
Dysregulation of the Ubiquitin Proteasome System in Human Malignancies: A Window for Therapeutic Intervention



Abstract
:Simple Summary
Abstract
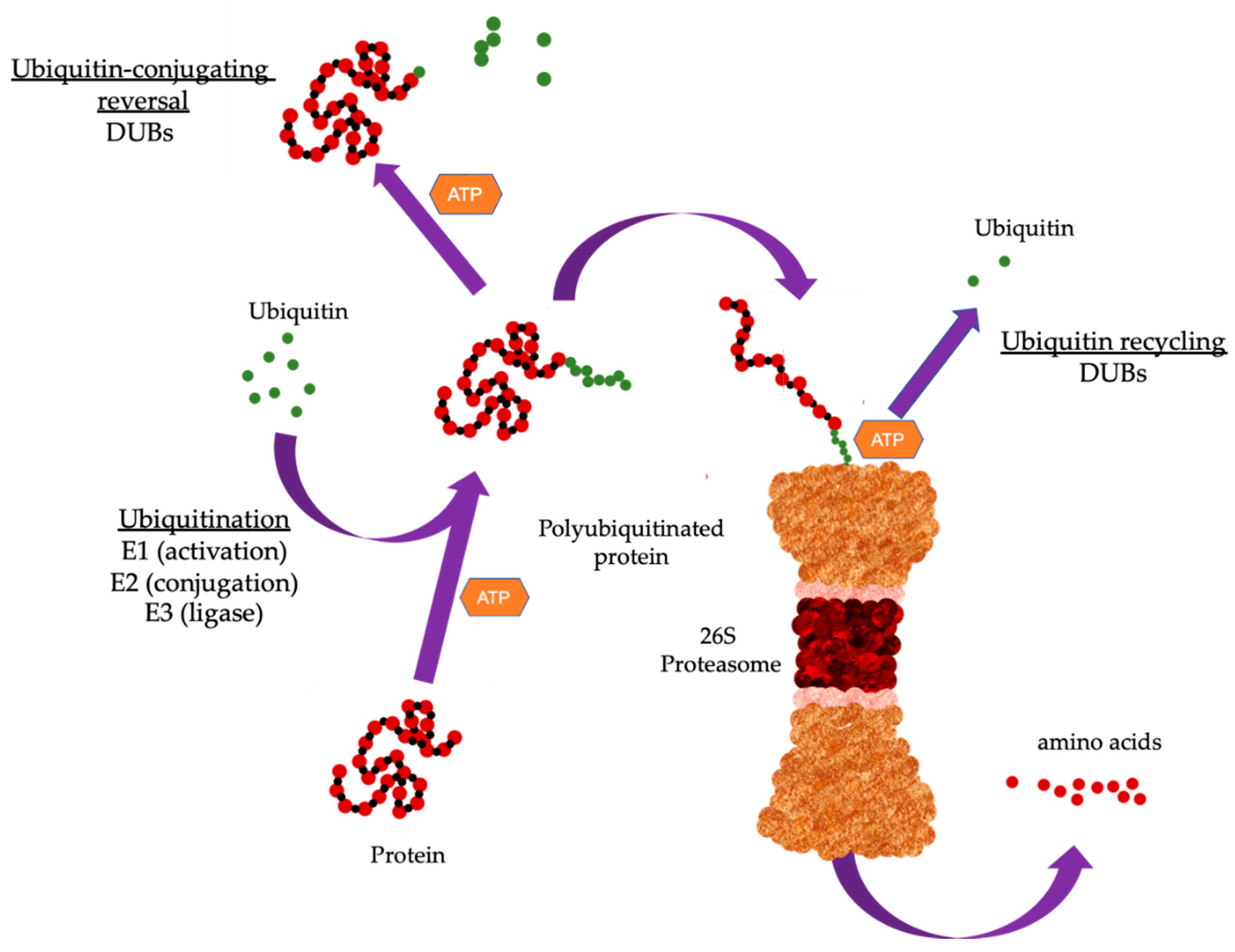
1. The Ubiquitin Proteasome System
2. Ubiquitin-Conjugating Enzymes
2.1. UBE2C

{kind=link}
| Family | Name | Role | Cancer Type | Function | Test Model | Reference |
|---|---|---|---|---|---|---|
| E2 | UBE2C | Oncogene | Gastric | Chromosomal stability, Proliferation, Migration, Invasion | In vitro, In vivo | [23] |
| Oncogene | Colon | Cell cycle, Proloferation | In vitro | [24] | ||
| Oncogene | Colorectal | Proliferation, Invasion | In vitro | [25] | ||
| Oncogene | Thyroid | Proliferation | In vitro | [26] | ||
| Oncogene | Breast | Proliferation, Drug resistance, Radiation resistance | In vitro | [29] | ||
| Oncogene | Liver | Proliferation, Drug resistance, Migration, Invasion | In vitro | [30] | ||
| Oncogene | Non-small cell lung | Drug resistance | In vitro | [31] | ||
| UBE2Q1 | Oncogene | Colorectal | Proliferation | [32] | ||
| Oncogene | Liver | p53 signaling, Cell cycle | In vitro | [33] | ||
| Oncogene | Breast | p53 signaling | In vitro | [34] | ||
| UBE2S | Oncogene | Endometrial | SOX6/β-catenin signaling, Proliferation | In vitro | [35] | |
| Oncogene | Lung adenocarcinoma | Proliferation, p53 signaling, Apoptosis | In vitro | [36] | ||
| Oncogene | Liver | p53 signaling, Cell cycle | In vitro | [37] | ||
| E3 | FBW7 | Tumor suppressor | Burkitt’s lymphoma | c-Myc signaling | In vitro | [38,39] |
| Tumor suppressor | Chronic myelogenous leukemia | c-Myc signaling | In vitro, In vivo | [40] | ||
| Lipogenesis | Lung, Melanoma, Thyroid, Cervical | mTORC2/SREBP1 signaling | In vitro | [41] | ||
| Tumor suppressor | T cell leukemia | Notch signaling | In vitro, In vivo | [42] | ||
| Tumor suppressor | Colorectal | c-Myc signaling, Cell cycle | In vitro | [43] | ||
| Tumor suppressor | Esophageal squamous cell | c-Myc signaling | In vitro | [44] | ||
| Tumor suppressor | Colorectal, Cervical, Ovarian, Non-small cell lung | Apoptosis (via Mcl1) | In vitro | [45] | ||
| MDM2 | Oncogene | Neuroblastoma | p53 signaling | In vitro, In vivo | [46] | |
| Oncogene | Cervical | Cell cycle, Apoptosis | In vitro | [47] | ||
| Oncogene | Liver | Metastasis, Drug response | In vitro, In vivo | [48] | ||
| Cdc20 | Oncogene | Breast | Metastasis, Drug response | In vitro | [49] | |
| Cdh1 | Tumor suppressor | Breast | Src signaling | In vitro | [50] | |
| β-TRCP | Tumor suppressor | Breast, Prostate | MTSS1 signaling | In vitro | [51] | |
| Oncogene | Lung | FOXN2 | In vitro, In vivo | [52] | ||
| Tumor suppressor | Papillary thyroid | VEGFR2 signaling | In vitro, In vivo | [53] | ||
| E6AP | Oncogene | Prostate | Radiation response | In vitro | [54] | |
| Oncogene | Prostate | p27 signaling | In vitro, In vivo | [55] | ||
| Oncogene | Prostate | Metastasis | In vitro, In vivo | [56] |
2.2. UBE2Q1
2.3. UBE2S
3. Ubiquitin Ligases
3.1. FBW7
3.2. MDM2
3.3. Cdc20 and Cdh1
3.4. βTrCP
3.5. E6AP
4. Deubiquitinases (DUBs)
| Name | Role | Cancer Type | Function | Test Model | Reference |
|---|---|---|---|---|---|
| BAP1 | Tumor suppressor | Lung, Osteosarcoma, Colon | DNA double-strand repair | In vitro | [80,81,82] |
| Tumor suppressor | Renal | Ferroptosis signaling | In vitro | [83] | |
| USP7 | Oncogene | Cervical | Self-renewal; Foxp3 signaling | In vitro | [84] |
| Oncogene | Non-small cell lung | Immune Response; Foxp3 signaling | In vitro | [85] | |
| Oncogene | Lung | p53 signaling | In vitro, in vivo | [86] | |
| USP22 | Oncogene | Lung | Cell Cycle | In vitro | [87] |
| Oncogene | Lung adenocarcinoma | EGFR-TKI resistance | In vitro, in vivo | [88] | |
| Oncogene | Colon | CCNB1 signaling | In vitro, in vivo | [89] | |
| Oncogene | Glioblastoma | KDM1A signaling | In vitro, in vivo | [90] | |
| UCHL1 | Oncogene | Breast | Drug resistance; Invasion/migration | In vitro | [91] |
| Ataxin 3 | Oncogene | Breast, Osteosarcoma, Cervical, Colorectal | DNA | In vitro | [92] |
| Oncogene | Testicular | mTOR/Akt signaling | In vitro | [93] | |
| PSMD11 | Oncogene | Cervical. Osteosarcoma | DNA damage response | In vitro | [94] |
| Oncogene | Lung, Prostate, Colorectal, Breast, Cervix | Cell cycle | In vitro | [95] | |
| Oncogene | Liver | E2F1 signaling | In vitro, in vivo | [96] | |
| A20 | Tumor suppressor | Colorectal | Apoptosis signaling | In vitro | [97] |
| Tumor suppressor | Diffuse large B-cell lymphoma | NF-κβ signaling | In vitro | [98] | |
| Tumor suppressor | Sarcoma | NF-κβ signaling | In vitro | [99] |
4.1. BAP1
4.2. USP7
4.3. USP22
4.4. UCHL1
4.5. Ataxin 3
4.6. PSMD14
4.7. A20
5. Other Ubiquitin Modifiers
6. UPS Inhibitors in Cancer Therapy
6.1. Targeting the Proteasome
6.2. Targeting Ubiquitinases
| Inhibitor | Target | Cancer Type | Clinical Trial | Reference |
|---|---|---|---|---|
| Bortezomib | Proteasomal inhibitor | Multiple myeloma, Mantle cell lymphoma, Leukemia, Neuroblastoma, Head and Neck, Thyroid, Hepatocellular | FDA approved | www.clinicaltrials.gov [126,166,167,169,170,171,172,177] |
| Carfilzomib | Proteasomal inhibitor | Multiple myeloma, Lymphoma, Relapsed and/or refractory multiple myeloma, Leukemia, Lung, Thyroid, Refractory renal cell carcinoma | FDA approved | www.clinicaltrials.gov [54,173,174,175,176,177] |
| Ixazomib | Proteasomal inhibitor | Multiple myeloma, Relapsed and/or refractory multiple myeloma, Lymphoma, Leukemia, Breast, Glioblastoma, Renal cell carcinoma, Hodgkin and T cell lymphoma | FDA approved | www.clinicaltrials.gov [178] |
| Delanzomib | Proteasomal inhibitor | Non-Hodgkin’s lymphoma | Phase I | www.clinicaltrials.gov |
| Marizomib | Proteasomal inhibitor | Multiple myeloma, Advanced solid tumors | Phase I/II | www.clinicaltrials.gov |
| Oprozomib | Proteasomal inhibitor | Multiple myeloma, Glioma, Pancreatic, Lung, Melanoma, Lymphoma, Glipblastoma | Phase I/II/III | www.clinicaltrials.gov |
| MLN4924 | NAE and UBA1(E1) | Advanced malignant solid tumors, Melanoma, Hepatocellular, B cell lymphoma, Hematologic malignancies, Acute myelocytic leukemia | Phase I/II/III | www.clinicaltrials.gov |
| TAK981 | SAE (E1) | B cell lymphoma, colorectal, non-Hodgkin’s, Advcnced/metasiatic solid tumors | Phase I/II | www.clinicaltrials.gov |
| TAS4464 | NAE (E1) | Multiple myeloma, non-Hodgkin lymphoma | Phase I/II | www.clinicaltrials.gov |
| SAR-405838 | MDM2 (E2) | Solid tumors | Phase I | www.clinicaltrials.gov [172,183] |
| CGM-097 | MDM2 (E2) | Advanced p53 wildtype solid tumors | Phase I | www.clinicaltrials.gov [184,185] |
| DS-3032b | MDM2 (E2) | Acute myelocytic leukemia | Phase I/II | www.clinicaltrials.gov [186,187] |
| Debio1143 (AT-406) | cIAP1/2 (E3) | Acute myeloid leukemia | Phase I | www.clinicaltrials.gov [188] |
| LC-161 | IAP (E3) | Advanced solid tumors | Phase I | www.clinicaltrials.gov [189] |
| Birinapant | IAP (E3) | Solid tumors | Phase I/II | www.clinicaltrials.gov [190] |
| Pimozide | USP1 | Glioma, Non-small cell lung cancer | FDA approced for Tourette’s syndrome; Preclinical | [191,192] |
| Mitoxantrone | USP11 | Metastatic crastrate -resistant prostate, Acute myeloid leukemia, Advanced breast cancer, non-Hodgkin’s lymphoma, Primary liver | FDA approved | [193,194,195,196,197,198,199,200,201] |
6.3. Targeting DUBs
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ciechanover, A. Intracellular protein degradation: From a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Best Pract. Res. Clin. Haematol. 2017, 30, 341–355. [Google Scholar] [CrossRef]
- Myung, J.; Kim, K.B.; Crews, C.M. The ubiquitin-proteasome pathway and proteasome inhibitors. Med. Res. Rev. 2001, 21, 245–273. [Google Scholar] [CrossRef]
