
Murlentamab, a Low Fucosylated Anti-Müllerian Hormone Type II Receptor (AMHRII) Antibody, Exhibits Anti-Tumor Activity through Tumor-Associated Macrophage Reprogrammation and T Cell Activation

 , and
, and
Abstract
:Simple Summary
Abstract
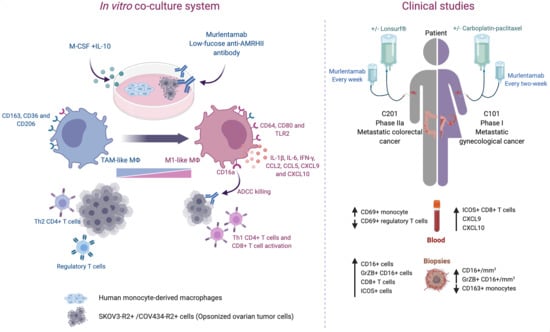
1. Introduction
2. Results
2.1. Murlentamab Treatment of Colorectal and Ovarian Cancer Patients Is Associated with the Activation of the Innate and Adaptive Immune Response
2.2. Murlentamab Enhances Naïve Macrophage and Tumor-Associated Macrophage Intrinsic Anti-Tumor Activity
2.3. Murlentamab Orients Naïve Macrophages and Reprograms Tumor-Associated Macrophages towards an M1-Like Profile
2.4. Murlentamab Promotes the Ability of Naïve and Tumor-Associated Macrophages to Activate the Anti-Tumor Adaptive Immune Response
2.5. Murlentamab and Pembrolizumab Association Enhances the Anti-Tumor Potential of Murlentamab Monotherapy
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Patients and Study Design
4.3. Tumor and Blood Specimens
4.4. Phenotype Analysis of Blood Circulating Immune Cells of Patients
4.5. Immunofluorescence
4.6. Preparation of Human Monocyte-Derived Macrophages (MDMs)
4.7. Opsonization and Co-Culture of Ovarian Carcinoma Tumor Cell Lines with Human MDMs
4.8. Evaluation of Ovarian Carcinoma Tumor Cell Number
4.9. Antibody-Dependent Cell-Mediated Cytotoxicity (ADCC) Assay
4.10. Phenotypic Characterization of Human MDMs
4.11. Production of Cytokines and Chemokines
4.12. Human T Cell Isolation
4.13. T Cell Polarization and Activation
4.14. Secretory Profile of M2-Like Macrophages in the Presence of Murlentamab
4.15. In Vivo Experiment
4.16. Flow Cytometry Antibodies
4.17. Statistical Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baarends, W.M.; van Helmond, M.J.; Post, M.; van der Schoot, P.J.; Hoogerbrugge, J.W.; de Winter, J.P.; Uilenbroek, J.T.; Karels, B.; Wilming, L.G.; Meijers, J.H. A Novel Member of the Transmembrane Serine/Threonine Kinase Receptor Family Is Specifically Expressed in the Gonads and in Mesenchymal Cells Adjacent to the Müllerian Duct. Dev. Camb. Engl. 1994, 120, 189–197. [Google Scholar]
- di Clemente, N.; Wilson, C.; Faure, E.; Boussin, L.; Carmillo, P.; Tizard, R.; Picard, J.Y.; Vigier, B.; Josso, N.; Cate, R. Cloning, Expression, and Alternative Splicing of the Receptor for Anti-Müllerian Hormone. Mol. Endocrinol. 1994, 8, 1006–1020. [Google Scholar] [CrossRef] [PubMed]
- Jamin, S.P.; Arango, N.A.; Mishina, Y.; Hanks, M.C.; Behringer, R.R. Genetic Studies of the AMH/MIS Signaling Pathway for Müllerian Duct Regression. Mol. Cell. Endocrinol. 2003, 211, 15–19. [Google Scholar] [CrossRef]
- Visser, J.A.; Schipper, I.; Laven, J.S.E.; Themmen, A.P.N. Anti-Müllerian Hormone: An Ovarian Reserve Marker in Primary Ovarian Insufficiency. Nat. Rev. Endocrinol. 2012, 8, 331–341. [Google Scholar] [CrossRef]
