
Evolutionary Trajectories and Genomic Divergence in Localized Breast Cancers after Ipsilateral Breast Tumor Recurrence

 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Overview of Patient Cohorts
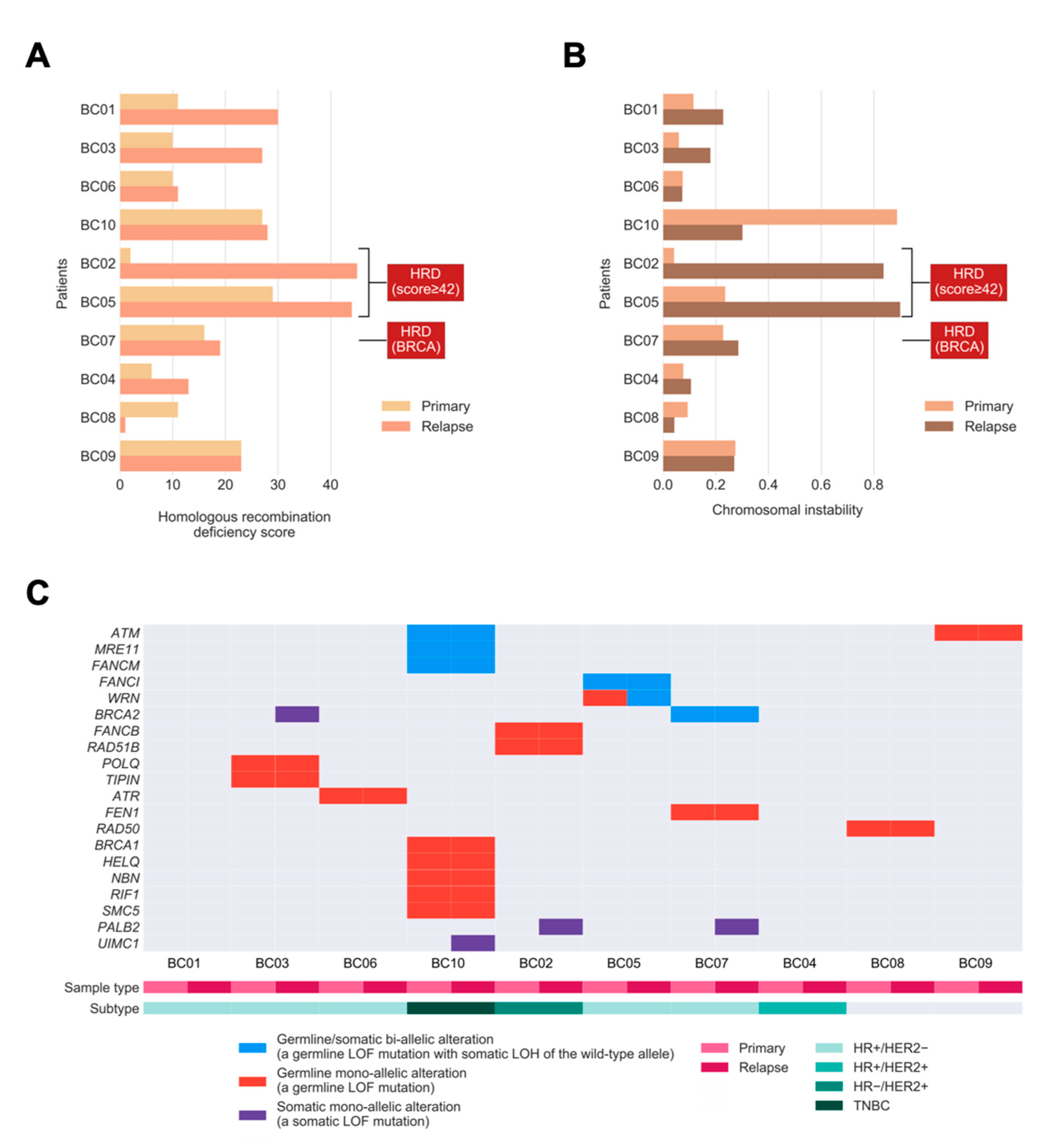
2.2. HRD and Chromosomal Instability (CIN) during BC Evolution
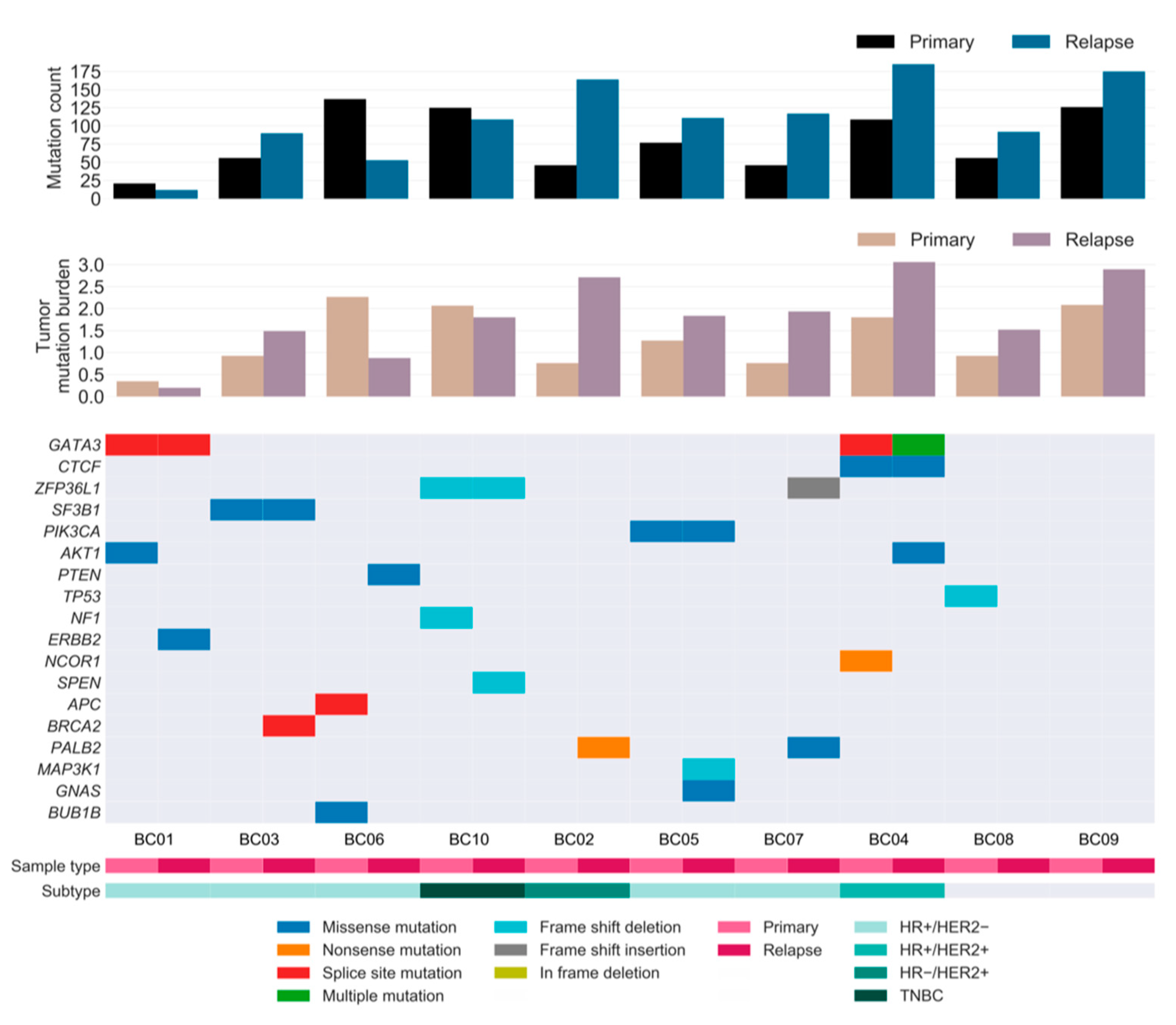
2.3. Somatic Drivers of Relapse
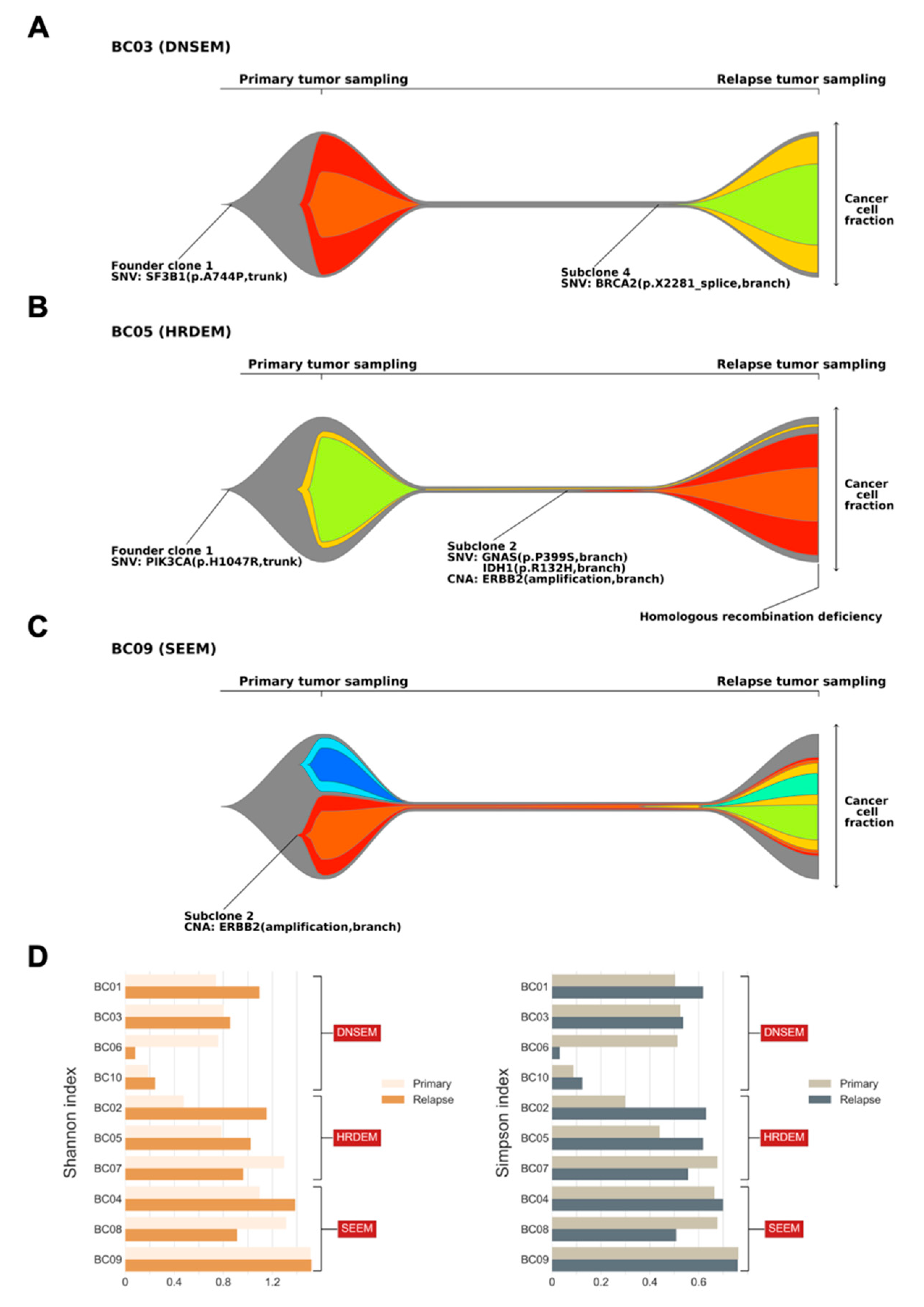
2.4. Clonal Architecture BC Progression
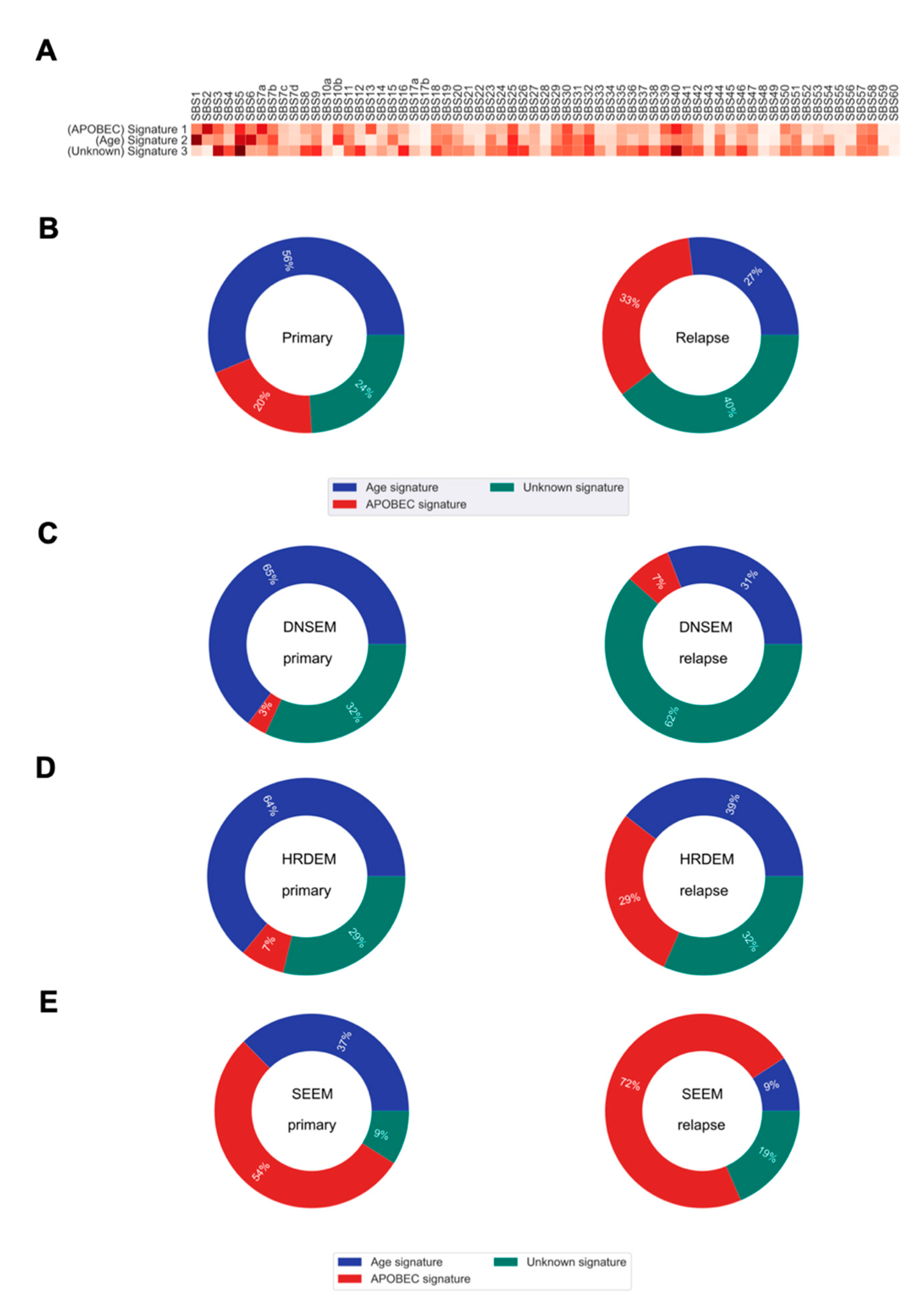
2.5. Evolution of the Mutational Processes over Time
2.6. Integration of Actionability and Tumor Evolution in Therapeutic Decisions
3. Discussion
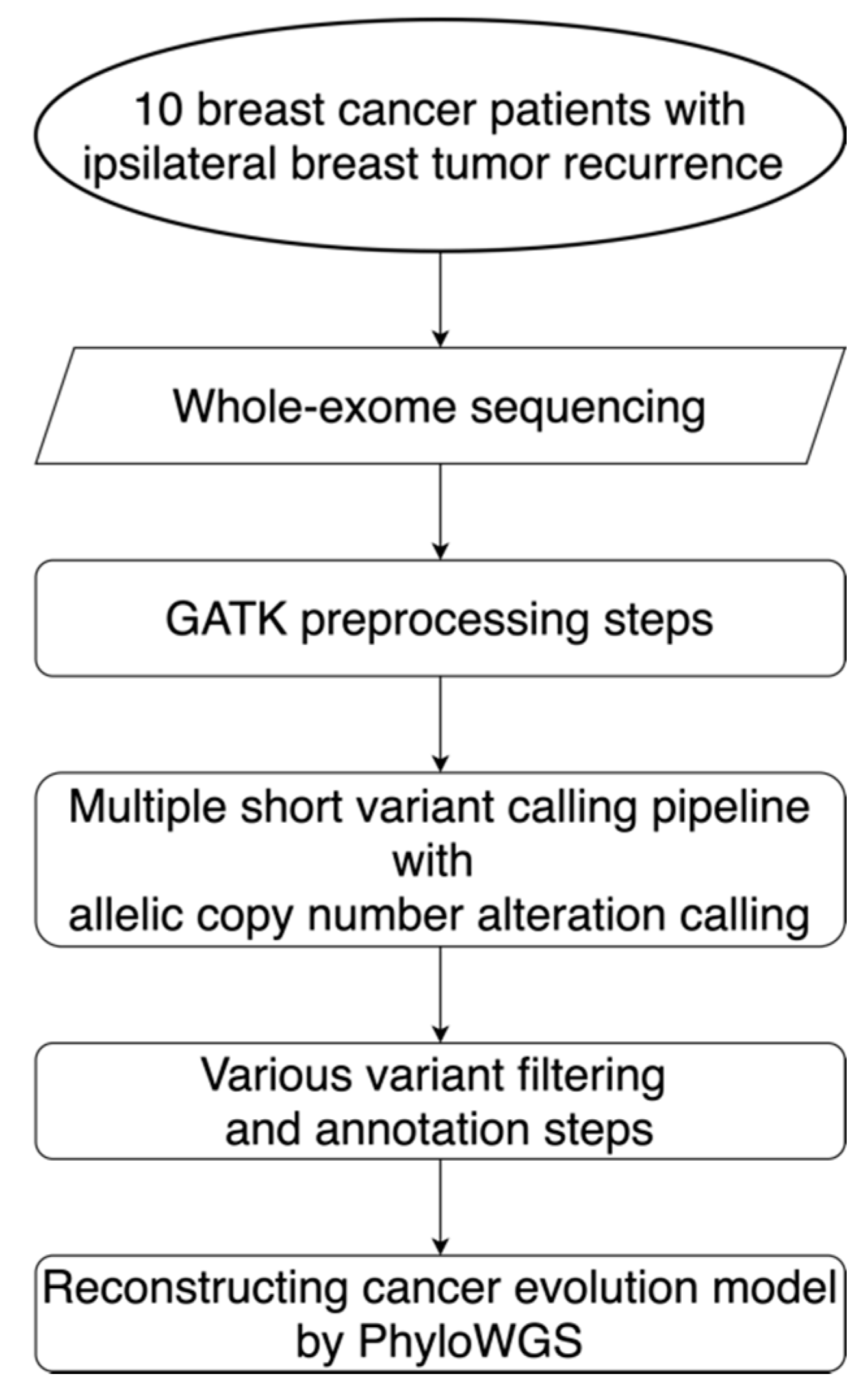
4. Materials and Methods
4.1. Study Population and Specimens
4.2. Exome Capture, Library Construction, and Sequencing
4.3. Sequencing Data Processing and Quality Assessment
4.4. Variant Calling and Post-Processing Strategies
4.5. Subclonal Copy Number Assessment
4.6. Large-Scale Genomic Events Analysis
4.6.1. Homologous Recombination Deficiency
4.6.2. Chromosomal Instability
4.6.3. Whole-Genome Doubling
4.7. Assessment of Bi-Allelic Alterations in HR-Related Genes
4.8. Potential Driver and Actionable Gene Identification in Primary and Relapsed Lesions
4.9. Reconstruction of Cancer Clonal Architecture
4.10. Trunk and Branch Alteration Classification and Clonal Architecture Labeling
4.11. Subclonal Diversity Assessment
4.12. Mutational Signature Analysis
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Polyak, K. Heterogeneity in breast cancer. J. Clin. Investig. 2011, 121, 3786–3788. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.J.; Wapnir, I.; Dignam, J.J.; Fisher, B.; Mamounas, E.P.; Jeong, J.H.; Geyer, C.E., Jr.; Wickerham, D.L.; Costantino, J.P.; Wolmark, N. Prognosis after ipsilateral breast tumor recurrence and locoregional recurrences in patients treated by breast-conserving therapy in five National Surgical Adjuvant Breast and Bowel Project protocols of node-negative breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 2466–2473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, B.; Anderson, S.; Bryant, J.; Margolese, R.G.; Deutsch, M.; Fisher, E.R.; Jeong, J.H.; Wolmark, N. Twenty-year follow-up of a randomized trial comparing total mastectomy, lumpectomy, and lumpectomy plus irradiation for the treatment of invasive breast cancer. N. Engl. J. Med. 2002, 347, 1233–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuman, H.B.; Schumacher, J.R.; Francescatti, A.B.; Adesoye, T.; Edge, S.B.; Vanness, D.J.; Yu, M.; McKellar, D.; Winchester, D.P.; Greenberg, C.C. Risk of Synchronous Distant Recurrence at Time of Locoregional Recurrence in Patients With Stage II and III Breast Cancer (AFT-01). J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 975–980. [Google Scholar] [CrossRef]
- Yates, L.R.; Knappskog, S.; Wedge, D.; Farmery, J.H.R.; Gonzalez, S.; Martincorena, I.; Alexandrov, L.B.; Van Loo, P.; Haugland, H.K.; Lilleng, P.K.; et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 2017, 32, 169–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [CrossRef] [PubMed] [Green Version]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nik-Zainal, S.; Morganella, S. Mutational Signatures in Breast Cancer: The Problem at the DNA Level. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 2617–2629. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Favero, F.; de Bruin, E.C.; Birkbak, N.J.; Szallasi, Z.; Swanton, C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med. 2015, 7, 283ra254. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.; Smeets, D.; Szekely, B.; Larsimont, D.; Szasz, A.M.; Adnet, P.Y.; Rothe, F.; Rouas, G.; Nagy, Z.I.; Farago, Z.; et al. Phylogenetic analysis of metastatic progression in breast cancer using somatic mutations and copy number aberrations. Nat. Commun. 2017, 8, 14944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, C.K.Y.; Bidard, F.C.; Piscuoglio, S.; Geyer, F.C.; Lim, R.S.; de Bruijn, I.; Shen, R.; Pareja, F.; Berman, S.H.; Wang, L.; et al. Genetic Heterogeneity in Therapy-Naive Synchronous Primary Breast Cancers and Their Metastases. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 4402–4415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrijver, W.; Selenica, P.; Lee, J.Y.; Ng, C.K.Y.; Burke, K.A.; Piscuoglio, S.; Berman, S.H.; Reis-Filho, J.S.; Weigelt, B.; van Diest, P.J.; et al. Mutation Profiling of Key Cancer Genes in Primary Breast Cancers and Their Distant Metastases. Cancer Res. 2018, 78, 3112–3121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savas, P.; Teo, Z.L.; Lefevre, C.; Flensburg, C.; Caramia, F.; Alsop, K.; Mansour, M.; Francis, P.A.; Thorne, H.A.; Silva, M.J.; et al. The Subclonal Architecture of Metastatic Breast Cancer: Results from a Prospective Community-Based Rapid Autopsy Program “CASCADE”. PLoS Med. 2016, 13, e1002204. [Google Scholar] [CrossRef] [Green Version]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef] [Green Version]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254. [Google Scholar] [CrossRef] [Green Version]
- Riaz, N.; Blecua, P.; Lim, R.S.; Shen, R.; Higginson, D.S.; Weinhold, N.; Norton, L.; Weigelt, B.; Powell, S.N.; Reis-Filho, J.S. Pan-cancer analysis of bi-allelic alterations in homologous recombination DNA repair genes. Nat. Commun. 2017, 8, 857. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385. [Google Scholar] [CrossRef] [Green Version]