- Yang, H.; Chen, X.; Li, K.; Cheaito, H.; Yang, Q.; Wu, G.; Liu, J.; Dou, Q.P. Repurposing old drugs as new inhibitors of the ubiquitin-proteasome pathway for cancer treatment. Semin. Cancer Biol. 2021, 68, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef] [PubMed]
- Nandi, D.; Tahiliani, P.; Kumar, A.; Chandu, D. The ubiquitin-proteasome system. J. Biosci. 2006, 31, 137–155. [Google Scholar] [CrossRef]
- Fang, S.; Weissman, A.M. Ubiquitin-proteasome system. Cell. Mol. Life Sci. 2004, 61, 1546–1561. [Google Scholar] [CrossRef]
- Adams, J. The proteasome: Structure, function, and role in the cell. Cancer Treat. Rev. 2003, 29 (Suppl. S1), 3–9. [Google Scholar] [CrossRef]
- Wang, G.; Gao, Y.; Li, L.; Jin, G.; Cai, Z.; Chao, J.-I.; Lin, H.-K. K63-Linked Ubiquitination in Kinase Activation and Cancer. Front. Oncol. 2012, 2, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, M.; Sorkin, A. Regulation of Receptors and Transporters by Ubiquitination: New Insights into Surprisingly Similar Mechanisms. Mol. Interv. 2007, 7, 157–167. [Google Scholar] [CrossRef]
- Hochstrasser, M. Ubiquitin-Dependent protein degradation. Annu. Rev. Genet. 1996, 30, 405–439. [Google Scholar] [CrossRef] [PubMed]
- Panier, S.; Durocher, D. Regulatory ubiquitylation in response to DNA double-strand breaks. DNA Repair 2009, 8, 436–443. [Google Scholar] [CrossRef]
- Kim, J.H.; Park, K.C.; Chung, S.S.; Bang, O.; Chung, C.H. Deubiquitinating Enzymes as Cellular Regulators. J. Biochem. 2003, 134, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M.; Luna-Vargas, M.P.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.; Sixma, T.K.; Bernards, R. A Genomic and Functional Inventory of Deubiquitinating Enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef] [Green Version]
- Clague, M.J.; Barsukov, I.; Coulson, J.M.; Liu, H.; Rigden, D.J.; Urbé, S. Deubiquitylases From Genes to Organism. Physiol. Rev. 2013, 93, 1289–1315. [Google Scholar] [CrossRef]
- Komander, D. The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 2009, 37 Pt 5, 937–953. [Google Scholar] [CrossRef] [Green Version]
- Ciechanover, A.; Brundin, P. The Ubiquitin Proteasome System in Neurodegenerative Diseases. Neuron 2003, 40, 427–446. [Google Scholar] [CrossRef] [Green Version]
- Ciechanover, A.; Schwartz, A.L. The ubiquitin system: Pathogenesis of human diseases and drug targeting. Biochim. Biophys. Acta 2004, 1695, 3–17. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Kitagaki, J.; Dai, R.-M.; Tsai, Y.C.; Lorick, K.L.; Ludwig, R.L.; Pierre, S.A.; Jensen, J.P.; Davydov, I.V.; Oberoi, P.; et al. Inhibitors of Ubiquitin-Activating Enzyme (E1), a New Class of Potential Cancer Therapeutics. Cancer Res. 2007, 67, 9472–9481. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Shaik, S.; Dai, X.; Wu, Q.; Zhou, X.; Wang, Z.; Wei, W. Targeting the ubiquitin pathway for cancer treatment. Biochim. Biophys. Acta 2015, 1855, 50–60. [Google Scholar] [CrossRef] [Green Version]
- Hosseini, S.M.; Okoye, I.; Chaleshtari, M.G.; Hazhirkarzar, B.; Mohamadnejad, J.; Azizi, G.; Hojjat-Farsangi, M.; Mohammadi, H.; Shotorbani, S.S.; Jadidi-Niaragh, F. E2 ubiquitin-conjugating enzymes in cancer: Implications for immunotherapeutic interventions. Clin. Chim. Acta 2019, 498, 126–134. [Google Scholar] [CrossRef]
- Clague, M.J.; Heride, C.; Urbé, S. The demographics of the ubiquitin system. Trends Cell Biol. 2015, 25, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 enzymes: More than just middle men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Liu, X.; Yu, G.; Liu, L.; Wang, J.; Chen, X.; Bian, Y.; Ji, Y.; Zhou, X.; Chen, Y.; et al. UBE2C Is a Potential Biomarker of Intestinal-Type Gastric Cancer With Chromosomal Instability. Front. Pharmacol. 2018, 9, 847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, T.; Ikeda, H.; Taira, N.; Hatoh, S.; Naito, M.; Doihara, H. Overexpression of UbcH10 alternates the cell cycle profile and accelerate the tumor proliferation in colon cancer. BMC Cancer 2009, 9, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Chen, Y.; Hu, C.; Jing, H.; Cao, Y.; Liu, X. Association of clinicopathological features with UbcH10 expression in colorectal cancer. J. Cancer Res. Clin. Oncol. 2009, 136, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Pallante, P.; Berlingieri, M.; Troncone, G.; Kruhoffer, M.; Orntoft, T.; Viglietto, G.; Caleo, A.; Migliaccio, I.; Decaussin-Petrucci, M.; Santoro, M.; et al. UbcH10 overexpression may represent a marker of anaplastic thyroid carcinomas. Br. J. Cancer 2005, 93, 464–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Psyrri, A.; Kalogeras, K.T.; Kronenwett, R.; Wirtz, R.M.; Batistatou, A.; Bournakis, E.; Timotheadou, E.; Gogas, H.; Aravantinos, G.; Christodoulou, C.; et al. Prognostic significance of UBE2C mRNA expression in high-risk early breast cancer. A Hellenic Cooperative Oncology Group (HeCOG) Study. Ann. Oncol. 2012, 23, 1422–1427. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.-K.; Wu, W.-G.; Chen, L.; Dong, P.; Gu, J.; Mu, J.-S.; Yang, J.-H.; Liu, Y.-B. Expression of UbcH10 in pancreatic ductal adenocarcinoma and its correlation with prognosis. Tumor Biol. 2013, 34, 1473–1477. [Google Scholar] [CrossRef]
- Rawat, A.; Gopal, G.; Selvaluxmy, G.; Rajkumar, T. Inhibition of ubiquitin conjugating enzyme UBE2C reduces proliferation and sensitizes breast cancer cells to radiation, doxorubicin, tamoxifen and letrozole. Cell. Oncol. 2013, 36, 459–467. [Google Scholar] [CrossRef]
- Xiong, Y.; Lu, J.; Fang, Q.; Lu, Y.; Xie, C.; Wu, H.; Yin, Z. UBE2C functions as a potential oncogene by enhancing cell proliferation, migration, invasion, and drug resistance in hepatocellular carcinoma cells. Biosci. Rep. 2019, 39, BSR20182384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Jin, D.; Wu, Y.; Yang, L.; Du, J.; Gong, K.; Chen, W.; Dai, J.; Miao, S.; Xi, S. The miR 495-UBE2C-ABCG2/ERCC1 axis reverses cisplatin resistance by downregulating drug resistance genes in cisplatin-resistant non-small cell lung cancer cells. EBioMedicine 2018, 35, 204–221. [Google Scholar] [CrossRef]
- Fahmidehkar, M.A.; Shafiee, S.M.; Eftekhar, E.; Mahbudi, L.; Seghatoleslam, A. Induction of cell proliferation, clonogenicity and cell accumulation in S phase as a consequence of human UBE2Q1 overexpression. Oncol. Lett. 2016, 12, 2169–2174. [Google Scholar] [CrossRef] [Green Version]
- Chang, R.; Wei, L.; Lu, Y.; Cui, X.; Lu, C.; Liu, L.; Jiang, D.; Xiong, Y.; Wang, G.; Wan, C.; et al. Upregulated expression of ubiquitin-conjugating enzyme E2Q1 (UBE2Q1) is associated with enhanced cell proliferation and poor prognosis in human hapatocellular carcinoma. J. Mol. Histol. 2014, 46, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Shafiee, S.M.; Rasti, M.; Seghatoleslam, A.; Azimi, T.; Owji, A.A. UBE2Q1 in a Human Breast Carcinoma Cell Line: Overexpression and Interaction with p53. Asian Pac. J. Cancer Prev. 2015, 16, 3723–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.; Lei, T.; Zheng, J.; Chen, S.; Du, L.; Xie, H. UBE2S mediates tumor progression via SOX6/β-Catenin signaling in endometrial cancer. Int. J. Biochem. Cell Biol. 2019, 109, 17–22. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, L. UBE2S promotes the proliferation and survival of human lung adenocarcinoma cells. BMB Rep. 2018, 51, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.-H.; Yang, M.; Liu, L.-P.; Wu, D.-C.; Li, M.-Y.; Su, S.-G. UBE2S enhances the ubiquitination of p53 and exerts oncogenic activities in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2018, 503, 895–902. [Google Scholar] [CrossRef]