- Sriraman, V.; Niu, E.; Matias, J.R.; Donahoe, P.K.; MacLaughlin, D.T.; Hardy, M.P.; Lee, M.M. Müllerian Inhibiting Substance Inhibits Testosterone Synthesis in Adult Rats. J. Androl. 2001, 22, 750–758. [Google Scholar]
- Bakkum-Gamez, J.N.; Aletti, G.; Lewis, K.A.; Keeney, G.L.; Thomas, B.M.; Navarro-Teulon, I.; Cliby, W.A. Müllerian Inhibiting Substance Type II Receptor (MISIIR): A Novel, Tissue-Specific Target Expressed by Gynecologic Cancers. Gynecol. Oncol. 2008, 108, 141–148. [Google Scholar] [CrossRef]
- Song, J.Y.; Chen, K.Y.; Kim, S.Y.; Kim, M.R.; Ryu, K.S.; Cha, J.H.; Kang, C.S.; MacLaughlin, D.T.; Kim, J.H. The Expression of Müllerian Inhibiting Substance/Anti-Müllerian Hormone Type II Receptor Protein and MRNA in Benign, Borderline and Malignant Ovarian Neoplasia. Int. J. Oncol. 2009, 34, 1583–1591. [Google Scholar] [CrossRef] [Green Version]
- Kersual, N.; Garambois, V.; Chardès, T.; Pouget, J.-P.; Salhi, I.; Bascoul-Mollevi, C.; Bibeau, F.; Busson, M.; Vié, H.; Clémenceau, B.; et al. The Human Müllerian Inhibiting Substance Type II Receptor as Immunotherapy Target for Ovarian Cancer: Validation Using the MAb 12G4. MABS 2014, 6, 1314–1326. [Google Scholar] [CrossRef] [Green Version]
- Pépin, D.; Sosulski, A.; Zhang, L.; Wang, D.; Vathipadiekal, V.; Hendren, K.; Coletti, C.M.; Yu, A.; Castro, C.M.; Birrer, M.J.; et al. AAV9 Delivering a Modified Human Mullerian Inhibiting Substance as a Gene Therapy in Patient-Derived Xenografts of Ovarian Cancer. Proc. Natl. Acad. Sci. USA 2015, 112, E4418–E4427. [Google Scholar] [CrossRef] [Green Version]
- Anttonen, M.; Färkkilä, A.; Tauriala, H.; Kauppinen, M.; MacLaughlin, D.T.; Unkila-Kallio, L.; Bützow, R.; Heikinheimo, M. Anti-Müllerian Hormone Inhibits Growth of AMH Type II Receptor-Positive Human Ovarian Granulosa Cell Tumor Cells by Activating Apoptosis. Lab. Invest. 2011, 91, 1605–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, T.N.; Korobeynikov, V.A.; Kudinov, A.E.; Georgopoulos, R.; Solanki, N.R.; Andrews-Hoke, M.; Kistner, T.M.; Pépin, D.; Donahoe, P.K.; Nicolas, E.; et al. Anti-Müllerian Hormone Signaling Regulates Epithelial Plasticity and Chemoresistance in Lung Cancer. Cell Rep. 2016, 16, 657–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barret, J.-M.M.; Meseure, D.; Bataillon, G.; Andersson, N.; Auguste, A.; Dubreuil, O.; Färkkilä, A.; Perrial, E.; Bossard, C.; Loison, E.; et al. Abstract 774: Anti-Müllerian Hormone Type II Receptor (AMHRII) Found Expressed in Human Non-Gynecological Solid Tumors, Suggesting Potential Broader Applications for Anti-AMHRII-Based Therapy. Cancer Res. 2018, 78, 774. [Google Scholar] [CrossRef]