- Bertucci, F.; Ng, C.K.Y.; Patsouris, A.; Droin, N.; Piscuoglio, S.; Carbuccia, N.; Soria, J.C.; Dien, A.T.; Adnani, Y.; Kamal, M.; et al. Genomic characterization of metastatic breast cancers. Nature 2019, 569, 560–564. [Google Scholar] [CrossRef]
- Angus, L.; Smid, M.; Wilting, S.M.; van Riet, J.; Van Hoeck, A.; Nguyen, L.; Nik-Zainal, S.; Steenbruggen, T.G.; Tjan-Heijnen, V.C.G.; Labots, M.; et al. The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nat. Genet. 2019, 51, 1450–1458. [Google Scholar] [CrossRef]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438. [Google Scholar] [CrossRef] [Green Version]
- Deshwar, A.G.; Vembu, S.; Yung, C.K.; Jang, G.H.; Stein, L.; Morris, Q. PhyloWGS: Reconstructing subclonal composition and evolution from whole-genome sequencing of tumors. Genome Biol. 2015, 16, 35. [Google Scholar] [CrossRef] [Green Version]
- Turajlic, S.; Sottoriva, A.; Graham, T.; Swanton, C. Resolving genetic heterogeneity in cancer. Nat. Rev. Genet. 2019, 20, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef] [Green Version]
- Simpson, E.H. Measurement of diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Von Wahlde, M.K.; Timms, K.M.; Chagpar, A.; Wali, V.B.; Jiang, T.; Bossuyt, V.; Saglam, O.; Reid, J.; Gutin, A.; Neff, C.; et al. Intratumor Heterogeneity of Homologous Recombination Deficiency in Primary Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 1193–1199. [Google Scholar] [CrossRef] [Green Version]