- Bahram, F.; von der Lehr, N.; Cetinkaya, C.; Larsson, L.G. c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood 2000, 95, 2104–2110. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.-H.; Bellon, M.; Nicot, C. FBXW7: A critical tumor suppressor of human cancers. Mol. Cancer 2018, 17, 115. [Google Scholar] [CrossRef]
- Reavie, L.; Buckley, S.M.; Loizou, E.; Takeishi, S.; Aranda-Orgilles, B.; Ndiaye-Lobry, D.; Abdel-Wahab, O.; Ibrahim, S.; Nakayama, K.I.; Aifantis, I. Regulation of c-Myc Ubiquitination Controls Chronic Myelogenous Leukemia Initiation and Progression. Cancer Cell 2013, 23, 362–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Oh, Y.T.; Yue, P.; Khuri, F.R.; Sun, S.-Y. Inhibition of mTOR complex 2 induces GSK3/FBXW7-dependent degradation of sterol regulatory element-binding protein 1 (SREBP1) and suppresses lipogenesis in cancer cells. Oncogene 2016, 35, 642–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, C.-H.; Bellon, M.; Pancewicz-Wojtkiewicz, J.; Nicot, C. Oncogenic mutations in the FBXW7 gene of adult T-cell leukemia patients. Proc. Natl. Acad. Sci. USA 2016, 113, 6731–6736. [Google Scholar] [CrossRef] [Green Version]
- Iwatsuki, M.; Mimori, K.; Ishii, H.; Yokobori, T.; Takatsuno, Y.; Sato, T.; Toh, H.; Onoyama, I.; Nakayama, K.I.; Baba, H.; et al. Loss of FBXW7, a cell cycle regulating gene, in colorectal cancer: Clinical significance. Int. J. Cancer 2010, 126, 1828–1837. [Google Scholar] [CrossRef]
- Mori, M.; Yokobori, T.; Mimori, K.; Iwatsuki, M.; Ishii, H.; Tanaka, F.; Sato, T.; Toh, H.; Sudo, T.; Iwaya, T.; et al. Copy number loss of FBXW7 is related to gene expression and poor prognosis in esophageal squamous cell carcinoma. Int. J. Oncol. 2012, 41, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Wertz, I.E.; Kusam, S.; Lam, C.; Okamoto, T.; Sandoval, W.; Anderson, D.J.; Helgason, E.; Ernst, J.A.; Eby, M.; Liu, J.; et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011, 471, 110–114. [Google Scholar] [CrossRef]
- Wang, W.; Wang, X.; Rajaei, M.; Youn, J.Y.; Zafar, A.; Deokar, H.; Buolamwini, J.K.; Yang, J.; Foster, J.H.; Zhou, J.; et al. Targeting MDM2 for Neuroblastoma Therapy: In Vitro and In Vivo Anticancer Activity and Mechanism of Action. Cancers 2020, 12, 3651. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, J.; Yuan, X.; Yang, S.; Xu, X.; Li, K.; He, Y.; Wei, L.; Zhang, J.; Tian, Y. IU1 suppresses proliferation of cervical cancer cells through MDM2 degradation. Int. J. Biol. Sci. 2020, 16, 2951–2963. [Google Scholar] [CrossRef]
- Wang, W.; Hu, B.; Qin, J.-J.; Cheng, J.-W.; Li, X.; Rajaei, M.; Fan, J.; Yang, X.-R.; Zhang, R. A novel inhibitor of MDM2 oncogene blocks metastasis of hepatocellular carcinoma and overcomes chemoresistance. Genes Dis. 2019, 6, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Castillo, V.; Sliva, D. CDC20 associated with cancer metastasis and novel mushroom-derived CDC20 inhibitors with antimetastatic activity. Int. J. Oncol. 2019, 54, 2250–2256. [Google Scholar] [CrossRef]
- Han, T.; Jiang, S.; Zheng, H.; Yin, Q.; Xie, M.; Little, M.R.; Yin, X.; Chen, M.; Song, S.J.; Beg, A.A.; et al. Interplay between c-Src and the APC/C co-activator Cdh1 regulates mammary tumorigenesis. Nat. Commun. 2019, 10, 3716. [Google Scholar] [CrossRef]
- Zhong, J.; Shaik, S.; Wan, L.; Tron, A.E.; Wang, Z.; Sun, L.; Inuzuka, H.; Wei, W. SCFβ-TRCP targets MTSS1 for ubiquitination-mediated destruction to regulate cancer cell proliferation and migration. Oncotarget 2013, 4, 2339–2353. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Lu, Y.; Zhang, S.; Li, Y.; Huang, J.; Yin, Z.; Ren, J.; Huang, K.; Liu, L.; Yang, K.; et al. β-Trcp ubiquitin ligase and RSK2 kinase-mediated degradation of FOXN2 promotes tumorigenesis and radioresistance in lung cancer. Cell Death Differ. 2018, 25, 1473–1485. [Google Scholar] [CrossRef]
- Shaik, S.; Nucera, C.; Inuzuka, H.; Gao, D.; Garnaas, M.; Frechette, G.; Harris, L.; Wan, L.; Fukushima, H.; Husain, A.; et al. SCFβ-TRCP suppresses angiogenesis and thyroid cancer cell migration by promoting ubiquitination and destruction of VEGF receptor 2. J. Exp. Med. 2012, 209, 1289–1307. [Google Scholar] [CrossRef]
- Paul, P.J.; Raghu, D.; Chan, A.-L.; Gulati, T.; Lambeth, L.; Takano, E.; Herold, M.J.; Hagekyriakou, J.; Vessella, R.L.; Fedele, C.; et al. Restoration of tumor suppression in prostate cancer by targeting the E3 ligase E6AP. Oncogene 2016, 35, 6235–6245. [Google Scholar] [CrossRef] [PubMed]
- Raghu, D.; Paul, P.J.; Gulati, T.; Deb, S.; Khoo, C.; Russo, A.; Gallo, E.; Blandino, G.; Chan, A.-L.; Takano, E.; et al. E6AP promotes prostate cancer by reducing p27 expression. Oncotarget 2017, 8, 42939–42948. [Google Scholar] [CrossRef] [PubMed]
- Gamell, C.; Bandilovska, I.; Gulati, T.; Kogan, A.; Lim, S.C.; Kovacevic, Z.; Takano, E.A.; Timpone, C.; Agupitan, A.D.; Litchfield, C.; et al. E6AP Promotes a Metastatic Phenotype in Prostate Cancer. iScience 2019, 22, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Taranets, L.; Popov, N. Regulating Fbw7 on the road to cancer. Semin. Cancer Biol. 2016, 36, 62–70. [Google Scholar] [CrossRef]
- Yumimoto, K.; Nakayama, K.I. Recent insight into the role of FBXW7 as a tumor suppressor. Semin. Cancer Biol. 2020, 67 Pt 2, 1–15. [Google Scholar] [CrossRef]
- Yada, M.; Hatakeyama, S.; Kamura, T.; Nishiyama, M.; Tsunematsu, R.; Imaki, H.; Ishida, N.; Okumura, F.; Nakayama, K.; Nakayama, K.I. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004, 23, 2116–2125. [Google Scholar] [CrossRef] [Green Version]
- Imura, S.; Tovuu, L.-O.; Utsunomiya, T.; Morine, Y.; Ikemoto, T.; Arakawa, Y.; Kanamoto, M.; Iwahashi, S.; Saito, Y.; Takasu, C.; et al. The role of Fbxw7 expression in hepatocellular carcinoma and adjacent non-tumor liver tissue. J. Gastroenterol. Hepatol. 2014, 29, 1822–1829. [Google Scholar] [CrossRef]
- Welcker, M.; Clurman, B.E. FBW7 ubiquitin ligase: A tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 2008, 8, 83–93. [Google Scholar] [CrossRef]
- Akhoondi, S.; Sun, D.; Von Der Lehr, N.; Apostolidou, S.; Klotz, K.; Maljukova, A.; Cepeda, D.; Fiegl, H.; Dofou, D.; Marth, C.; et al. FBXW7/hCDC4 Is a General Tumor Suppressor in Human Cancer. Cancer Res. 2007, 67, 9006–9012. [Google Scholar] [CrossRef] [Green Version]
- DeVine, T.; Dai, M.S. Targeting the Ubiquitin-Mediated Proteasome Degradation of p53 for Cancer Therapy. Curr. Pharm. Des. 2013, 19, 3248–3262. [Google Scholar] [CrossRef] [Green Version]
- Wade, M.; Li, Y.-C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaikh, M.F.; Morano, W.F.; Lee, J.; Gleeson, E.; Babcock, B.D.; Michl, J.; Sarafraz-Yazdi, E.; Pincus, M.R.; Bowne, W.B. Emerging Role of MDM2 as Target for Anti-Cancer Therapy: A Review. Ann. Clin. Lab. Sci. 2016, 46, 627–634. [Google Scholar] [PubMed]
- Yang, L.; Song, T.; Cheng, Q.; Chen, L.; Chen, J. Mutant p53 Sequestration of the MDM2 Acidic Domain Inhibits E3 Ligase Activity. Mol. Cell. Biol. 2019, 39, e00375-18. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-E.; Koay, T.S.; Ho, Y.-H.; Lovat, P.; Lunec, J. TP53 mutant cell lines selected for resistance to MDM2 inhibitors retain growth inhibition by MAPK pathway inhibitors but a reduced apoptotic response. Cancer Cell Int. 2019, 19, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riscal, R.; Schrepfer, E.; Arena, G.; Cissé, M.Y.; Bellvert, F.; Heuillet, M.; Rambow, F.; Bonneil, E.; Sabourdy, F.; Vincent, C.; et al. Chromatin-Bound MDM2 Regulates Serine Metabolism and Redox Homeostasis Independently of p53. Mol. Cell 2016, 62, 890–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfarsi, L.H.; El Ansari, R.; Craze, M.L.; Toss, M.S.; Masisi, B.; Ellis, I.O.; Rakha, E.A.; Green, A.R. CDC20 expression in oestrogen receptor positive breast cancer predicts poor prognosis and lack of response to endocrine therapy. Breast Cancer Res. Treat. 2019, 178, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Schrock, M.S.; Stromberg, B.R.; Scarberry, L.; Summers, M.K. APC/C ubiquitin ligase: Functions and mechanisms in tumorigenesis. Semin. Cancer Biol. 2020, 67 Pt 2, 80–91. [Google Scholar] [CrossRef]
