
O-GlcNAcylation and O-GlcNAc Cycling Regulate Gene Transcription: Emerging Roles in Cancer


Abstract
:Simple Summary
Abstract
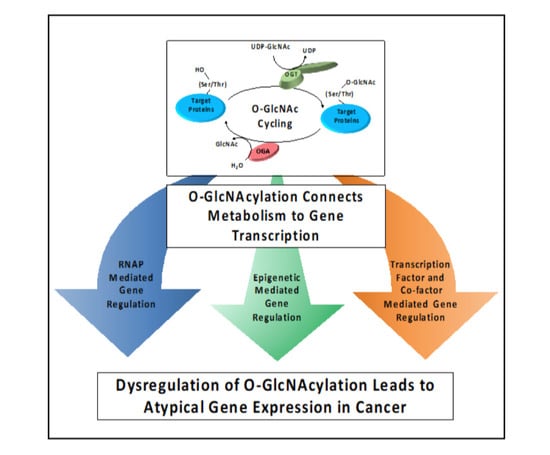
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. O-GlcNAcylation Is a Post-Translational Modification (PTM) That Has Regulatory Roles in Gene Transcription
2. O-GlcNAcylation Is Sensitive to Metabolite Pools via HBP
3. O-GlcNAcylation Regulates RNA Polymerase Function
3.1. O-GlcNAcylation and RNAP II Function
3.2. O-GlcNAcylation of TATA-Box Binding Protein (TBP) Alters Metabolic Gene Expression
4. O-GlcNAcylation and Epigenetic Gene Regulators
4.1. O-GlcNAcylation of Ten-Eleven Translocation Protein Family
4.2. O-GlcNAcylation of Polycomb Group Proteins
4.3. O-GlcNAcylation of GATA1 Target Genes and the Sin3A Corepressor Complex
4.4. O-GlcNAcylation of Nucleosome Remodeling Deacetylase
5. Defects in O-GlcNAcylation of Transcription Factors Promote Cancer
5.1. O-GlcNAcylation of Transcription Factor Sp1
5.2. O-GlcNAcylation of Pluripotent Transcription Factors Sox2 and Oct4
5.3. O-GlcNAcylation of Transcription Factors in Breast Cancer
5.4. O-GlcNAcylation Regulates the Hippo Pathway Co-Activator YAP
6. Challenges Associated with O-GlcNAc Research
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hardiville, S.; Hart, G.W. Nutrient Regulation of Signaling, Transcription, and Cell Physiology by O-GlcNAcylation. Cell Metab. 2014, 20, 208–213. [Google Scholar] [CrossRef] [Green Version]
- Brimble, S.; Wollaston-Hayden, E.E.; Teo, C.F.; Morris, A.C.; Wells, L. The Role of the O-GlcNAc Modification in Regulating Eukaryotic Gene Expression. Curr. Signal. Transduct. Ther. 2010, 5, 12–24. [Google Scholar] [CrossRef] [Green Version]
- Torres, C.R.; Hart, G.W. Topography and Polypeptide Distribution of Terminal N-acetylglucosamine Residues on the Surfaces of Intact Lymphocytes. Evidence for O-linked GlcNAc. J. Biol. Chem. 1984, 259, 3308–3317. [Google Scholar] [CrossRef]
- Hart, G.W.; Housley, M.P.; Slawson, C. Cycling of O-linked beta-N-acetylglucosamine on Nucleocytoplasmic Proteins. Nature 2007, 446, 1017–1022. [Google Scholar] [CrossRef]
- Hart, G.W.; Slawson, C.; Ramirez-Correa, G.; Lagerlof, O. Cross Talk between O-GlcNAcylation and Phosphorylation: Roles in Signaling, Transcription, and Chronic Disease. Annu. Rev. Biochem. 2011, 80, 825–858. [Google Scholar] [CrossRef] [Green Version]
- Dong, D.L.; Hart, G.W. Purification and Characterization of an O-GlcNAc Selective N-acetyl-beta-D-glucosaminidase from Rat Spleen Cytosol. J. Biol. Chem. 1994, 269, 19321–19330. [Google Scholar] [CrossRef]
- Wang, T.; Birsoy, K.; Hughes, N.W.; Krupczak, K.M.; Post, Y.; Wei, J.J.; Lander, E.S.; Sabatini, D.M. Identification and Characterization of Essential Genes in the Human Genome. Science 2015, 350, 1096–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafi, R.; Iyer, S.P.; Ellies, L.G.; O’Donnell, N.; Marek, K.W.; Chui, D.; Hart, G.W.; Marth, J.D. The O-GlcNAc Transferase Gene Resides on the X Chromosome and is Essential for Embryonic Stem Cell Viability and Mouse Ontogeny. Proc. Natl. Acad. Sci. USA 2000, 97, 5735–5739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keembiyehetty, C.; Love, D.C.; Harwood, K.R.; Gavrilova, O.; Comly, M.E.; Hanover, J.A. Conditional Knock-out Reveals a Requirement for O-linked N-Acetylglucosaminase (O-GlcNAcase) in Metabolic Homeostasis. J. Biol. Chem. 2015, 290, 7097–7113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.L.; Wybenga-Groot, L.E.; Tong, J.; Taylor, P.; Minden, M.D.; Trudel, S.; McGlade, C.J.; Moran, M.F. Tyrosine Phosphorylation of the Lyn Src Homology 2 (SH2) Domain Modulates Its Binding Affinity and Specificity. Mol. Cell. Proteom. MCP 2015, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toleman, C.A.; Schumacher, M.A.; Yu, S.-H.; Zeng, W.; Cox, N.J.; Smith, T.J.; Soderblom, E.J.; Wands, A.M.; Kohler, J.J.; Boyce, M. Structural Basis of O-GlcNAc Recognition by Mammalian 14-3-3 Proteins. Proc. Natl. Acad. Sci. USA 2018, 115. [Google Scholar] [CrossRef] [Green Version]