- Estupina, P.; Fontayne, A.; Barret, J.-M.; Kersual, N.; Dubreuil, O.; Blay, M.L.; Pichard, A.; Jarlier, M.; Pugnière, M.; Chauvin, M.; et al. The Anti-Tumor Efficacy of 3C23K, a Glyco-Engineered Humanized Anti-MISRII Antibody, in an Ovarian Cancer Model Is Mainly Mediated by Engagement of Immune Effector Cells. Oncotarget 2017, 8, 37061–37079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bougherara, H.; Némati, F.; Nicolas, A.; Massonnet, G.; Pugnière, M.; Ngô, C.; Le Frère-Belda, M.-A.; Leary, A.; Alexandre, J.; Meseure, D.; et al. The Humanized Anti-Human AMHRII MAb 3C23K Exerts an Anti-Tumor Activity against Human Ovarian Cancer through Tumor-Associated Macrophages. Oncotarget 2017, 8, 99950–99965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jefferis, R.; Lund, J.; Pound, J.D. IgG-Fc-Mediated Effector Functions: Molecular Definition of Interaction Sites for Effector Ligands and the Role of Glycosylation. Immunol. Rev. 1998, 163, 59–76. [Google Scholar] [CrossRef]
- Shields, R.L.; Lai, J.; Keck, R.; O’Connell, L.Y.; Hong, K.; Meng, Y.G.; Weikert, S.H.A.; Presta, L.G. Lack of Fucose on Human IgG1 N-Linked Oligosaccharide Improves Binding to Human Fcgamma RIII and Antibody-Dependent Cellular Toxicity. J. Biol. Chem. 2002, 277, 26733–26740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinkawa, T.; Nakamura, K.; Yamane, N.; Shoji-Hosaka, E.; Kanda, Y.; Sakurada, M.; Uchida, K.; Anazawa, H.; Satoh, M.; Yamasaki, M.; et al. The Absence of Fucose but Not the Presence of Galactose or Bisecting N-Acetylglucosamine of Human IgG1 Complex-Type Oligosaccharides Shows the Critical Role of Enhancing Antibody-Dependent Cellular Cytotoxicity. J. Biol. Chem. 2003, 278, 3466–3473. [Google Scholar] [CrossRef] [Green Version]
- Herter, S.; Birk, M.C.; Klein, C.; Gerdes, C.; Umana, P.; Bacac, M. Glycoengineering of Therapeutic Antibodies Enhances Monocyte/Macrophage-Mediated Phagocytosis and Cytotoxicity. J. Immunol. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, D.M. Venetoclax and Obinutuzumab for Frontline Treatment of Chronic Lymphocytic Leukemia. Blood 2019, 134, 1691–1696. [Google Scholar] [CrossRef]
- O’Nions, J.; Townsend, W. The Role of Obinutuzumab in the Management of Follicular Lymphoma. Future Oncol. Lond. Engl. 2019, 15, 3565–3578. [Google Scholar] [CrossRef]
- Paz-Ares, L.G.; Gomez-Roca, C.; Delord, J.-P.; Cervantes, A.; Markman, B.; Corral, J.; Soria, J.-C.; Bergé, Y.; Roda, D.; Russell-Yarde, F.; et al. Phase I Pharmacokinetic and Pharmacodynamic Dose-Escalation Study of RG7160 (GA201), the First Glycoengineered Monoclonal Antibody against the Epidermal Growth Factor Receptor, in Patients with Advanced Solid Tumors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 3783–3790. [Google Scholar] [CrossRef]
- Delord, J.-P.; Tabernero, J.; García-Carbonero, R.; Cervantes, A.; Gomez-Roca, C.; Bergé, Y.; Capdevila, J.; Paz-Ares, L.; Roda, D.; Delmar, P.; et al. Open-Label, Multicentre Expansion Cohort to Evaluate Imgatuzumab in Pre-Treated Patients with KRAS-Mutant Advanced Colorectal Carcinoma. Eur. J. Cancer 2014, 50, 496–505. [Google Scholar] [CrossRef]