- Beaudenon, S.; Huibregtse, J.M. HPV E6, E6AP and cervical cancer. BMC Biochem. 2008, 9 (Suppl. S1), S4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amerik, A.Y.; Hochstrasser, M. Mechanism and function of deubiquitinating enzymes. Biochim. Biophys. Acta 2004, 1695, 189–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glickman, M.H.; Ciechanover, A. The Ubiquitin-Proteasome Proteolytic Pathway: Destruction for the Sake of Construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Schnell, J.D.; Hicke, L. Non-traditional Functions of Ubiquitin and Ubiquitin-binding Proteins. J. Biol. Chem. 2003, 278, 35857–35860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, D.; Riezman, H. Proteasome-Independent Functions of Ubiquitin in Endocytosis and Signaling. Science 2007, 315, 201–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.; Li, P.; Li, M.; Li, W.; Yao, T.; Wu, J.-W.; Gu, W.; Cohen, R.E.; Shi, Y. Crystal Structure of a UBP-Family Deubiquitinating Enzyme in Isolation and in Complex with Ubiquitin Aldehyde. Cell 2002, 111, 1041–1054. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Li, P.; Song, L.; Jeffrey, P.D.; Chernova, T.A.; Wilkinson, K.D.; Cohen, R.E.; Shi, Y. Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO J. 2005, 24, 3747–3756. [Google Scholar] [CrossRef]
- Avvakumov, G.V.; Walker, J.R.; Xue, S.; Finerty, P.J.; MacKenzie, F.; Newman, E.M.; Dhe-Paganon, S. Amino-terminal Dimerization, NRDP1-Rhodanese Interaction, and Inhibited Catalytic Domain Conformation of the Ubiquitin-specific Protease 8 (USP8). J. Biol. Chem. 2006, 281, 38061–38070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renatus, M.; Gil Parrado, S.; D’Arcy, A.; Eidhoff, U.; Gerhartz, B.; Hassiepen, U.; Pierrat, B.; Riedl, R.; Vinzenz, D.; Worpenberg, S.; et al. Structural Basis of Ubiquitin Recognition by the Deubiquitinating Protease USP2. Structure 2006, 14, 1293–1302. [Google Scholar] [CrossRef] [Green Version]
- Ismail, I.H.; Davidson, R.; Gagné, J.-P.; Xu, Z.Z.; Poirier, G.G.; Hendzel, M.J. Germline Mutations in BAP1 Impair Its Function in DNA Double-Strand Break Repair. Cancer Res. 2014, 74, 4282–4294. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Pak, H.; Hammond-Martel, I.; Ghram, M.; Rodrigue, A.; Daou, S.; Barbour, H.; Corbeil, L.; Hébert, J.; Drobetsky, E.; et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 2014, 111, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-S.; Seo, H.-R.; Lee, S.-A.; Choi, S.; Kang, D.; Kwon, J. BAP1 promotes stalled fork restart and cell survival via INO80 in response to replication stress. Biochem. J. 2019, 476, 3053–3066. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, J.; Liu, X.; Feng, L.; Gong, Z.; Koppula, P.; Sirohi, K.; Li, X.; Wei, Y.; Lee, H.; et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat. Cell Biol. 2018, 20, 1181–1192. [Google Scholar] [CrossRef]
- Wang, L.; Guan, R.; Xie, L.; Liao, X.; Xiong, K.; Rees, T.W.; Chen, Y.; Ji, L.; Chao, H. An ER-Targeting Iridium(III) Complex That Induces Immunogenic Cell Death in Non-Small-Cell Lung Cancer. Angew. Chem. Int. Ed. 2021, 60, 4657–4665. [Google Scholar] [CrossRef] [PubMed]
- Van Loosdregt, J.; Coffer, P.J. Post-translational modification networks regulating FOXP3 function. Trends Immunol. 2014, 35, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, D.; Shiloh, A.; Luo, J.; Nikolaev, A.Y.; Qin, J.; Gu, W. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 2002, 416, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-Y.; Varthi, M.; Sykes, S.M.; Phillips, C.; Warzecha, C.; Zhu, W.; Wyce, A.; Thorne, A.W.; Berger, S.L.; McMahon, S.B. The Putative Cancer Stem Cell Marker USP22 Is a Subunit of the Human SAGA Complex Required for Activated Transcription and Cell-Cycle Progression. Mol. Cell 2008, 29, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Han, B.; Lu, H.; Zhao, Y.; Chen, X.; Meng, Q.; Cao, M.; Cai, L.; Hu, J. USP22 promotes resistance to EGFR-TKIs by preventing ubiquitination-mediated EGFR degradation in EGFR-mutant lung adenocarcinoma. Cancer Lett. 2018, 433, 186–198. [Google Scholar] [CrossRef]
- Lin, Z.; Tan, C.; Qiu, Q.; Kong, S.; Yang, H.; Zhao, F.; Liu, Z.; Li, J.; Kong, Q.; Gao, B.; et al. Ubiquitin-specific protease 22 is a deubiquitinase of CCNB1. Cell Discov. 2015, 1, 15028. [Google Scholar] [CrossRef]
- Zhou, A.; Lin, K.; Zhang, S.; Chen, Y.; Zhang, N.; Xue, J.; Wang, Z.; Aldape, K.D.; Xie, K.; Woodgett, J.R.; et al. Nuclear GSK3β promotes tumorigenesis by phosphorylating KDM1A and inducing its deubiquitylation by USP22. Nat. Cell Biol. 2016, 18, 954–966. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zou, L.; Zhou, D.; Zhou, Z.; Tang, F.; Xu, Z.; Liu, X. Overexpression of ubiquitin carboxyl terminal hydrolase-L1 enhances multidrug resistance and invasion/metastasis in breast cancer by activating the MAPK/Erk signaling pathway. Mol. Carcinog. 2015, 55, 1329–1342. [Google Scholar] [CrossRef]
- Singh, A.N.; Oehler, J.; Torrecilla, I.; Kilgas, S.; Li, S.; Vaz, B.; Guérillon, C.; Fielden, J.; Hernandez-Carralero, E.; Cabrera, E.; et al. The p97–Ataxin 3 complex regulates homeostasis of the DNA damage response E3 ubiquitin ligase RNF 8. EMBO J. 2019, 38, e102361. [Google Scholar] [CrossRef]
- Shi, Z.; Chen, J.; Zhang, X.; Chu, J.; Han, Z.; Xu, D.; Gan, S.; Pan, X.; Ye, J.; Cui, X. Ataxin-3 promotes testicular cancer cell proliferation by inhibiting anti-oncogene PTEN. Biochem. Biophys. Res. Commun. 2018, 503, 391–396. [Google Scholar] [CrossRef]
- Butler, L.R.; Densham, R.M.; Jia, J.; Garvin, A.J.; Stone, H.R.; Shah, V.; Weekes, D.; Festy, F.; Beesley, J.; Morris, J.R. The proteasomal de-ubiquitinating enzyme POH1 promotes the double-strand DNA break response. EMBO J. 2012, 31, 3918–3934. [Google Scholar] [CrossRef]
- Byrne, A.; McLaren, R.P.; Mason, P.; Chai, L.; Dufault, M.R.; Huang, Y.; Liang, B.; Gans, J.D.; Zhang, M.; Carter, K. Knockdown of human deubiquitinase PSMD14 induces cell cycle arrest and senescence. Exp. Cell Res. 2010, 316, 258–271. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Ma, A.; Zhang, L.; Jin, W.-L.; Qiang, X.; Xu, G.; Qiu, B.; Yang, Z.; Liu, Y.; Xia, Q.; et al. POH1 deubiquitylates and stabilizes E2F1 to promote tumour formation. Nat. Commun. 2015, 6, 8704. [Google Scholar] [CrossRef]
- Jin, Z.; Li, Y.; Pitti, R.; Lawrence, D.; Pham, V.C.; Lill, J.R.; Ashkenazi, A. Cullin3-Based Polyubiquitination and p62-Dependent Aggregation of Caspase-8 Mediate Extrinsic Apoptosis Signaling. Cell 2009, 137, 721–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-W.; Ramasamy, K.; Bouamar, H.; Lin, A.-P.; Jiang, D.; Aguiar, R.C.T. MicroRNAs miR-125a and miR-125b constitutively activate the NF- B pathway by targeting the tumor necrosis factor alpha-induced protein 3 (TNFAIP3, A20). Proc. Natl. Acad. Sci. USA 2012, 109, 7865–7870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balkhi, M.Y.; Iwenofu, O.H.; Bakkar, N.; Ladner, K.J.; Chandler, D.S.; Houghton, P.J.; London, C.A.; Kraybill, W.; Perrotti, D.; Croce, C.M.; et al. miR-29 Acts as a Decoy in Sarcomas to Protect the Tumor Suppressor A20 mRNA from Degradation by HuR. Sci. Signal. 2013, 6, ra63. [Google Scholar] [CrossRef] [Green Version]