- Hanover, J.A.; Krause, M.W.; Love, D.C. The Hexosamine Signaling Pathway: O-GlcNAc Cycling in Feast or Famine. Biochim. Biophys. Acta 2010, 1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bond, M.R.; Hanover, J.A. O-GlcNAc Cycling: A Link between Metabolism and Chronic Disease. Annu. Rev. Nutr. 2013, 33, 205–229. [Google Scholar] [CrossRef] [PubMed]
- Mondoux, M.A.; Love, D.C.; Ghosh, S.K.; Fukushige, T.; Bond, M.; Weerasinghe, G.R.; Hanover, J.A.; Krause, M.W. O-linked-N-acetylglucosamine Cycling and Insulin Signaling are Required for the Glucose Stress Response in Caenorhabditis Elegans. Genetics 2011, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, B.A.; Burlingame, A.L.; Myers, S.A. Human RNA Polymerase II Promoter Recruitment in vitro Is Regulated by O-Linked N-Acetylglucosaminyltransferase (OGT). J. Biol. Chem. 2016, 291, 14056–14061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lothrop, A.P.; Torres, M.P.; Fuchs, S.M. Deciphering Post-translational Modification Codes. FEBS Lett. 2013, 587. [Google Scholar] [CrossRef] [Green Version]
- Chiaradonna, F.; Ricciardiello, F.; Palorini, R. The Nutrient-Sensing Hexosamine Biosynthetic Pathway as the Hub of Cancer Metabolic Rewiring. Cells 2018, 7, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haltiwanger, R.S.; Holt, G.D.; Hart, G.W. Enzymatic Addition of O-GlcNAc to Nuclear and Cytoplasmic Proteins. Identification of a Uridine Diphospho-N-acetylglucosamine:Peptide Beta-N-acetylglucosaminyltransferase. J. Biol. Chem. 1990, 265, 2563–2568. [Google Scholar] [CrossRef]
- Chaveroux, C.; Sarcinelli, C.; Barbet, V.; Belfeki, S.; Barthelaix, A.; Ferraro-Peyret, C.; Lebecque, S.; Renno, T.; Bruhat, A.; Fafournoux, P.; et al. Nutrient Shortage Triggers the Hexosamine Biosynthetic Pathway via the GCN2-ATF4 Signalling Pathway. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.V.; Deng, Y.; Gao, N.; Pedrozo, Z.; Li, D.L.; Morales, C.R.; Criollo, A.; Luo, X.; Tan, W.; Jiang, N.; et al. Spliced X-box Binding Protein 1 Couples the Unfolded Protein Response to Hexosamine Biosynthetic Pathway. Cell 2014, 156, 1179–1192. [Google Scholar] [CrossRef] [Green Version]
- Ishino, K.; Kudo, M.; Peng, W.X.; Kure, S.; Kawahara, K.; Teduka, K.; Kawamoto, Y.; Kitamura, T.; Fujii, T.; Yamamoto, T.; et al. 2-Deoxy-d-glucose Increases GFAT1 Phosphorylation Resulting in Endoplasmic Reticulum-related Apoptosis via Disruption of Protein N-glycosylation in Pancreatic Cancer Cells. Biochem. Biophys. Res. Commun. 2018, 501, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S.; Nadeau, O.; Yamasaki, K. Dynamic Actions of Glucose and Glucosamine on Hexosamine Biosynthesis in Isolated Adipocytes: Differential Effects on Glucosamine 6-phosphate, UDP-N-acetylglucosamine, and ATP Levels. J. Biol. Chem. 2004, 279. [Google Scholar] [CrossRef] [Green Version]
- Schleicher, E.D.; Weigert, C. Role of the Hexosamine Biosynthetic Pathway in Diabetic Nephropathy. Kidney Int. Suppl. 2000, 77. [Google Scholar] [CrossRef] [Green Version]
- Vasconcelos-Dos-Santos, A.; Loponte, H.F.; Mantuano, N.R.; Oliverira, I.A.; De Paula, I.F.; Teixeira, L.K.; De-Freitas-Junior, J.C.; Gondim, K.C.; Heise, N.; Mohana-Borges, R.; et al. Hyperglycemia Exacerbates Colon Cancer Malignancy through Hexosamine Biosynthetic Pathway. Oncogenesis 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Abdel Rahman, A.M.; Ryczko, M.; Pawling, J.; Dennis, J.W. Probing the Hexosamine Biosynthetic Pathway in Human Tumor Cells by Multitargeted Tandem Mass Spectrometry. ACS Chem. Biol. 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Akella, N.M.; Ciraku, L.; Reginato, M.J. Fueling the Fire: Emerging Role of the Hexosamine Biosynthetic Pathway in Cancer. BMC Biol. 2019, 17, 52. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Vosseller, K. O-GlcNAc in Cancer Biology. Amino Acids 2013, 45, 719–733. [Google Scholar] [CrossRef]