- Temam, S.; Spicer, J.; Farzaneh, F.; Soria, J.C.; Oppenheim, D.; McGurk, M.; Hollebecque, A.; Sarini, J.; Hussain, K.; Soehrman Brossard, S.; et al. An Exploratory, Open-Label, Randomized, Multicenter Study to Investigate the Pharmacodynamics of a Glycoengineered Antibody (Imgatuzumab) and Cetuximab in Patients with Operable Head and Neck Squamous Cell Carcinoma. Ann. Oncol. 2017, 28, 2827–2835. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma Receptors as Regulators of Immune Responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef]
- De Henau, O.; Rausch, M.; Winkler, D.; Campesato, L.F.; Liu, C.; Cymerman, D.H.; Budhu, S.; Ghosh, A.; Pink, M.; Tchaicha, J.; et al. Overcoming Resistance to Checkpoint Blockade Therapy by Targeting PI3Kγ in Myeloid Cells. Nature 2016, 539, 443–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanueva, M.T. Macrophages Steal the Show. Nat. Rev. Drug Discov. 2017, 16, 455. [Google Scholar] [CrossRef]
- Tan, B.; Shi, X.; Zhang, J.; Qin, J.; Zhang, N.; Ren, H.; Qian, M.; Siwko, S.; Carmon, K.; Liu, Q.; et al. Inhibition of Rspo-Lgr4 Facilitates Checkpoint Blockade Therapy by Switching Macrophage Polarization. Cancer Res. 2018, 78, 4929–4942. [Google Scholar] [CrossRef] [Green Version]
- Le Naour, A.; Prat, M.; Thibault, B.; Mével, R.; Lemaitre, L.; Leray, H.; Joubert, M.-V.; Coulson, K.; Golzio, M.; Lefevre, L.; et al. Tumor Cells Educate Mesenchymal Stromal Cells to Release Chemoprotective and Immunomodulatory Factors. J. Mol. Cell Biol. 2020, 12, 202–215. [Google Scholar] [CrossRef]
- Duluc, D.; Delneste, Y.; Tan, F.; Moles, M.-P.; Grimaud, L.; Lenoir, J.; Preisser, L.; Anegon, I.; Catala, L.; Ifrah, N.; et al. Tumor-Associated Leukemia Inhibitory Factor and IL-6 Skew Monocyte Differentiation into Tumor-Associated Macrophage-like Cells. Blood 2007, 110, 4319–4330. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-Y.; Huang, T.-W.; Hsieh, Y.-T.; Wang, Y.-F.; Yen, C.-C.; Lee, G.-L.; Yeh, C.-C.; Peng, Y.-J.; Kuo, Y.-Y.; Wen, H.-T.; et al. Cancer-Derived Succinate Promotes Macrophage Polarization and Cancer Metastasis via Succinate Receptor. Mol. Cell 2020, 77, 213–227.e5. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Luo, G.; Zhang, K.; Cao, J.; Huang, C.; Jiang, T.; Liu, B.; Su, L.; Qiu, Z. Hypoxic Tumor-Derived Exosomal MiR-301a Mediates M2 Macrophage Polarization via PTEN/PI3Kγ to Promote Pancreatic Cancer Metastasis. Cancer Res. 2018, 78, 4586–4598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassetta, L.; Pollard, J.W. Targeting Macrophages: Therapeutic Approaches in Cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 123–147. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The Chemokine System in Diverse Forms of Macrophage Activation and Polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Laviron, M.; Boissonnas, A. Ontogeny of Tumor-Associated Macrophages. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Prat, M.; Le Naour, A.; Coulson, K.; Lemée, F.; Leray, H.; Jacquemin, G.; Rahabi, M.C.; Lemaitre, L.; Authier, H.; Ferron, G.; et al. Circulating CD14high CD16low Intermediate Blood Monocytes as a Biomarker of Ascites Immune Status and Ovarian Cancer Progression. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Kistner, L.; Doll, D.; Holtorf, A.; Nitsche, U.; Janssen, K.-P. Interferon-Inducible CXC-Chemokines Are Crucial Immune Modulators and Survival Predictors in Colorectal Cancer. Oncotarget 2017, 8, 89998–90012. [Google Scholar] [CrossRef] [Green Version]