- Jensen, D.E.; Proctor, M.; Marquis, S.T.; Gardner, H.P.; Ha, S.I.; Chodosh, L.A.; Ishov, A.M.; Tommerup, N.; Vissing, H.; Sekido, Y.; et al. BAP1: A novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene 1998, 16, 1097–1112. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, H.; Wu, W.; Koike, A.; Kojima, R.; Gomi, H.; Fukuda, M.; Ohta, T. BRCA1-Associated Protein 1 Interferes with BRCA1/BARD1 RING Heterodimer Activity. Cancer Res. 2009, 69, 111–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauri, S.; Comoglio, F.; Seimiya, M.; Gerstung, M.; Glatter, T.; Hansen, K.; Aebersold, R.; Paro, R.; Gstaiger, M.; Beisel, C. A High-Density Map for Navigating the Human Polycomb Complexome. Cell Rep. 2016, 17, 583–595. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-S.; Lee, S.-A.; Hur, S.-K.; Seo, J.-W.; Kwon, J. Stabilization and targeting of INO80 to replication forks by BAP1 during normal DNA synthesis. Nat. Commun. 2014, 5, 5128. [Google Scholar] [CrossRef] [Green Version]
- Machida, Y.J.; Machida, Y.; Vashisht, A.A.; Wohlschlegel, J.A.; Dutta, A. The Deubiquitinating Enzyme BAP1 Regulates Cell Growth via Interaction with HCF-1. J. Biol. Chem. 2009, 284, 34179–34188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Mashtalir, N.; Daou, S.; Hammond-Martel, I.; Ross, J.; Sui, G.; Hart, G.W.; Rauscher, F.J.; Drobetsky, E.; Milot, E.; et al. The Ubiquitin Carboxyl Hydrolase BAP1 Forms a Ternary Complex with YY1 and HCF-1 and Is a Critical Regulator of Gene Expression. Mol. Cell. Biol. 2010, 30, 5071–5085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Baughman, J.M.; Rose, C.M.; Kolumam, G.; Webster, J.D.; Wilkerson, E.M.; Merrill, A.E.; Rhoads, T.W.; Noubade, R.; Katavolos, P.; Lesch, J.; et al. NeuCode Proteomics Reveals Bap1 Regulation of Metabolism. Cell Rep. 2016, 16, 583–595. [Google Scholar] [CrossRef] [Green Version]
- Ruan, H.-B.; Han, X.; Li, M.-D.; Singh, J.P.; Qian, K.; Azarhoush, S.; Zhao, L.; Bennett, A.M.; Samuel, V.T.; Wu, J.; et al. O-GlcNAc Transferase/Host Cell Factor C1 Complex Regulates Gluconeogenesis by Modulating PGC-1α Stability. Cell Metab. 2012, 16, 226–237. [Google Scholar] [CrossRef] [Green Version]
- Carbone, M.; Adusumilli, P.S.; Alexander, H.R., Jr.; Baas, P.; Bardelli, F.; Bononi, A.; Bueno, R.; Felley-Bosco, E.; Galateau-Salle, F.; Jablons, D.; et al. Mesothelioma: Scientific clues for prevention, diagnosis, and therapy. CA Cancer J. Clin. 2019, 69, 402–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, K.R.; Pilarski, R.; Cebulla, C.M.; Abdelrahman, M.H. Comprehensive review ofBAP1tumor predisposition syndrome with report of two new cases. Clin. Genet. 2016, 89, 285–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kittaneh, M.; Berkelhammer, C. Detecting germline BAP1 mutations in patients with peritoneal mesothelioma: Benefits to patient and family members. J. Transl. Med. 2018, 16, 194. [Google Scholar] [CrossRef] [Green Version]
- Panou, V.; Gadiraju, M.; Wolin, A.; Weipert, C.M.; Skarda, E.; Husain, A.N.; Patel, J.D.; Rose, B.; Zhang, S.R.; Weatherly, M.; et al. Frequency of Germline Mutations in Cancer Susceptibility Genes in Malignant Mesothelioma. J. Clin. Oncol. 2018, 36, 2863–2871. [Google Scholar] [CrossRef] [PubMed]
- Kobrinski, D.A.; Yang, H.; Kittaneh, M. BAP1: Role in carcinogenesis and clinical implications. Transl. Lung Cancer Res. 2020, 9 (Suppl. S1), S60–S66. [Google Scholar] [CrossRef]
- Yoshikawa, Y.; Emi, M.; Nakano, T.; Gaudino, G. Mesothelioma developing in carriers of inherited genetic mutations. Transl. Lung Cancer Res. 2020, 9 (Suppl. S1), S67–S76. [Google Scholar] [CrossRef]
- Kuznetsov, J.N.; Aguero, T.H.; Owens, D.A.; Kurtenbach, S.; Field, M.G.; Durante, M.A.; Rodriguez, D.A.; King, M.L.; Harbour, J.W. BAP1 regulates epigenetic switch from pluripotency to differentiation in developmental lineages giving rise to BAP1-mutant cancers. Sci. Adv. 2019, 5, eaax1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.O.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent Mutation of BAP1 in Metastasizing Uveal Melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Njauw, C.-N.J.; Kim, I.; Piris, A.; Gabree, M.; Taylor, M.; Lane, A.M.; DeAngelis, M.M.; Gragoudas, E.; Duncan, L.M.; Tsao, H. Germline BAP1 Inactivation Is Preferentially Associated with Metastatic Ocular Melanoma and Cutaneous-Ocular Melanoma Families. PLoS ONE 2012, 7, e35295. [Google Scholar] [CrossRef] [Green Version]
- Peña-Llopis, S.; Vega-Rubín-De-Celis, S.; Liao, A.; Leng, N.; Pavía-Jiménez, A.; Wang, S.; Yamasaki, T.; Zhrebker, L.; Sivanand, S.; Spence, P.; et al. BAP1 loss defines a new class of renal cell carcinoma. Nat. Genet. 2012, 44, 751–759. [Google Scholar] [CrossRef]
- Popova, T.; Hebert, L.; Jacquemin, V.; Gad, S.; Caux-Moncoutier, V.; Dubois-D’Enghien, C.; Richaudeau, B.; Renaudin, X.; Sellers, J.; Nicolas, A.; et al. Germline BAP1 Mutations Predispose to Renal Cell Carcinomas. Am. J. Hum. Genet. 2013, 92, 974–980. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Zhang, L.; Hildebrandt, M.A.T.; Huang, M.; Wu, X.; Ye, Y. Common, germline genetic variations in the novel tumor suppressor BAP1 and risk of developing different types of cancer. Oncotarget 2017, 8, 74936–74946. [Google Scholar] [CrossRef] [Green Version]
- Carbone, M.; Yang, H.; Pass, H.I.; Krausz, T.; Testa, J.R.; Gaudino, G. BAP1 and cancer. Nat. Rev. Cancer 2013, 13, 153–159. [Google Scholar] [CrossRef]
- Testa, J.R.; Cheung, M.; Pei, J.; Below, J.E.; Tan, Y.; Sementino, E.; Cox, N.J.; Dogan, A.U.; Pass, H.I.; Trusa, S.; et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat. Genet. 2011, 43, 1022–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, M.; Ferris, L.K.; Baumann, F.; Napolitano, A.; Lum, C.A.; Flores, E.G.; Gaudino, G.; Powers, A.; Bryant-Greenwood, P.; Krausz, T.; et al. BAP1 cancer syndrome: Malignant mesothelioma, uveal and cutaneous melanoma, and MBAITs. J. Transl. Med. 2012, 10, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everett, R.D.; Meredith, M.; Orr, A.; Cross, A.; Kathoria, M.; Parkinson, J. A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J. 1997, 16, 1519–1530. [Google Scholar] [CrossRef] [Green Version]
- Holowaty, M.N.; Zeghouf, M.; Wu, H.; Tellam, J.; Athanasopoulos, V.; Greenblatt, J.; Frappier, L. Protein Profiling with Epstein-Barr Nuclear Antigen-1 Reveals an Interaction with the Herpesvirus-associated Ubiquitin-specific Protease HAUSP/USP7. J. Biol. Chem. 2003, 278, 29987–29994. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Orr, A.; Everett, R.D. Stimulation of the Replication of ICP0-Null Mutant Herpes Simplex Virus 1 and pp71-Deficient Human Cytomegalovirus by Epstein-Barr Virus Tegument Protein BNRF1. J. Virol. 2016, 90, 9664–9673. [Google Scholar] [CrossRef] [Green Version]
- Everett, R.D. HSV-1 Biology and Life Cycle. Methods Mol. Biol. 2014, 1144, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Laurence, A.; Belkaid, Y.; O’Shea, J.J. A Degrading View of Regulatory T Cells. Immunity 2013, 39, 201–203. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Guo, C.; Ji, T.; Chen, X. SOX2 Regulates lncRNA CCAT1/MicroRNA-185-3p/FOXP3 Axis to Affect the Proliferation and Self-Renewal of Cervical Cancer Stem Cells. Nanoscale Res. Lett. 2021, 16, 2. [Google Scholar] [CrossRef]
- Cai, J.; Chen, H.; Peng, S.; Meng, J.; Wang, Y.; Zhou, Y.; Qian, X.; Sun, X.; Pang, X.; Zhang, Y.; et al. USP7-TRIM27 axis negatively modulates antiviral type I IFN signaling. FASEB J. 2018, 32, 5238–5249. [Google Scholar] [CrossRef] [Green Version]