- Espiritu, S.M.G.; Liu, L.Y.; Rubanova, Y.; Bhandari, V.; Holgersen, E.M.; Szyca, L.M.; Fox, N.S.; Chua, M.L.K.; Yamaguchi, T.N.; Heisler, L.E.; et al. The Evolutionary Landscape of Localized Prostate Cancers Drives Clinical Aggression. Cell 2018, 173, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- Almendro, V.; Kim, H.J.; Cheng, Y.K.; Gonen, M.; Itzkovitz, S.; Argani, P.; van Oudenaarden, A.; Sukumar, S.; Michor, F.; Polyak, K. Genetic and phenotypic diversity in breast tumor metastases. Cancer Res. 2014, 74, 1338–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, A.K.; Fink, J.L.; Grady, J.P.; Morgan, G.J.; Mullighan, C.G.; To, L.B.; Hewett, D.R.; Zannettino, A.C.W. Subclonal evolution in disease progression from MGUS/SMM to multiple myeloma is characterised by clonal stability. Leukemia 2019, 33, 457–468. [Google Scholar] [CrossRef]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Kallberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and accurate calling of germline and somatic variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef]
- Fan, Y.; Xi, L.; Hughes, D.S.; Zhang, J.; Zhang, J.; Futreal, P.A.; Wheeler, D.A.; Wang, W. MuSE: Accounting for tumor heterogeneity using a sample-specific error model improves sensitivity and specificity in mutation calling from sequencing data. Genome Biol. 2016, 17, 178. [Google Scholar] [CrossRef] [Green Version]
- Larson, D.E.; Harris, C.C.; Chen, K.; Koboldt, D.C.; Abbott, T.E.; Dooling, D.J.; Ley, T.J.; Mardis, E.R.; Wilson, R.K.; Ding, L. SomaticSniper: Identification of somatic point mutations in whole genome sequencing data. Bioinformatics 2012, 28, 311–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Ellrott, K.; Bailey, M.H.; Saksena, G.; Covington, K.R.; Kandoth, C.; Stewart, C.; Hess, J.; Ma, S.; Chiotti, K.E.; McLellan, M.; et al. Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst. 2018, 6, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.C.; Fan, C.T.; Liao, C.C.; Chen, Y.S. Taiwan Biobank: Making cross-database convergence possible in the Big Data era. GigaScience 2018, 7, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Diossy, M.; Reiniger, L.; Sztupinszki, Z.; Krzystanek, M.; Timms, K.M.; Neff, C.; Solimeno, C.; Pruss, D.; Eklund, A.C.; Toth, E.; et al. Breast cancer brain metastases show increased levels of genomic aberration-based homologous recombination deficiency scores relative to their corresponding primary tumors. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 1948–1954. [Google Scholar] [CrossRef]
- Taylor-Weiner, A.; Stewart, C.; Giordano, T.; Miller, M.; Rosenberg, M.; Macbeth, A.; Lennon, N.; Rheinbay, E.; Landau, D.A.; Wu, C.J.; et al. DeTiN: Overcoming tumor-in-normal contamination. Nat. Methods 2018, 15, 531–534. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Shen, R.; Seshan, V.E. FACETS: Allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016, 44, e131. [Google Scholar] [CrossRef] [Green Version]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birkbak, N.J.; Wang, Z.C.; Kim, J.Y.; Eklund, A.C.; Li, Q.; Tian, R.; Bowman-Colin, C.; Li, Y.; Greene-Colozzi, A.; Iglehart, J.D.; et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012, 2, 366–375. [Google Scholar] [CrossRef] [Green Version]
- Popova, T.; Manie, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef] [Green Version]
- Sztupinszki, Z.; Diossy, M.; Krzystanek, M.; Reiniger, L.; Csabai, I.; Favero, F.; Birkbak, N.J.; Eklund, A.C.; Syed, A.; Szallasi, Z. Migrating the SNP array-based homologous recombination deficiency measures to next generation sequencing data of breast cancer. NPJ Breast Cancer 2018, 4, 16. [Google Scholar] [CrossRef]