- Skytthe, M.K.; Graversen, J.H.; Moestrup, S.K. Targeting of CD163+ Macrophages in Inflammatory and Malignant Diseases. Int. J. Mol. Sci. 2020, 21, 5497. [Google Scholar] [CrossRef]
- Mallmann, M.; Schmidt, S.; Schultze, J. Macrophages in Human Cancer: Current and Future Aspects. Atlas Genet. Cytogenet. Oncol. Haematol. 2012. [Google Scholar] [CrossRef] [Green Version]
- Heusinkveld, M.; van der Burg, S.H. Identification and Manipulation of Tumor Associated Macrophages in Human Cancers. J. Transl. Med. 2011, 9, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niwa, R.; Shoji-Hosaka, E.; Sakurada, M.; Shinkawa, T.; Uchida, K.; Nakamura, K.; Matsushima, K.; Ueda, R.; Hanai, N.; Shitara, K. Defucosylated Chimeric Anti-CC Chemokine Receptor 4 IgG1 with Enhanced Antibody-Dependent Cellular Cytotoxicity Shows Potent Therapeutic Activity to T-Cell Leukemia and Lymphoma. Cancer Res. 2004, 64, 2127–2133. [Google Scholar] [CrossRef] [Green Version]
- Horton, H.M.; Bernett, M.J.; Pong, E.; Peipp, M.; Karki, S.; Chu, S.Y.; Richards, J.O.; Vostiar, I.; Joyce, P.F.; Repp, R.; et al. Potent in Vitro and in Vivo Activity of an Fc-Engineered Anti-CD19 Monoclonal Antibody against Lymphoma and Leukemia. Cancer Res. 2008, 68, 8049–8057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leary, A.; Aftimos, P.G.; Delord, J.-P.; Le Tourneau, C.; Ray-Coquard, I.L.; Jungels, C.; Varga, A.I.; Ricci, F.; Gomez-Roca, C.A.; Bataillon, G.; et al. A First-in-Human Study of Monoclonal Antibody GM102 in Patients with Anti-Mullerian-Hormone-Receptor II (AMHRII) Positive Gynecological Cancers. J. Clin. Oncol. 2018, 36, 5542. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Melichar, B.; Van den Eynde, M.; Prausova, J.; Geboes, K.; Dekervel, J.; Vitaskova, D.; Cuyper, A.D.; Linke, Z.; Acker, N.V.; et al. Phase 2 Study Results of Murlentamab, a Monoclonal Antibody Targeting the Anti-Mullerian-Hormone-Receptor II (AMHRII), Acting through Tumor-Associated Macrophage Engagement in Advanced/Metastatic Colorectal Cancers. Ann. Oncol. 2019, 30, iv153–iv154. [Google Scholar] [CrossRef]
- Mantovani, A. Tumour-Associated Macrophages as Treatment Targets in Oncology. Clin. Oncol. 2017, 14, 399. [Google Scholar] [CrossRef]
- Bournazos, S.; Gupta, A.; Ravetch, J.V. The Role of IgG Fc Receptors in Antibody-Dependent Enhancement. Nat. Rev. Immunol. 2020, 20, 633–643. [Google Scholar] [CrossRef]
- Yeap, W.H.; Wong, K.L.; Shimasaki, N.; Teo, E.C.Y.; Quek, J.K.S.; Yong, H.X.; Diong, C.P.; Bertoletti, A.; Linn, Y.C.; Wong, S.C. CD16 Is Indispensable for Antibody-Dependent Cellular Cytotoxicity by Human Monocytes. Sci. Rep. 2016, 6, 34310. [Google Scholar] [CrossRef]