- Oliner, J.D.; Kinzler, K.W.; Meltzer, P.S.; George, D.L.; Vogelstein, B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 1992, 358, 80–83. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Gu, L.; Li, M.; Jeffrey, P.D.; Gu, W.; Shi, Y. Structural Basis of Competitive Recognition of p53 and MDM2 by HAUSP/USP7: Implications for the Regulation of the p53–MDM2 Pathway. PLoS Biol. 2006, 4, e27. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zang, H.; Luo, Y.; Wu, J.; Fang, Z.; Zhu, W.; Li, Y. High expression of USP22 predicts poor prognosis and advanced clinicopathological features in solid tumors: A meta-analysis. OncoTargets Ther. 2018, 11, 3035–3046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, I.N.; Thompson, R.J. UCHL1 (PGP 9.5): Neuronal biomarker and ubiquitin system protein. Prog. Neurobiol. 2010, 90, 327–362. [Google Scholar] [CrossRef] [PubMed]
- Tezel, E.; Hibi, K.; Nagasaka, T.; Nakao, A. PGP9.5 as a prognostic factor in pancreatic cancer. Clin. Cancer Res. 2000, 6, 4764–4767. [Google Scholar] [PubMed]
- Otsuki, T.; Yata, K.; Takata-Tomokuni, A.; Hyodoh, F.; Miura, Y.; Sakaguchi, H.; Hatayama, T.; Hatada, S.; Tsujioka, T.; Sato, Y.; et al. Expression of protein gene product 9·5 (PGP9·5)/ubiquitin-C-terminal hydrolase 1 (UCHL-1) in human myeloma cells. Br. J. Haematol. 2004, 127, 292–298. [Google Scholar] [CrossRef]
- Leiblich, A.; Cross, S.S.; Catto, J.W.; Pesce, G.; Hamdy, F.C.; Rehman, I. Human prostate cancer cells express neuroendocrine cell markers PGP 9.5 and chromogranin A. Prostate 2007, 67, 1761–1769. [Google Scholar] [CrossRef]
- Liu, X.; Zeng, B.; Ma, J.; Wan, C. Comparative Proteomic Analysis of Osteosarcoma Cell and Human Primary Cultured Osteoblastic Cell. Cancer Investig. 2009, 27, 345–352. [Google Scholar] [CrossRef]
- Hibi, K.; Westra, W.H.; Borges, M.; Goodman, S.; Sidransky, D.; Jen, J. PGP9.5 As a Candidate Tumor Marker for Non-Small-Cell Lung Cancer. Am. J. Pathol. 1999, 155, 711–715. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Magesh, V.; Lee, J.-J.; Kim, S.; Knaus, U.G.; Lee, K.-J. Ubiquitin C-terminal hydrolase-L1 increases cancer cell invasion by modulating hydrogen peroxide generated via NADPH oxidase 4. Oncotarget 2015, 6, 16287–16303. [Google Scholar] [CrossRef]
- Brichory, F.; Beer, D.; Le Naour, F.; Giordano, T.; Hanash, S. Proteomics-based identification of protein gene product 9.5 as a tumor antigen that induces a humoral immune response in lung cancer. Cancer Res. 2001, 61, 7908–7912. [Google Scholar] [PubMed]
- Nicastro, G.; Menon, R.P.; Masino, L.; Knowles, P.P.; McDonald, N.Q.; Pastore, A. The solution structure of the Josephin domain of ataxin-3: Structural determinants for molecular recognition. Proc. Natl. Acad. Sci. USA 2005, 102, 10493–10498. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Senic-Matuglia, F.; Di Fiore, P.P.; Polo, S.; Hodsdon, M.E.; De Camilli, P. Deubiquitinating function of ataxin-3: Insights from the solution structure of the Josephin domain. Proc. Natl. Acad. Sci. USA 2005, 102, 12700–12705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnett, B.; Li, F.; Pittman, R.N. The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum. Mol. Genet. 2003, 12, 3195–3205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakarougkas, A.; Ismail, A.; Katsuki, Y.; Freire, R.; Shibata, A.; Jeggo, P.A. Co-operation of BRCA1 and POH1 relieves the barriers posed by 53BP1 and RAP80 to resection. Nucleic Acids Res. 2013, 41, 10298–10311. [Google Scholar] [CrossRef]
- Stitzel, M.L.; Durso, R.; Reese, J.C. The proteasome regulates the UV-induced activation of the AP-1-like transcription factor Gcn4. Genes Dev. 2001, 15, 128–133. [Google Scholar] [CrossRef] [Green Version]
- Opipari, A.W.; Hu, H.M.; Yabkowitz, R.; Dixit, V.M. The A20 zinc finger protein protects cells from tumor necrosis factor cytotoxicity. J. Biol. Chem. 1992, 267, 12424–12427. [Google Scholar] [CrossRef] [PubMed]
- He, K.-L.; Ting, A.T. A20 Inhibits Tumor Necrosis Factor (TNF) Alpha-Induced Apoptosis by Disrupting Recruitment of TRADD and RIP to the TNF Receptor 1 Complex in Jurkat T Cells. Mol. Cell. Biol. 2002, 22, 6034–6045. [Google Scholar] [CrossRef] [Green Version]
- Chanudet, E.; Huang, Y.; Ichimura, K.; Dong, G.; Hamoudi, R.A.; Radford, J.; Wotherspoon, A.C.; Isaacson, P.G.; Ferry, J.; Du, M.-Q. A20 is targeted by promoter methylation, deletion and inactivating mutation in MALT lymphoma. Leukemia 2010, 24, 483–487. [Google Scholar] [CrossRef]
- Novak, U.; Rinaldi, A.; Kwee, I.; Nandula, S.V.; Rancoita, P.M.V.; Compagno, M.; Cerri, M.; Rossi, D.; Murty, V.V.; Zucca, E.; et al. The NF-κB negative regulator TNFAIP3 (A20) is inactivated by somatic mutations and genomic deletions in marginal zone lymphomas. Blood 2009, 113, 4918–4921. [Google Scholar] [CrossRef]
- Hochstrasser, M. Origin and function of ubiquitin-like proteins. Nature 2009, 458, 422–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulman, B.A.; Harper, J.W. Ubiquitin-like protein activation by E1 enzymes: The apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 2009, 10, 319–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barghout, S.H.; Schimmer, A.D. E1 Enzymes as Therapeutic Targets in Cancer. Pharmacol. Rev. 2021, 73, 1–56. [Google Scholar] [CrossRef] [PubMed]
- Groen, E.J.; Gillingwater, T.H. UBA1: At the Crossroads of Ubiquitin Homeostasis and Neurodegeneration. Trends Mol. Med. 2015, 21, 622–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of Cancer Therapy: Oncogene and Non-oncogene Addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Dobbelstein, M.; Moll, U. Targeting tumour-supportive cellular machineries in anticancer drug development. Nat. Rev. Drug Discov. 2014, 13, 179–196. [Google Scholar] [CrossRef]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nat. Cell Biol. 2009, 458, 732–736. [Google Scholar] [CrossRef]
- Hyer, M.L.; Milhollen, M.A.; Ciavarri, J.; Fleming, P.; Traore, T.; Sappal, D.; Huck, J.; Shi, J.; Gavin, J.; Brownell, J.; et al. A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat. Med. 2018, 24, 186–193. [Google Scholar] [CrossRef]
- Duda, D.M.; Borg, L.A.; Scott, D.C.; Hunt, H.W.; Hammel, M.; Schulman, B.A. Structural Insights into NEDD8 Activation of Cullin-RING Ligases: Conformational Control of Conjugation. Cell 2008, 134, 995–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osaka, F.; Saeki, M.; Katayama, S.; Aida, N.; Toh-E, A.; Kominami, K.; Toda, T.; Suzuki, T.; Chiba, T.; Tanaka, K.; et al. Covalent modifier NEDD8 is essential for SCF ubiquitin-ligase in fission yeast. EMBO J. 2000, 19, 3475–3484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toth, J.I.; Yang, L.; Dahl, R.; Petroski, M.D. A Gatekeeper Residue for NEDD8-Activating Enzyme Inhibition by MLN4924. Cell Rep. 2012, 1, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Tisdale, M.J. Cachexia in cancer patients. Nat. Rev. Cancer 2002, 2, 862–871. [Google Scholar] [CrossRef]
- Adams, J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell 2004, 5, 417–421. [Google Scholar] [CrossRef] [Green Version]
- Hideshima, T.; Richardson, P.; Chauhan, D.; Palombella, V.J.; Elliott, P.J.; Adams, J.; Anderson, K.C. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001, 61, 3071–3076. [Google Scholar] [PubMed]
- Chauhan, D.; Hideshima, T.; Mitsiades, C.; Richardson, P.; Anderson, K.C. Proteasome inhibitor therapy in multiple myeloma. Mol. Cancer Ther. 2005, 4, 686–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hideshima, T.; Mitsiades, C.; Akiyama, M.; Hayashi, T.; Chauhan, D.; Richardson, P.; Schlossman, R.; Podar, K.; Munshi, N.C.; Mitsiades, N.; et al. Molecular mechanisms mediating antimyeloma activity of proteasome inhibitor PS-341. Blood 2003, 101, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.-T.; Agrawal, S.G.; Gribben, J.G.; Ye, H.; Du, M.-Q.; Newland, A.C.; Jia, L. Bortezomib blocks Bax degradation in malignant B cells during treatment with TRAIL. Blood 2008, 111, 2797–2805. [Google Scholar] [CrossRef] [Green Version]