- Miller, C.A.; McMichael, J.; Dang, H.X.; Maher, C.A.; Ding, L.; Ley, T.J.; Mardis, E.R.; Wilson, R.K. Visualizing tumor evolution with the fishplot package for R. BMC Genom. 2016, 17, 880. [Google Scholar] [CrossRef] [Green Version]
- Gaujoux, R.; Seoighe, C. A flexible R package for nonnegative matrix factorization. BMC Bioinform. 2010, 11, 367. [Google Scholar] [CrossRef] [Green Version]
- Blokzijl, F.; Janssen, R.; van Boxtel, R.; Cuppen, E. MutationalPatterns: Comprehensive genome-wide analysis of mutational processes. Genome Med. 2018, 10, 33. [Google Scholar] [CrossRef]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case ID | Age at Diagnosis (Years) | Laterality | Grade | Tumor Size (Invasive, mm) | Lymph Node Status | ER | PR | HER2 | Subtype | Relapse after Primary Diagnosis (Months) |
|---|---|---|---|---|---|---|---|---|---|---|
| BC01 | 48 | Right | 1 | 15 | − | + | + | − | Luminal A-like | |
| BC02 | 70 | Left | <1 | − | − | − | + | HER2-enriched | 6 | |
| BC03 | 39 | Left | 2 | 40 | − | + | + | − | Luminal A-like | 45 |
| BC04 | 47 | Left | 2 | − | + | + | + | Luminal B-like | 61 | |
| BC05 | 70 | Left | 2 | 18 | − | + | − | − | Luminal A-like | 59 |
| BC06 | 29 | Right | 1 | 47 | − | + | + | − | Luminal A-like | |
| BC07 | 36 | Left | 2 | 24 | − | + | + | − | Luminal A-like | |
| BC08 | 47 | Right | 2 | 13 | − | − | − | 24 | ||
| BC09 | 72 | Right | 2 | 25 | − | + | − | 23 | ||
| BC10 | 27 | Right | 2 | 32 | − | − | − | − | Triple negative | 107 |
| Sample ID | Actionable Biomarker | Actionable Alteration | Clone | Cancer Cell Fraction | Classification |
|---|---|---|---|---|---|
| BC01 (relapse) | OncoKB | CDK4 amplification | Clone 1 | 1 | Trunk |
| BC01 (relapse) | OncoKB | ERBB2 mutation | Clone 5 | 0.522 | Branch |