- Ikeda, H.; Old, L.J.; Schreiber, R.D. The Roles of IFNγ in Protection against Tumor Development and Cancer Immunoediting. Cytokine Growth Factor Rev. 2002, 13, 95–109. [Google Scholar] [CrossRef]
- Tugues, S.; Burkhard, S.H.; Ohs, I.; Vrohlings, M.; Nussbaum, K.; vom Berg, J.; Kulig, P.; Becher, B. New Insights into IL-12-Mediated Tumor Suppression. Cell Death Differ. 2015, 22, 237–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, J.; Hamaguchi, Y.; Oliver, J.A.; Ravetch, J.V.; Poe, J.C.; Haas, K.M.; Tedder, T.F. The Innate Mononuclear Phagocyte Network Depletes B Lymphocytes through Fc Receptor-Dependent Mechanisms during Anti-CD20 Antibody Immunotherapy. J. Exp. Med. 2004, 199, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.M.; Lee, S.C.; Andrade Filho, P.A.; Lord, C.A.; Jie, H.-B.; Davidson, H.C.; López-Albaitero, A.; Gibson, S.P.; Gooding, W.E.; Ferrone, S.; et al. Cetuximab-Activated Natural Killer and Dendritic Cells Collaborate to Trigger Tumor Antigen-Specific T-Cell Immunity in Head and Neck Cancer Patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 1858–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- House, I.G.; Savas, P.; Lai, J.; Chen, A.X.Y.; Oliver, A.J.; Teo, Z.L.; Todd, K.L.; Henderson, M.A.; Giuffrida, L.; Petley, E.V.; et al. Macrophage-Derived CXCL9 and CXCL10 Are Required for Antitumor Immune Responses Following Immune Checkpoint Blockade. Clin. Cancer Res. 2020, 26, 487–504. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Vollmer, M.; De Geyter, M.; Litzistorf, Y.; Ladewig, A.; Dürrenberger, M.; Guggenheim, R.; Miny, P.; Holzgreve, W.; De Geyter, C. Characterization of an Immortalized Human Granulosa Cell Line (COV434). Mol. Hum. Reprod. 2000, 6, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Jeannin, P.; Paolini, L.; Adam, C.; Delneste, Y. The Roles of CSFs on the Functional Polarization of Tumor-Associated Macrophages. FEBS J. 2018, 285, 680–699. [Google Scholar] [CrossRef] [Green Version]
- Némati, F.; Sastre-Garau, X.; Laurent, C.; Couturier, J.; Mariani, P.; Desjardins, L.; Piperno-Neumann, S.; Lantz, O.; Asselain, B.; Plancher, C.; et al. Establishment and Characterization of a Panel of Human Uveal Melanoma Xenografts Derived from Primary and/or Metastatic Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 2352–2362. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibodies | Supplier | Catalog No. | Clone No. | Experiment |
|---|---|---|---|---|
| CD4-AF700 | Thermofisher | 56-0049-42 | RPA-T4 | CD69+ detection |
| CD15-eF450 | Thermofisher | 48-0158-42 | MMA | |
| CD16-PC7 | Thermofisher | 25-0168-42 | CB16 | |
| CD25-PerCP | Thermofisher | 46-0257-42 | CD45-4E3 | |
| CD56-FITC | Thermofisher | 61-0567-42 | CMSSB | |
| CD64-PerCPeF710 | Thermofisher | 46-0649-42 | 10.1 | |
| CD69-PE | Thermofisher | 12-0699-42 | FN50 | |
| CD127-APC | Thermofisher | 17-1278-42 | eBioRD5 | |