- Fardini, Y.; Dehennaut, V.; Lefebvre, T.; Issad, T. O-GlcNAcylation: A New Cancer Hallmark? Front. Endocrinol. 2013, 4, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slawson, C.; Hart, G.W. O-GlcNAc Signalling: Implications for Cancer Cell Biology. Nat. Rev. Cancer 2011, 11, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Carter, R.; Drouin, G. Structural Differentiation of the Three Eukaryotic RNA Polymerases. Genomics 2009, 94, 388–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koster, M.J.; Snel, B.; Timmers, H.T. Genesis of Chromatin and Transcription Dynamics in the Origin of Species. Cell 2015, 161, 724–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, M.; Cattoglio, C.; Tjian, R. Looping Back to Leap Forward: Transcription Enters a New Era. Cell 2014, 157, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, B.A.; Hanover, J.A. O-GlcNAc and the Epigenetic Regulation of Gene Expression. J. Biol. Chem. 2014, 289, 34440–34448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahmus, M.E. Reversible Phosphorylation of the C-terminal Domain of RNA Polymerase II. J. Biol. Chem. 1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phatnani, H.P.; Greenleaf, A.L. Phosphorylation and Functions of the RNA Polymerase II CTD. Genes Dev. 2006, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Flores, O.; Weinmann, R.; Reinberg, D. The Nonphosphorylated form of RNA Polymerase II Preferentially Associates with the Preinitiation Complex. Proc. Natl. Acad. Sci. USA 1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, W.G.; Dahmus, M.E.; Hart, G.W. RNA Polymerase II is a Glycoprotein. Modification of the COOH-terminal Domain by O-GlcNAc. J. Biol. Chem. 1993, 268, 10416–10424. [Google Scholar] [CrossRef]
- Ranuncolo, S.M.; Ghosh, S.; Hanover, J.A.; Hart, G.W.; Lewis, B.A. Evidence of the Involvement of O-GlcNAc-modified Human RNA Polymerase II CTD in Transcription in vitro and in vivo. J. Biol. Chem. 2012, 287, 23549–23561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resto, M.; Kim, B.H.; Fernandez, A.G.; Abraham, B.J.; Zhao, K.; Lewis, B.A. O-GlcNAcase Is an RNA Polymerase II Elongation Factor Coupled to Pausing Factors SPT5 and TIF1β. J. Biol. Chem. 2016, 291. [Google Scholar] [CrossRef] [Green Version]
- Love, D.C.; Ghosh, S.; Mondoux, M.A.; Fukushige, T.; Wang, P.; Wilson, M.A.; Iser, W.B.; Wolkow, C.A.; Krause, M.W.; Hanover, J.A. Dynamic O-GlcNAc Cycling at Promoters of Caenorhabditis elegans Genes Regulating Longevity, Stress, and Immunity. Proc. Natl. Acad. Sci. USA 2010. [Google Scholar] [CrossRef] [Green Version]
- Lewis, B.A.; Levens, D. O-GlcNAc Transferase Activity is Essential for RNA Pol II Pausing in a Human Cell-Free Transcription System. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Dynlacht, B.D.; Hoey, T.; Tjian, R. Isolation of Coactivators Associated with the TATA-binding Protein That Mediate Transcriptional Activation. Cell 1991, 66, 563–576. [Google Scholar] [CrossRef]
- Hardiville, S.; Banerjee, P.S.; Selen Alpergin, E.S.; Smith, D.M.; Han, G.; Ma, J.; Talbot, C.C., Jr.; Hu, P.; Wolfgang, M.J.; Hart, G.W. TATA-Box Binding Protein O-GlcNAcylation at T114 Regulates Formation of the B-TFIID Complex and Is Critical for Metabolic Gene Regulation. Mol. Cell 2020, 77, 1143–1152.e1147. [Google Scholar] [CrossRef]
- Leney, A.C.; Atmioui, D.E.; Wu, W.; Ovaa, H.; Heck, A.J.R. Elucidating Crosstalk Mechanisms between Phosphorylation and O-GlcNAcylation. Proc. Natl. Acad. Sci. USA 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Gucek, M.; Hart, G.W. Cross-talk between GlcNAcylation and Phosphorylation: Site-specific Phosphorylation Dynamics in Response to Globally Elevated O-GlcNAc. Proc. Natl. Acad. Sci. USA 2008, 105, 13793–13798. [Google Scholar] [CrossRef] [Green Version]