- Pei, X.-Y.; Dai, Y.; Grant, S. Synergistic Induction of Oxidative Injury and Apoptosis in Human Multiple Myeloma Cells by the Proteasome Inhibitor Bortezomib and Histone Deacetylase Inhibitors. Clin. Cancer Res. 2004, 10, 3839–3852. [Google Scholar] [CrossRef] [Green Version]
- Mata-Cantero, L.; Lobato-Gil, S.; Aillet, F.; Lang, V.; Rodriguez, M.S. The Ubiquitin-Proteasome System (UPS) as a Cancer Drug Target: Emerging Mechanisms and Therapeutics. In Stress Response Pathways in Cancer; Wondrak, G., Ed.; Springer: Dordrecht, The Netherlands, 2015; pp. 225–264. [Google Scholar]
- Zhang, X.; Linder, S.; Bazzaro, M. Drug Development Targeting the Ubiquitin–Proteasome System (UPS) for the Treatment of Human Cancers. Cancers 2020, 12, 902. [Google Scholar] [CrossRef] [Green Version]
- Kortuem, K.M.; Stewart, A.K. Carfilzomib. Blood 2013, 121, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Mohan, R.; Kwok, B.H.B.; Elofsson, M.; Sin, N.; Crews, C.M. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc. Nat. Acad. Sci. USA 1999, 96, 10403–10408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demo, S.D.; Kirk, C.J.; Aujay, M.A.; Buchholz, T.J.; Dajee, M.; Ho, M.N.; Jiang, J.; Laidig, G.J.; Lewis, E.R.; Parlati, F.; et al. Antitumor Activity of PR-171, a Novel Irreversible Inhibitor of the Proteasome. Cancer Res. 2007, 67, 6383–6391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arastu-Kapur, S.; Anderl, J.L.; Kraus, M.; Parlati, F.; Shenk, K.D.; Lee, S.J.; Muchamuel, T.; Bennett, M.K.; Driessen, C.; Ball, A.J.; et al. Nonproteasomal Targets of the Proteasome Inhibitors Bortezomib and Carfilzomib: A Link to Clinical Adverse Events. Clin. Cancer Res. 2011, 17, 2734–2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, A.K.; Rajkumar, S.V.; Dimopoulos, M.A.; Masszi, T.; Špička, I.; Oriol, A.; Hájek, R.; Rosiñol, L.; Siegel, D.S.; Mihaylov, G.G.; et al. Carfilzomib, Lenalidomide, and Dexamethasone for Relapsed Multiple Myeloma. N. Engl. J. Med. 2015, 372, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Tian, Z.; Zhou, B.; Kuhn, D.; Orlowski, R.; Raje, N.; Richardson, P.; Anderson, K.C. In Vitro and In Vivo Selective Antitumor Activity of a Novel Orally Bioavailable Proteasome Inhibitor MLN9708 against Multiple Myeloma Cells. Clin. Cancer Res. 2011, 17, 5311–5321. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.; Pan, Y.; Jeong, L.S.; Liu, J.; Jia, L. Inactivation of the Cullin (CUL)-RING E3 ligase by the NEDD8-activating enzyme inhibitor MLN4924 triggers protective autophagy in cancer cells. Autophagy 2012, 8, 1677–1679. [Google Scholar] [CrossRef]
- Aubry, A.; Yu, T.; Bremner, R. Preclinical studies reveal MLN4924 is a promising new retinoblastoma therapy. Cell Death Discov. 2020, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Swords, R.T.; Kelly, K.R.; Smith, P.G.; Garnsey, J.J.; Mahalingam, D.; Medina, E.; Oberheu, K.; Padmanabhan, S.; O’Dwyer, M.; Nawrocki, S.T.; et al. Inhibition of NEDD8-activating enzyme: A novel approach for the treatment of acute myeloid leukemia. Blood 2010, 115, 3796–3800. [Google Scholar] [CrossRef] [Green Version]
- Yao, W.-T.; Wu, J.-F.; Yu, G.-Y.; Wang, R.; Wang, K.; Li, L.-H.; Chen, P.; Jiang, Y.-N.; Cheng, H.; Lee, H.W.; et al. Suppression of tumor angiogenesis by targeting the protein neddylation pathway. Cell Death Dis. 2014, 5, e1059. [Google Scholar] [CrossRef] [Green Version]
- Pulvino, M.; Liang, Y.; Oleksyn, D.; DeRan, M.; Van Pelt, E.; Shapiro, J.; Sanz, I.; Chen, L.; Zhao, J. Inhibition of proliferation and sur- vival of diffuse large B-cell lymphoma cells by a small-molecule inhib-itor of the ubiquitin-conjugating enzyme Ubc13-Uev1A. Blood 2012, 120, 1668–1677. [Google Scholar] [CrossRef]
- Bauer, S.; Demetri, G.; Jeay, S.; Dummer, R.; Guerreiro, N.; Tan, D.; Kumar, A.; Meille, C.; Van Bree, L.; Halilovic, E.; et al. A phase I, open-label, multi-center, dose escalation study of oral NVP-CGM097, a p53/HDM2-protein-protein interaction inhibitor, in adult patients with selected advanced solid tumors. Ann. Oncol. 2016, 27 (Suppl. S6), vi114–vi135. [Google Scholar] [CrossRef] [Green Version]
- Holzer, P.; Masuya, K.; Furet, P.; Kallen, J.; Valat-Stachyra, T.; Ferretti, S.; Berghausen, J.; Bouisset-Leonard, M.; Buschmann, N.; Pissot-Soldermann, C.; et al. Discovery of a Dihydroisoquinolinone Derivative (NVP-CGM097): A Highly Potent and Selective MDM2 Inhibitor Undergoing Phase 1 Clinical Trials in p53wt Tumors. J. Med. Chem. 2015, 58, 6348–6358. [Google Scholar] [CrossRef]
- Bauer, T.; Hong, D.; Somaiah, N.; Cai, C.; Song, S.; Kumar, P.; Gajee, R.; Rosen, M.; Kochan, J.; Chen, S.; et al. Abstract B27: A phase I dose escalation study of the MDM2 inhibitor DS-3032b in patients with advanced solid tumors and lymphomas. Mol. Cancer Ther. 2015, 14 (Suppl. S2), Abstract nr B27. [Google Scholar] [CrossRef]
- Gounder, M.M.; Bauer, T.M.; Schwartz, G.K.; Masters, T.; Carvajal, R.D.; Song, S.; Kumar, P.; Gajee, R.; Zernovak, O.; Rosen, M.M.; et al. A phase 1 study of the MDM2 inhibitor DS-3032b in patients (pts) with advanced solid tumors and lymphomas. J. Clin. Oncol. 2016, 34 (Suppl. S15), 2581. [Google Scholar] [CrossRef]
- DiPersio, J.F.; Erba, H.P.; Larson, R.A.; Luger, S.M.; Tallman, M.S.; Brill, J.M.; Vuagniaux, G.; Rouits, E.; Sorensen, J.M.; Zanna, C. Oral Debio1143 (AT406), an Antagonist of Inhibitor of Apoptosis Proteins, Combined With Daunorubicin and Cytarabine in Patients With Poor-Risk Acute Myeloid Leukemia—Results of a Phase I Dose-Escalation Study. Clin. Lymphoma Myeloma Leuk. 2015, 15, 443–449. [Google Scholar] [CrossRef] [Green Version]
- Infante, J.R.; Dees, E.C.; Olszanski, A.J.; Dhuria, S.V.; Sen, S.; Cameron, S.; Cohen, R.B. Phase I Dose-Escalation Study of LCL161, an Oral Inhibitor of Apoptosis Proteins Inhibitor, in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2014, 32, 3103–3110. [Google Scholar] [CrossRef] [PubMed]
- Schilder, R.J.; Albertella, M.; Strauss, J.; Sydvander, M.; Nair, S.M.; Urakpo, K.; Norin, S.; Öhd, J. A phase 1/2 study with birinapant in combination with pembrolizumab. J. Clin. Oncol. 2020, 36 (Suppl. S15), TPS3131. [Google Scholar] [CrossRef]
- Chen, J.; Dexheimer, T.S.; Ai, Y.; Liang, Q.; Villamil, M.A.; Inglese, J.; Maloney, D.J.; Jadhav, A.; Simeonov, A.; Zhuang, Z. Selective and Cell-Active Inhibitors of the USP1/ UAF1 Deubiquitinase Complex Reverse Cisplatin Resistance in Non-small Cell Lung Cancer Cells. Chem. Biol. 2011, 18, 1390–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rath, B.H.; Camphausen, K.; Tofilon, P.J. Glioblastoma radiosensitization by pimozide. Transl. Cancer Res. 2016, 5 (Suppl. S6), S1029–S1032. [Google Scholar] [CrossRef] [PubMed]
- Green, A.K.; Corty, R.W.; Wood, W.A.; Meeneghan, M.; Reeder-Hayes, K.E.; Basch, E.; Milowsky, M.I.; Dusetzina, S.B. Comparative Effectiveness of Mitoxantrone Plus Prednisone Versus Prednisone Alone in Metastatic Castrate-Resistant Prostate Cancer After Docetaxel Failure. Oncolologist 2015, 20, 516–522. [Google Scholar] [CrossRef] [Green Version]
- Abbi, K.K.; Rybka, W.; Ehmann, W.C.; Claxton, D.F. Phase I/II Study of Clofarabine, Etoposide, and Mitoxantrone in Patients with Refractory or Relapsed Acute Leukemia. Clin. Lymphoma Myeloma Leuk. 2015, 15, 41–46. [Google Scholar] [CrossRef]