| BC02 (relapse) | HRD | (HRD score ≥ 42) | N/A | N/A | N/A |
| BC03 (relapse) | OncoKB | BRCA2 mutation | Clone 4 | 0.943 | Branch |
| BC05 (primary) | OncoKB | PIK3CA mutation | Clone 1 | 1 | Trunk |
| BC05 (relapse) | OncoKB | PIK3CA mutation | Clone 1 | 1 | Trunk |
| BC05 (relapse) | OncoKB | IDH1 mutation | Clone 2 | 0.838 | Branch |
| BC05 (relapse) | OncoKB | ERBB2 amplification | Clone 2 | 0.838 | Branch |
| BC05 (relapse) | HRD | (HRD score ≥ 42) | N/A | N/A | N/A |
| BC07 (primary) | HRD | Bi-allelic BRCA2 | N/A | N/A | N/A |
| BC07 (relapse) | HRD | Bi-allelic BRCA2 | N/A | N/A | N/A |
| BC09 (primary) | OncoKB | ERBB2 amplification | Clone 2 | 0.554 | Branch |
| BC09 (relapse) | OncoKB | ERBB2 amplification | Clone 2 | 0.682 | Branch |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.-H.; Yeh, H.-T.; Hsieh, C.-S.; Huang, C.-C.; Chattopadhyay, A.; Chung, Y.-C.; Tu, S.-H.; Li, Y.-H.; Lu, T.-P.; Lai, L.-C.; et al. Evolutionary Trajectories and Genomic Divergence in Localized Breast Cancers after Ipsilateral Breast Tumor Recurrence. Cancers 2021, 13, 1821. https://doi.org/10.3390/cancers13081821
Wu C-H, Yeh H-T, Hsieh C-S, Huang C-C, Chattopadhyay A, Chung Y-C, Tu S-H, Li Y-H, Lu T-P, Lai L-C, et al. Evolutionary Trajectories and Genomic Divergence in Localized Breast Cancers after Ipsilateral Breast Tumor Recurrence. Cancers. 2021; 13(8):1821. https://doi.org/10.3390/cancers13081821
Chicago/Turabian StyleWu, Chia-Hsin, Hsien-Tang Yeh, Chia-Shan Hsieh, Chi-Cheng Huang, Amrita Chattopadhyay, Yuan-Chiang Chung, Shih-Hsin Tu, Yung-Hua Li, Tzu-Pin Lu, Liang-Chuan Lai, and et al. 2021. "Evolutionary Trajectories and Genomic Divergence in Localized Breast Cancers after Ipsilateral Breast Tumor Recurrence" Cancers 13, no. 8: 1821. https://doi.org/10.3390/cancers13081821