| CD3-VB | Miltenyi | 130-133-133 | BW264/56 | |
| CD45-VG | Miltenyi | 130-113-124 | 5B1 | |
| CD14- AF714 | Miltenyi | 130-113-144 | TUK4 | |
| CD8-PEeF610 | BD BioSciences | 563919 | SK1 | |
| CD3-AA700 | Beckman | B10823 | UCHT1 | ICOS+ detection |
| CD8-KrO | Beckman | B00067 | B9.11 | |
| CD4-PB | Beckman | A99020 | RMO52 | |
| HLA-DR-ECD | Beckman | A66330 | 3G8 | |
| CD45RA-PE | AbD Serotec | MCA1075F | AT10 | |
| ICOS-APC | Beckman | IM1239U | 84H10 | |
| PD1 | Cell Signaling | 13684S | EIL3N | In vivo experiment |
| CD45-BV421 | BD BioSciences | 563879 | HI30 | |
| CD3-FITC | Miltenyi | 130-098-162 | REA613 | |
| CD14-PE | Miltenyi | 130-100-676 | REA599 | |
| CD163-APC | Miltenyi | 130-112-129 | GHI:61.1 | |
| CD86-Alexa700 | BD BioSciences | 561124 | 2331 | |
| CD11b-APCVio770 | Miltenyi | 130-110-556 | REA713 | |
| ViobilityTM Fixable Dyes | Miltenyi | 130-109-816 | - | In vitro experiments |
| CD11b-FITC | Miltenyi | 130-110-552 | REA713 | |
| CD163-PE | Miltenyi | 130-112-286 | REA812 | |
| CD36-PEVio770 | Miltenyi | 130-110-742 | REA770 | |
| CD206-APC | Miltenyi | 130-124-012 | DCN 228 | |
| CD64-PerCPVio770 | Miltenyi | 130-116-303 | REA978 | |
| CD80-PE | Miltenyi | 130-123-253 | REA661 | |
| CD32-PEVio770 | Miltenyi | 130-097-506 | 2E1 | |
| CD282-APC | Miltenyi | 130-120-138 | REA109 | |
| CD14-APCVio770 | Miltenyi | 130-110-552 | REA599 | |
| CD183-APC | Miltenyi | 130-120-450 | REA232 | |
| CD3-APCVio770 | Miltenyi | 130-113-136 | REA613 | |
| CD4-VioBright FITC | Miltenyi | 130-113-791 | REA623 | |
| CD25-PE | Miltenyi | 130-115-534 | REA945 | |
| CD8-PEVio770 | Miltenyi | 130-110-680 | REA734 | |
| CD45-VioGreen | Miltenyi | 130-110-638 | REA747 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prat, M.; Salon, M.; Allain, T.; Dubreuil, O.; Noël, G.; Preisser, L.; Jean, B.; Cassard, L.; Lemée, F.; Tabah-Fish, I.; et al. Murlentamab, a Low Fucosylated Anti-Müllerian Hormone Type II Receptor (AMHRII) Antibody, Exhibits Anti-Tumor Activity through Tumor-Associated Macrophage Reprogrammation and T Cell Activation. Cancers 2021, 13, 1845. https://doi.org/10.3390/cancers13081845
Prat M, Salon M, Allain T, Dubreuil O, Noël G, Preisser L, Jean B, Cassard L, Lemée F, Tabah-Fish I, et al. Murlentamab, a Low Fucosylated Anti-Müllerian Hormone Type II Receptor (AMHRII) Antibody, Exhibits Anti-Tumor Activity through Tumor-Associated Macrophage Reprogrammation and T Cell Activation. Cancers. 2021; 13(8):1845. https://doi.org/10.3390/cancers13081845
Chicago/Turabian StylePrat, Mélissa, Marie Salon, Thibault Allain, Olivier Dubreuil, Grégory Noël, Laurence Preisser, Bérangère Jean, Lydie Cassard, Fanny Lemée, Isabelle Tabah-Fish, and et al. 2021. "Murlentamab, a Low Fucosylated Anti-Müllerian Hormone Type II Receptor (AMHRII) Antibody, Exhibits Anti-Tumor Activity through Tumor-Associated Macrophage Reprogrammation and T Cell Activation" Cancers 13, no. 8: 1845. https://doi.org/10.3390/cancers13081845