.; et al. Deciphering MET-Dependent Modulation of Global Cellular Responses to DNA Damage by Quantitative Phosphoproteomics. Mol. Oncol. 2020, 14, 1185–1206. [Google Scholar] [CrossRef]
- Seiwert, T.; Sarantopoulos, J.; Kallender, H.; McCallum, S.; Keer, H.N.; Blumenschein, G. Phase II Trial of Single-Agent Foretinib (GSK1363089) in Patients with Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck. Investig. New Drugs 2013, 31, 417–424. [Google Scholar] [CrossRef] [Green Version]
- Blumenschein, G.R.; Mills, G.B.; Gonzalez-Angulo, A.M. Targeting the Hepatocyte Growth Factor-CMET Axis in Cancer Therapy. J. Clin. Oncol. 2012, 30, 3287–3296. [Google Scholar] [CrossRef] [Green Version]
- Raghav, K.P.; Wang, W.; Liu, S.; Chavez-MacGregor, M.; Meng, X.; Hortobagyi, G.N.; Mills, G.B.; Meric-Bernstam, F.; Blumenschein, G.R.; Gonzalez-Angulo, A.M. CMET and Phospho-CMET Protein Levels in Breast Cancers and Survival Outcomes. Clin. Cancer Res. 2012, 18, 2269–2277. [Google Scholar] [CrossRef] [Green Version]
- Spigel, D.R.; Reynolds, C.; Waterhouse, D.; Garon, E.B.; Chandler, J.; Babu, S.; Thurmes, P.; Spira, A.; Jotte, R.; Zhu, J.; et al. Phase 1/2 Study of the Safety and Tolerability of Nivolumab Plus Crizotinib for the First-Line Treatment of Anaplastic Lymphoma Kinase Translocation—Positive Advanced Non-Small Cell Lung Cancer (CheckMate 370). J. Thorac. Oncol. 2018, 13, 682–688. [Google Scholar] [CrossRef] [Green Version]
- Gherardi, E.; Youles, M.E.; Miguel, R.N.; Blundell, T.L.; Iamele, L.; Gough, J.; Bandyopadhyay, A.; Hartmann, G.; Butler, P.J.G. Functional Map and Domain Structure of MET, the Product of the c-Met Protooncogene and Receptor for Hepatocyte Growth Factor/Scatter Factor. Proc. Natl. Acad. Sci. USA 2003, 100, 12039–12044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baschnagel, A.M.; Galoforo, S.; Thibodeau, B.J.; Ahmed, S.; Nirmal, S.; Akervall, J.; Wilson, G.D. Crizotinib Fails to Enhance the Effect of Radiation in Head and Neck Squamous Cell Carcinoma Xenografts. Anticancer Res. 2015, 35, 5973–5982. [Google Scholar] [PubMed]
- Kriegs, M.; Kasten-Pisula, U.; Riepen, B.; Hoffer, K.; Struve, N.; Myllynen, L.; Braig, F.; Binder, M.; Rieckmann, T.; Grénman, R.; et al. Radiosensitization of HNSCC Cells by EGFR Inhibition Depends on the Induction of Cell Cycle Arrests. Oncotarget 2016, 7, 45122–45133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, L.M.; Stabile, L.P.; Egloff, A.M.; Rothstein, M.E.; Thomas, S.M.; Gubish, C.T.; Lerner, E.C.; Seethala, R.R.; Suzuki, S.; Quesnelle, K.M.; et al. HGF and C-Met Participate in Paracrine Tumorigenic Pathways in Head and Neck Squamous Cell Cancer. Clin. Cancer Res. 2009, 15, 3740–3750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.-H.; Lee, J.S.; Kang, S.-O.; Bae, J.-H.; Hong, S.P.; Kahng, H. Serum Hepatocyte Growth Factor as a Marker of Tumor Activity in Head and Neck Squamous Cell Carcinoma. Oral. Oncol. 2007, 43, 1021–1025. [Google Scholar] [CrossRef]