- Bourre, G.; Cantrelle, F.X.; Kamah, A.; Chambraud, B.; Landrieu, I.; Smet-Nocca, C. Direct Crosstalk between O-GlcNAcylation and Phosphorylation of Tau Protein Investigated by NMR Spectroscopy. Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Morachis, J.M.; Huang, R.; Emerson, B.M. Identification of Kinase Inhibitors That Target Transcription Initiation by RNA Polymerase II. Oncotarget 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Segil, N.; Guermah, M.; Hoffmann, A.; Roeder, R.G.; Heintz, N. Mitotic Regulation of TFIID: Inhibition of Activator-dependent Transcription and Changes in Subcellular Localization. Genes Dev. 1996, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baylin, S.B.; Jones, P.A. A Decade of Exploring the Cancer Epigenome-Biological and Translational Implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L. The Complex Language of Chromatin Regulation during Transcription. Nature 2007, 447, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Hart, G.W. Nutrient Regulation of Signaling and Transcription. J. Biol. Chem. 2019, 294, 2211–2231. [Google Scholar] [CrossRef] [Green Version]
- Olivier-Van Stichelen, S.; Hanover, J.A. You Are What You Eat: O-linked N-acetylglucosamine in Disease, Development and Epigenetics. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18. [Google Scholar] [CrossRef]
- Sakabe, K.; Wang, Z.; Hart, G.W. β-N-acetylglucosamine (O-GlcNAc) is Part of the Histone Code. Proc. Natl. Acad. Sci. USA 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakabe, K.; Hart, G.W. O-GlcNAc Transferase Regulates Mitotic Chromatin Dynamics. J. Biol. Chem. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schübeler, D. Function and Information Content of DNA Methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-J.; Wang, Y.; Li, B.-X.; Yang, Y.; Guan, F.; Pang, X.-C. Roles of Ten-eleven Translocation Family Proteins and Their O-linked β-N-acetylglucosaminylated Forms in Cancer Development (Review). Oncol. Lett. 2020, 21, 1. [Google Scholar] [CrossRef]
- Chen, Q.; Chen, Y.; Bian, C.; Fujiki, R.; Yu, X. TET2 Promotes Histone O-GlcNAcylation during Gene Transcription. Nature 2013, 493, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Deplus, R.; Delatte, B.; Schwinn, M.K.; Defrance, M.; Mendez, J.; Murphy, N.; Dawson, M.A.; Volkmar, M.; Putmans, P.; Calonne, E.; et al. TET2 and TET3 Regulate GlcNAcylation and H3K4 Methylation through OGT and SET1/COMPASS. EMBO J. 2013, 32, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Bauer, C.; Gobel, K.; Nagaraj, N.; Colantuoni, C.; Wang, M.; Muller, U.; Kremmer, E.; Rottach, A.; Leonhardt, H. Phosphorylation of TET proteins is regulated via O-GlcNAcylation by the O-linked N-acetylglucosamine transferase (OGT). J. Biol. Chem. 2015, 290, 4801–4812. [Google Scholar] [CrossRef] [Green Version]
- Schuettengruber, B.; Bourbon, H.M.; Di Croce, L.; Cavalli, G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 2017, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The Polycomb Group Protein EZH2 is Involved in Progression of Prostate Cancer. Nature 2002, 419. [Google Scholar] [CrossRef] [PubMed]
- Benard, A.; Goossens-Beumer, I.J.; Van Hoesel, A.Q.; Horati, H.; Putter, H.; Zeestraten, E.C.; Van De Velde, C.J.; Kuppen, P.J. Prognostic Value of Polycomb Proteins EZH2, BMI1 and SUZ12 and Histone Modification H3K27me3 in Colorectal cancer. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W. A Gene That Regulates the Bithorax Complex Differentially in Larval and Adult Cells of Drosophila. Cell 1984, 37, 815–823. [Google Scholar] [CrossRef]
- Gambetta, M.C.; Oktaba, K.; Muller, J. Essential Role of the Glycosyltransferase sxc/Ogt in Polycomb Repression. Science 2009, 325, 93–96. [Google Scholar] [CrossRef]