- Gill, H.; Yim, R.; Pang, H.H.; Lee, P.; Chan, T.S.Y.; Hwang, Y.; Leung, G.M.K.; Ip, H.; Leung, R.Y.Y.; Yip, S.; et al. Clofarabine, cytarabine, and mitoxantrone in refractory/relapsed acute myeloid leukemia: High response rates and effective bridge to allogeneic hematopoietic stem cell transplantation. Cancer Med. 2020, 9, 3371–3382. [Google Scholar] [CrossRef] [Green Version]
- Cornbleet, M.A.; Stuart-Harris, R.C.; Smith, I.E.; Coleman, R.E.; Rubens, R.D.; McDonald, M.; Mouridsen, H.T.; Rainer, H.; Van Oosterom, A.T.; Smyth, J.F. Mitoxantrone for the treatment of advanced breast cancer: Single-agent therapy in previously untreated patients. Eur. J. Cancer Clin. Oncol. 1984, 20, 1141–1146. [Google Scholar] [CrossRef]
- Neidhart, J.A.; Gochnour, D.; Roach, R.; Hoth, D.; Young, D. A comparison of mitoxantrone and doxorubicin in breast cancer. J. Clin. Oncol. 1986, 4, 672–677. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Holmes, F.A.; Esparza, L.; Valero, V.; Buzdar, A.U.; Neidhart, J.A.; Hortobagyi, G.N. Phase I/II trial of high dose mitoxantrone in metastatic breast cancer: The M.D. Anderson Cancer Center experience. Breast Cancer Res. Treat. 1999, 54, 225–233. [Google Scholar] [CrossRef]
- Silver, R.T.; Case, D.C.; Wheeler, R.H.; Miller, T.P.; Stein, R.S.; Stuart, J.J.; Peterson, B.A.; Rivkin, S.E.; Golomb, H.M.; Costanzi. Multicenter clinical trial of mitoxantrone in non-Hodgkin’s lymphoma and Hodgkin’s disease. J. Clin. Oncol. 1991, 9, 754–761. [Google Scholar] [CrossRef]
- Bajetta, E.; Buzzoni, R.; Valagussa, P.; Bonadonna, G. Mitoxantrone: An active agent in refractory non-Hodgkin’s lymphomas. Am. J. Clin. Oncol. 1988, 11, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.B.; Van Echo, D.A.; Leone, L.A.; Henderson, E.S. Phase II trial of mitoxantrone in advanced primary liver cancer: A Cancer and Leukemia Group B Study. Cancer Treat. Rep. 1986, 70, 1125–1126. [Google Scholar] [PubMed]
- De Weger, V.A.; De Jonge, M.; Langenberg, M.H.G.; Schellens, J.H.M.; Lolkema, M.; Varga, A.; Demers, B.; Thomas, K.; Hsu, K.; Tuffal, G.; et al. A phase I study of the HDM2 antagonist SAR405838 combined with the MEK inhibitor pimasertib in patients with advanced solid tumours. Br. J. Cancer 2019, 120, 286–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feltham, R.; Bettjeman, B.; Budhidarmo, R.; Mace, P.D.; Shirley, S.; Condon, S.M.; Chunduru, S.K.; McKinlay, M.A.; Vaux, D.L.; Silke, J.; et al. Smac Mimetics Activate the E3 Ligase Activity of cIAP1 Protein by Promoting RING Domain Dimerization. J. Biol. Chem. 2011, 286, 17015–17028. [Google Scholar] [CrossRef] [Green Version]
- Dubrez, L.; Berthelet, J.; Glorian, V. IAP proteins as targets for drug development in oncology. Onco Targets Ther. 2013, 9, 1285–1304. [Google Scholar] [CrossRef] [Green Version]
- Allensworth, J.L.; Sauer, S.J.; Lyerly, H.K.; Morse, M.A.; Devi, G.R. Smac mimetic Birinapant induces apoptosis and enhances TRAIL potency in inflammatory breast cancer cells in an IAP-dependent and TNF-α-independent mechanism. Breast Cancer Res. Treat. 2013, 137, 359–371. [Google Scholar] [CrossRef]
- Cai, Q.; Sun, H.; Peng, Y.; Lu, J.; Nikolovska-Coleska, Z.; McEachern, D.; Liu, L.; Qiu, S.; Yang, C.-Y.; Miller, R.; et al. A Potent and Orally Active Antagonist (SM-406/AT-406) of Multiple Inhibitor of Apoptosis Proteins (IAPs) in Clinical Development for Cancer Treatment. J. Med. Chem. 2011, 54, 2714–2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flygare, J.A.; Beresini, M.; Budha, N.; Chan, H.; Chan, I.T.; Cheeti, S.; Cohen, F.; Deshayes, K.; Doerner, K.; Eckhardt, S.G.; et al. Discovery of a Potent Small-Molecule Antagonist of Inhibitor of Apoptosis (IAP) Proteins and Clinical Candidate for the Treatment of Cancer (GDC-0152). J. Med. Chem. 2012, 55, 4101–4113. [Google Scholar] [CrossRef]
- Varfolomeev, E.; Blankenship, J.W.; Wayson, S.M.; Fedorova, A.V.; Kayagaki, N.; Garg, P.; Zobel, K.; Dynek, J.N.; Elliott, L.O.; Wallweber, H.J.; et al. IAP Antagonists Induce Autoubiquitination of c-IAPs, NF-κB Activation, and TNFα-Dependent Apoptosis. Cell 2007, 131, 669–681. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Yang, K.; Dejsuphong, D.; Hirota, K.; Takeda, S.; D’Andrea, A.D. The USP1/UAF1 Complex Promotes Double-Strand Break Repair through Homologous Recombination. Mol. Cell. Biol. 2011, 31, 2462–2469. [Google Scholar] [CrossRef] [Green Version]
- Burkhart, R.A.; Peng, Y.; Norris, Z.A.; Tholey, R.M.; Talbott, V.A.; Liang, Q.; Ai, Y.; Miller, K.; Lal, S.; Cozzitorto, J.A.; et al. Mitoxantrone targets human ubiquitin-specific peptidase 11 (USP11) and is a potent inhibitor of pan-creatic cancer cell survival. Mol. Cancer Res. 2013, 11, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Schoenfeld, A.R.; Apgar, S.; Dolios, G.; Wang, R.; Aaronson, S.A. BRCA2 Is Ubiquitinated In Vivo and Interacts with USP11, a Deubiquitinating Enzyme That Exhibits Prosurvival Function in the Cellular Response to DNA Damage. Mol. Cell. Biol. 2004, 24, 7444–7455. [Google Scholar] [CrossRef] [Green Version]
- Jacko, A.M.; Nan, L.; Li, S.; Tan, J.; Zhao, J.; Kass, D.J.; Zhao, Y. De-ubiquitinating enzyme, USP11, promotes transforming growth factor β-1 signaling through stabilization of transforming growth factor β receptor II. Cell Death Dis. 2016, 7, e2474. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, D.; Tian, Z.; Nicholson, B.; Kumar, G.S.; Zhou, B.; Carrasco, R.; McDermott, J.L.; Leach, C.A.; Fulcinniti, M.; Kodrasov, M.P.; et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 2012, 22, 345–358. [Google Scholar] [CrossRef] [Green Version]
- D’Arcy, P.; Brnjic, S.; Olofsson, M.H.; Fryknäs, M.; Lindsten, K.; De Cesare, M.; Perego, P.; Sadeghi, B.; Hassan, M.; Larsson, R.; et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat. Med. 2011, 17, 1636–1640. [Google Scholar] [CrossRef]
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-Dependent and Independent Signals In Selective Autophagy. Trends Cell Biol. 2016, 26, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Duncan, K.; Schäfer, G.; Vava, A.; Parker, M.I.; Zerbini, L.F. Targeting neddylation in cancer therapy. Future Oncol. 2012, 8, 1461–1470. [Google Scholar] [CrossRef]
- Fouladkou, F.; Landry, T.; Kawabe, H.; Neeb, A.; Lu, C.; Brose, N.; Stambolic, V.; Rotin, D. The ubiquitin ligase Nedd4-1 is dispensable for the regulation of PTEN stability and localization. Proc. Natl. Acad. Sci. USA 2008, 105, 8585–8590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Trotman, L.C.; Koppie, T.; Alimonti, A.; Chen, Z.; Gao, Z.; Wang, J.; Erdjument-Bromage, H.; Tempst, P.; Cordon-Cardo, C.; et al. NEDD4-1 Is a Proto-Oncogenic Ubiquitin Ligase for PTEN. Cell 2007, 128, 129–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trotman, L.C.; Wang, X.; Alimonti, A.; Chen, Z.; Teruya-Feldstein, J.; Yang, H.; Pavletich, N.P.; Carver, B.S.; Cordon-Cardo, C.; Erdjument-Bromage, H.; et al. Ubiquitination Regulates PTEN Nuclear Import and Tumor Suppression. Cell 2007, 128, 141–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fhu, C.W.; Ali, A. Dysregulation of the Ubiquitin Proteasome System in Human Malignancies: A Window for Therapeutic Intervention. Cancers 2021, 13, 1513. https://doi.org/10.3390/cancers13071513
Fhu CW, Ali A. Dysregulation of the Ubiquitin Proteasome System in Human Malignancies: A Window for Therapeutic Intervention. Cancers. 2021; 13(7):1513. https://doi.org/10.3390/cancers13071513
Chicago/Turabian StyleFhu, Chee Wai, and Azhar Ali. 2021. "Dysregulation of the Ubiquitin Proteasome System in Human Malignancies: A Window for Therapeutic Intervention" Cancers 13, no. 7: 1513. https://doi.org/10.3390/cancers13071513