- Kim, I.-G.; Lee, J.-H.; Kim, S.-Y.; Kim, J.-Y.; Cho, E.-W. Fibulin-3 Negatively Regulates ALDH1 via c-MET Suppression and Increases γ-Radiation-Induced Sensitivity in Some Pancreatic Cancer Cell Lines. Biochem. Biophys. Res. Commun. 2014, 454, 369–375. [Google Scholar] [CrossRef] [Green Version]
- Seiwert, T.Y.; Jagadeeswaran, R.; Faoro, L.; Janamanchi, V.; Nallasura, V.; El Dinali, M.; Yala, S.; Kanteti, R.; Cohen, E.E.W.; Lingen, M.W.; et al. The MET Receptor Tyrosine Kinase Is a Potential Novel Therapeutic Target for Head and Neck Squamous Cell Carcinoma. Cancer Res. 2009, 69, 3021–3031. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Li, Q.; Sun, Q.; Zhang, Y.; Yang, H.; Wang, R.; Chen, L.; Wang, W. MET Inhibitor PHA-665752 Suppresses the Hepatocyte Growth Factor-Induced Cell Proliferation and Radioresistance in Nasopharyngeal Carcinoma Cells. Biochem. Biophys. Res. Commun. 2014, 449, 49–54. [Google Scholar] [CrossRef]
- Dy, G.K.; Adjei, A.A. Understanding, Recognizing, and Managing Toxicities of Targeted Anticancer Therapies. CA A Cancer J. Clin. 2013, 63, 249–279. [Google Scholar] [CrossRef]
- Grandis, J.R.; Tweardy, D.J. TGF-Alpha and EGFR in Head and Neck Cancer. J. Cell Biochem. Suppl. 1993, 17F, 188–191. [Google Scholar] [CrossRef]
- Bussink, J.; van der Kogel, A.J.; Kaanders, J.H.A.M. Activation of the PI3-K/AKT Pathway and Implications for Radioresistance Mechanisms in Head and Neck Cancer. Lancet Oncol. 2008, 9, 288–296. [Google Scholar] [CrossRef]
- Medová, M.; Aebersold, D.M.; Zimmer, Y. MET Inhibition in Tumor Cells by PHA665752 Impairs Homologous Recombination Repair of DNA Double Strand Breaks. Int. J. Cancer 2012, 130, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.P.; Franck, D.; Parachoniak, C.A.; Gregg, J.P.; Moore, M.G.; Farwell, D.G.; Rao, S.; Heilmann, A.M.; Erlich, R.L.; Ross, J.S.; et al. MET Genomic Alterations in Head and Neck Squamous Cell Carcinoma (HNSCC): Rapid Response to Crizotinib in a Patient with HNSCC with a Novel MET R1004G Mutation. Oncologist 2019, 24, 1305–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lüttich, L.; Besso, M.J.; Heiden, S.; Koi, L.; Baumann, M.; Krause, M.; Dubrovska, A.; Linge, A.; Kurth, I.; Peitzsch, C. Tyrosine Kinase c-MET as Therapeutic Target for Radiosensitization of Head and Neck Squamous Cell Carcinomas. Cancers 2021, 13, 1865. https://doi.org/10.3390/cancers13081865
Lüttich L, Besso MJ, Heiden S, Koi L, Baumann M, Krause M, Dubrovska A, Linge A, Kurth I, Peitzsch C. Tyrosine Kinase c-MET as Therapeutic Target for Radiosensitization of Head and Neck Squamous Cell Carcinomas. Cancers. 2021; 13(8):1865. https://doi.org/10.3390/cancers13081865
Chicago/Turabian StyleLüttich, Lina, María José Besso, Stephan Heiden, Lydia Koi, Michael Baumann, Mechthild Krause, Anna Dubrovska, Annett Linge, Ina Kurth, and Claudia Peitzsch. 2021. "Tyrosine Kinase c-MET as Therapeutic Target for Radiosensitization of Head and Neck Squamous Cell Carcinomas" Cancers 13, no. 8: 1865. https://doi.org/10.3390/cancers13081865