- Akan, I.; Love, D.C.; Harwood, K.R.; Bond, M.R.; Hanover, J.A. Drosophila O-GlcNAcase Deletion Globally Perturbs Chromatin O-GlcNAcylation. J. Biol. Chem. 2016, 291, 9906–9919. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.-W.; Myschyshyn, M.; Sinclair, D.A.; Cecioni, S.; Beja, K.; Honda, B.M.; Morin, R.D.; Vocadlo, D.J. Genome-wide Chemical Mapping of O-GlcNAcylated Proteins in Drosophila melanogaster. Nat. Chem. Biol. 2016, 13, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Yang, Y.; Qiu, R.; Zhang, K.; Teng, X.; Liu, R.; Wang, Y. Proteomic analysis of the OGT Interactome: Novel Links to Epithelial-mesenchymal Transition and Metastasis of Cervical Cancer. Carcinogenesis 2018, 39, 1222–1234. [Google Scholar] [CrossRef] [PubMed]
- Hauri, S.; Comoglio, F.; Seimiya, M.; Gerstung, M.; Glatter, T.; Hansen, K.; Aebersold, R.; Paro, R.; Gstaiger, M.; Beisel, C. A High-Density Map for Navigating the Human Polycomb Complexome. Cell Rep. 2016, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forma, E.; Jozwiak, P.; Ciesielski, P.; Zaczek, A.; Starska, K.; Brys, M.; Krzeslak, A. Impact of OGT Deregulation on EZH2 Target Genes FOXA1 and FOXC1 Expression in Breast Cancer Cells. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Xu, B.; Li, X.; Shang, Y.; Chu, Y.; Wang, W.; Chen, D.; Wu, N.; Hu, S.; Zhang, S.; et al. Correction: O-GlcNAcylation Promotes Colorectal Cancer Metastasis via the miR-101-O-GlcNAc/EZH2 Regulatory Feedback Circuit. Oncogene 2019, 38, 5744–5745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decourcelle, A.; Very, N.; Djouina, M.; Loison, I.; Thevenet, J.; Body-Malapel, M.; Lelievre, E.; Coqueret, O.; Leprince, D.; El Yazidi-Belkoura, I.; et al. O-GlcNAcylation Links Nutrition to the Epigenetic Downregulation of UNC5A during Colon Carcinogenesis. Cancers 2020, 12, 3168. [Google Scholar] [CrossRef] [PubMed]
- Lo, P.-W.; Shie, J.-J.; Chen, C.-H.; Wu, C.-Y.; Hsu, T.-L.; Wong, C.-H. O-GlcNAcylation Regulates the Stability and Enzymatic Activity of the Histone Methyltransferase EZH2. Proc. Natl. Acad. Sci. USA 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, C.-S.; Lo, P.-W.; Yeh, Y.-H.; Hsu, P.-H.; Peng, S.-H.; Teng, Y.-C.; Kang, M.-L.; Wong, C.-H.; Juan, L.-J. O-GlcNAcylation Regulates EZH2 Protein Stability and Function. Proc. Natl. Acad. Sci. USA 2014. [Google Scholar] [CrossRef] [Green Version]
- Maury, J.J.; El Farran, C.A.; Ng, D.; Loh, Y.H.; Bi, X.; Bardor, M.; Choo, A.B. RING1B O-GlcNAcylation Regulates Gene Targeting of Polycomb Repressive Complex 1 in Human Embryonic Stem Cells. Stem Cell Res. 2015, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Parker, M.P.; Graw, S.; Novikova, L.V.; Fedosyuk, H.; Fontes, J.D.; Koestler, D.C.; Peterson, K.R.; Slawson, C. O-GlcNAc Homeostasis Contributes to Cell Fate Decisions during Hematopoiesis. J. Biol. Chem. 2019, 294, 1363–1379. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhang, F.; Kudlow, J.E. Recruitment of O-GlcNAc Transferase to Promoters by Corepressor mSin3A: Coupling Protein O-GlcNAcylation to Transcriptional Repression. Cell 2002, 110, 69–80. [Google Scholar] [CrossRef] [Green Version]
- McDonel, P.; Costello, I.; Hendrich, B. Keeping Things Quiet: Roles of NuRD and Sin3 Co-repressor Complexes during Mammalian Development. Int. J. Biochem. Cell Biol. 2009, 41, 108–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, S.A.; Panning, B.; Burlingame, A.L. Polycomb Repressive Complex 2 is Necessary for the Normal Site-specific O-GlcNAc Distribution in Mouse Embryonic Stem Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 9490–9495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bornelov, S.; Reynolds, N.; Xenophontos, M.; Gharbi, S.; Johnstone, E.; Floyd, R.; Ralser, M.; Signolet, J.; Loos, R.; Dietmann, S.; et al. The Nucleosome Remodeling and Deacetylation Complex Modulates Chromatin Structure at Sites of Active Transcription to Fine-Tune Gene Expression. Mol. Cell 2018, 71. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, A.; Spengler, D. Chromatin Remodeling Complex NuRD in Neurodevelopment and Neurodevelopmental Disorders. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Costa, F.C.; Tan, E.P.; Bushue, N.; DiTacchio, L.; Costello, C.E.; McComb, M.E.; Whelan, S.A.; Peterson, K.R.; Slawson, C. O-Linked N-Acetylglucosamine (O-GlcNAc) Transferase and O-GlcNAcase Interact with Mi2beta Protein at the Agamma-Globin Promoter. J. Biol. Chem. 2016, 291, 15628–15640. [Google Scholar] [CrossRef] [Green Version]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; Barrio, M.G.d.; Portillo, F.; Nieto, M.A. The Transcription Factor Snail Controls Epithelial–mesenchymal Transitions by Repressing E-cadherin Expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Sancho, E.; Franci, C.; Dominguez, D.; Monfar, M.; Baulida, J.; Garcia De Herreros, A. The Transcription Factor Snail is a Repressor of E-cadherin Gene Expression in Epithelial Tumour Cells. Nat. Cell Biol. 2000, 2. [Google Scholar] [CrossRef]
- Fujita, N.; Jaye, D.L.; Kajita, M.; Geigerman, C.; Moreno, C.S.; Wade, P.A. MTA3, a Mi-2/NuRD complex Subunit, Regulates an Invasive Growth Pathway in Breast Cancer. Cell 2003, 113. [Google Scholar] [CrossRef] [Green Version]
- Basta, J.; Rauchman, M. The Nucleosome Remodeling and Deacetylase Complex in Development and Disease. Transl. Res. J. Lab. Clin. Med. 2015, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A Landscape Takes Shape. Cell 2007, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in Cancer. Carcinogenesis 2010, 31. [Google Scholar] [CrossRef]
- Deniaud, E.; Baguet, J.; Mathieu, A.L.; Pages, G.; Marvel, J.; Leverrier, Y. Overexpression of Sp1 Transcription Factor Induces Apoptosis. Oncogene 2006, 25. [Google Scholar] [CrossRef] [Green Version]
- Han, I.; Kudlow, J.E. Reduced O Glycosylation of Sp1 is Associated with Increased Proteasome Susceptibility. Mol. Cell. Biol. 1997, 17, 2550–2558. [Google Scholar] [CrossRef] [Green Version]
- Vellingir, B.; Iyer, M.; Devi Subramaniam, M.; Jayaramayya, K.; Siama, Z.; Giridharan, B.; Narayanasamy, A.; Abdal Dayem, A.; Cho, S.G. Understanding the Role of the Transcription Factor Sp1 in Ovarian Cancer: From Theory to Practice. Int. J. Mol. Sci. 2020, 21, 1153. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Sangwan, V.; McGinn, O.; Chugh, R.; Dudeja, V.; Vickers, S.M.; Saluja, A.K. Triptolide-induced Cell Death in Pancreatic Cancer is Mediated by O-GlcNAc Modification of Transcription Factor Sp1. J. Biol. Chem. 2013, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Qian, K. Protein O-GlcNAcylation: Emerging Mechanisms and Functions. Nat. Rev. Mol. Cell Biol. 2017, 18, 452–465. [Google Scholar] [CrossRef]
- Yang, X.; Su, K.; Roos, M.D.; Chang, Q.; Paterson, A.J.; Kudlow, J.E. O-linkage of N-acetylglucosamine to Sp1 Activation Domain Inhibits Its Transcriptional Capability. Proc. Natl. Acad. Sci. USA 2001, 98. [Google Scholar] [CrossRef] [Green Version]
- Olivier-Van Stichelen, S.; Wang, P.; Comly, M.; Love, D.C.; Hanover, J.A. Nutrient-driven O-linked N-acetylglucosamine (O-GlcNAc) Cycling Impacts Neurodevelopmental Timing and Metabolism. J. Biol. Chem. 2017, 292, 6076–6085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, B.; Duan, M.; Xing, Y.; Liu, C.; Yang, F.; Li, Y.; Yang, T.; Wei, Y.; Gao, Q.; Jiang, J. O-GlcNAc Transferase Activates Stem-like Cell Potential in Hepatocarcinoma through O-GlcNAcylation of Eukaryotic Initiation Factor 4E. J. Cell. Mol. Med. 2019, 23, 2384–2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.S.; Gupta, V.K.; Dauer, P.; Kesh, K.; Hadad, R.; Giri, B.; Chandra, A.; Dudeja, V.; Slawson, C.; Banerjee, S.; et al. O-GlcNAc Modification of Sox2 Regulates Self-renewal in Pancreatic Cancer by Promoting Its Stability. Theranostics 2019, 9, 3410–3424. [Google Scholar] [CrossRef] [PubMed]
- Constable, S.; Lim, J.M.; Vaidyanathan, K.; Wells, L. O-GlcNAc Transferase Regulates Transcriptional Activity of Human Oct4. Glycobiology 2017, 27, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Mi, W.; Ge, Y.; Liu, H.; Fan, Q.; Han, C.; Yang, J.; Han, F.; Lu, X.; Yu, W. GlcNAcylation Plays an Essential Role in Breast Cancer Metastasis. Cancer Res. 2010, 70. [Google Scholar] [CrossRef] [Green Version]
- Champattanachai, V.; Netsirisawan, P.; Chaiyawat, P.; Phueaouan, T.; Charownwattanasatien, R.; Chokchaichamnankit, D.; Punyarit, P.; Srisomsap, C.; Svasti, J. Proteomic Analysis and Abrogated Expression of O-GlcNAcylated Proteins Associated with Primary Breast Cancer. Proteomics 2013, 13. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, C.M.; Lynch, T.P.; Sodi, V.L.; Falcone, J.N.; Schwab, L.P.; Peacock, D.L.; Vocadlo, D.J.; Seagroves, T.N.; Reginato, M.J. O-GlcNAcylation Regulates Cancer Metabolism and Survival Stress Signaling via Regulation of the HIF-1 Pathway. Mol. Cell 2014, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzeslak, A.; Forma, E.; Bernaciak, M.; Romanowicz, H.; Brys, M. Gene Expression of O-GlcNAc Cycling Enzymes in Human Breast Cancers. Clin. Exp. Med. 2012, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldwell, S.A.; Jackson, S.R.; Shahriari, K.S.; Lynch, T.P.; Sethi, G.; Walker, S.; Vosseller, K.; Reginato, M.J. Nutrient Sensor O-GlcNAc Transferase Regulates Breast Cancer Tumorigenesis through Targeting of the Oncogenic Transcription Factor FoxM1. Oncogene 2010, 29, 2831–2842. [Google Scholar] [CrossRef] [Green Version]
- Sodi, V.L.; Khaku, S.; Krutilina, R.; Schwab, L.P.; Vocadlo, D.J.; Seagroves, T.N.; Reginato, M.J. mTOR/MYC Axis Regulates O-GlcNAc Transferase Expression and O-GlcNAcylation in Breast Cancer. Mol. Cancer Res. 2015, 13, 923–933. [Google Scholar] [CrossRef] [Green Version]
- Trinca, G.M.; Goodman, M.L.; Papachristou, E.K.; D’Santos, C.S.; Chalise, P.; Madan, R.; Slawson, C.; Hagan, C.R. O-GlcNAc-Dependent Regulation of Progesterone Receptor Function in Breast Cancer. Horm. Cancer 2018, 9, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.S.; Hart, G.W. A Subpopulation of Estrogen Receptors are Modified by O-linked N-acetylglucosamine. J. Biol. Chem. 1997, 272. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Hart, G.W. Glycosylation of the Murine Estrogen Receptor-alpha. J. Steroid Biochem. Mol. Biol. 2000, 75, 147–158. [Google Scholar] [CrossRef]
- Cheng, X.; Hart, G.W. Alternative O-glycosylation/O-phosphorylation of Serine-16 in Murine Estrogen Receptor Beta: Post-translational Regulation of Turnover and Transactivation Activity. J. Biol. Chem. 2001, 276, 10570–10575. [Google Scholar] [CrossRef] [Green Version]
- Ozcan, S.; Andrali, S.S.; Cantrell, J.E. Modulation of Transcription Factor Function by O-GlcNAc Modification. Biochim. Biophys. Acta 2010, 1799. [Google Scholar] [CrossRef] [Green Version]
- Carroll, J.S.; Hickey, T.E.; Tarulli, G.A.; Williams, M.; Tilley, W.D. Deciphering the Divergent Roles of Progestogens in Breast Cancer. Nat. Rev. Cancer 2016, 17, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, X.; Zhu, T.; Zhang, N.; Wang, L.; Huang, T.; Cao, Y.; Li, W.; Zhang, J. Resistance to Bortezomib in Breast Cancer Cells That Downregulate Bim through FOXA1 O-GlcNAcylation. J. Cell. Physiol. 2019, 234, 17527–17537. [Google Scholar] [CrossRef]
- Das, S.; Bailey, S.K.; Metge, B.J.; Hanna, A.; Hinshaw, D.C.; Mota, M.; Forero-Torres, A.; Chatham, J.C.; Samant, R.S.; Shevde, L.A. O-GlcNAcylation of GLI Transcription Factors in Hyperglycemic Conditions Augments Hedgehog Activity. Lab. Invest. 2019, 99, 260–270. [Google Scholar] [CrossRef]
- Peng, C.; Zhu, Y.; Zhang, W.; Liao, Q.; Chen, Y.; Zhao, X.; Guo, Q.; Shen, P.; Zhen, B.; Qian, X.; et al. Regulation of the Hippo-YAP Pathway by Glucose Sensor O-GlcNAcylation. Mol. Cell 2017, 68. [Google Scholar] [CrossRef] [PubMed]
- Halder, G.; Johnson, R.L. Hippo Signaling: Growth Control and Beyond. Development 2011. [Google Scholar] [CrossRef] [Green Version]
- Pan, D. The Hippo Signaling Pathway in Development and Cancer. Dev. Cell 2010, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Li, L.; Lei, Q.; Guan, K.L. The Hippo-YAP Pathway in Organ Size Control and Tumorigenesis: An Updated Version. Genes Dev. 2010, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Tumaneng, K.; Guan, K.-L. The Hippo Pathway in Organ Size Control, Tissue Regeneration and Stem Cell Self-renewal. Nat. Cell Biol. 2011, 13, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kin, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP Oncoprotein by the Hippo Pathway is Involved in Cell Contact Inhibition and Tissue Growth Control. Genes Dev. 2007, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhoa, B.; Li, L.; Tumaneng, K.; Wang, C.Y.; Guan, K.L. A Coordinated Phosphorylation by Lats and CK1 Regulates YAP Stability through SCF(beta-TRCP). Genes Dev. 2010, 24. [Google Scholar] [CrossRef] [Green Version]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo Pathway Regulation. Genes Dev. 2016, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamar, J.M.; Stern, P.; Liu, H.; Schindler, J.W.; Jiang, Z.-G.; Hynes, R.O. The Hippo Pathway Target, YAP, Promotes Metastasis through Its TEAD-interaction Domain. Proc. Natl. Acad. Sci. USA 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gumbiner, B.M.; Kim, N.-G. The Hippo-YAP Signaling Pathway and Contact Inhibition of Growth. J. Cell Sci. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Qiao, Y.; Wu, Q.; Chen, Y.; Zou, S.; Liu, X.; Zhu, G.; Zhao, Y.; Chen, Y.; Yu, Y.; et al. The Essential Role of YAP O-GlcNAcylation in High-glucose-stimulated Liver Tumorigenesis. Nat. Commun. 2017, 8, 15280. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, N.; Zachara, N.E.; Hart, G.W.; Marth, J.D. Ogt-dependent X-chromosome-linked Protein Glycosylation is a Requisite Modification in Somatic Cell Function and Embryo Viability. Mol. Cell. Biol. 2004, 24, 1680–1690. [Google Scholar] [CrossRef] [Green Version]
- Slawson, C.; Copeland, R.J.; Hart, G.W. O-GlcNAc Signaling: A Metabolic Link between Diabetes and Cancer? Trends Biochem. Sci. 2010, 35, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Tan, E.P.; VandenHull, N.J.; Peterson, K.R.; Slawson, C. O-GlcNAcase Expression is Sensitive to Changes in O-GlcNAc Homeostasis. Front. Endocrinol. 2014, 5, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazemi, Z.; Chang, H.; Haserodt, S.; McKen, C.; Zachara, N.E. O-linked Beta-N-acetylglucosamine (O-GlcNAc) Regulates Stress-induced Heat Shock Protein Expression in a GSK-3beta-Dependent Manner. J. Biol. Chem. 2010, 285, 39096–39107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slawson, C.; Zachara, N.E.; Vosseller, K.; Cheung, W.D.; Lane, M.D.; Hart, G.W. Perturbations in O-linked Beta-N-acetylglucosamine Protein Modification Cause Severe Defects in Mitotic Progression and Cytokinesis. J. Biol. Chem. 2005, 280, 32944–32956. [Google Scholar] [CrossRef] [PubMed]
- Boulard, M.; Rucli, S.; Edwards, J.R.; Bestor, T.H. Methylation-directed Glycosylation of Chromatin Factors Represses Retrotransposon Promoters. Proc. Natl. Acad. Sci. USA 2020, 117, 14292–14298. [Google Scholar] [CrossRef] [PubMed]
- Forma, E.; Jóźwiak, P.; Bryś, M.; Krześlak, A. The Potential Role of O-GlcNAc Modification in Cancer Epigenetics. Cell. Mol. Biol. Lett. 2014, 19, 438–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parker, M.P.; Peterson, K.R.; Slawson, C. O-GlcNAcylation and O-GlcNAc Cycling Regulate Gene Transcription: Emerging Roles in Cancer. Cancers 2021, 13, 1666. https://doi.org/10.3390/cancers13071666
Parker MP, Peterson KR, Slawson C. O-GlcNAcylation and O-GlcNAc Cycling Regulate Gene Transcription: Emerging Roles in Cancer. Cancers. 2021; 13(7):1666. https://doi.org/10.3390/cancers13071666
Chicago/Turabian StyleParker, Matthew P., Kenneth R. Peterson, and Chad Slawson. 2021. "O-GlcNAcylation and O-GlcNAc Cycling Regulate Gene Transcription: Emerging Roles in Cancer" Cancers 13, no. 7: 1666. https://doi.org/10.3390/cancers13071666