
Classification of Molecular Subtypes of High-Grade Serous Ovarian Cancer by MALDI-Imaging

 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
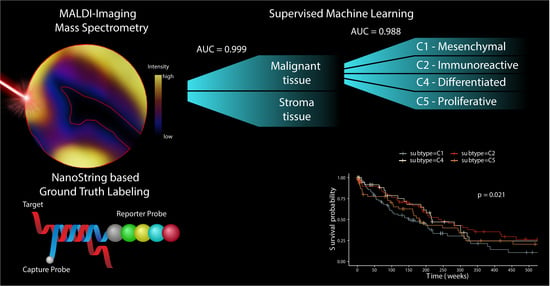
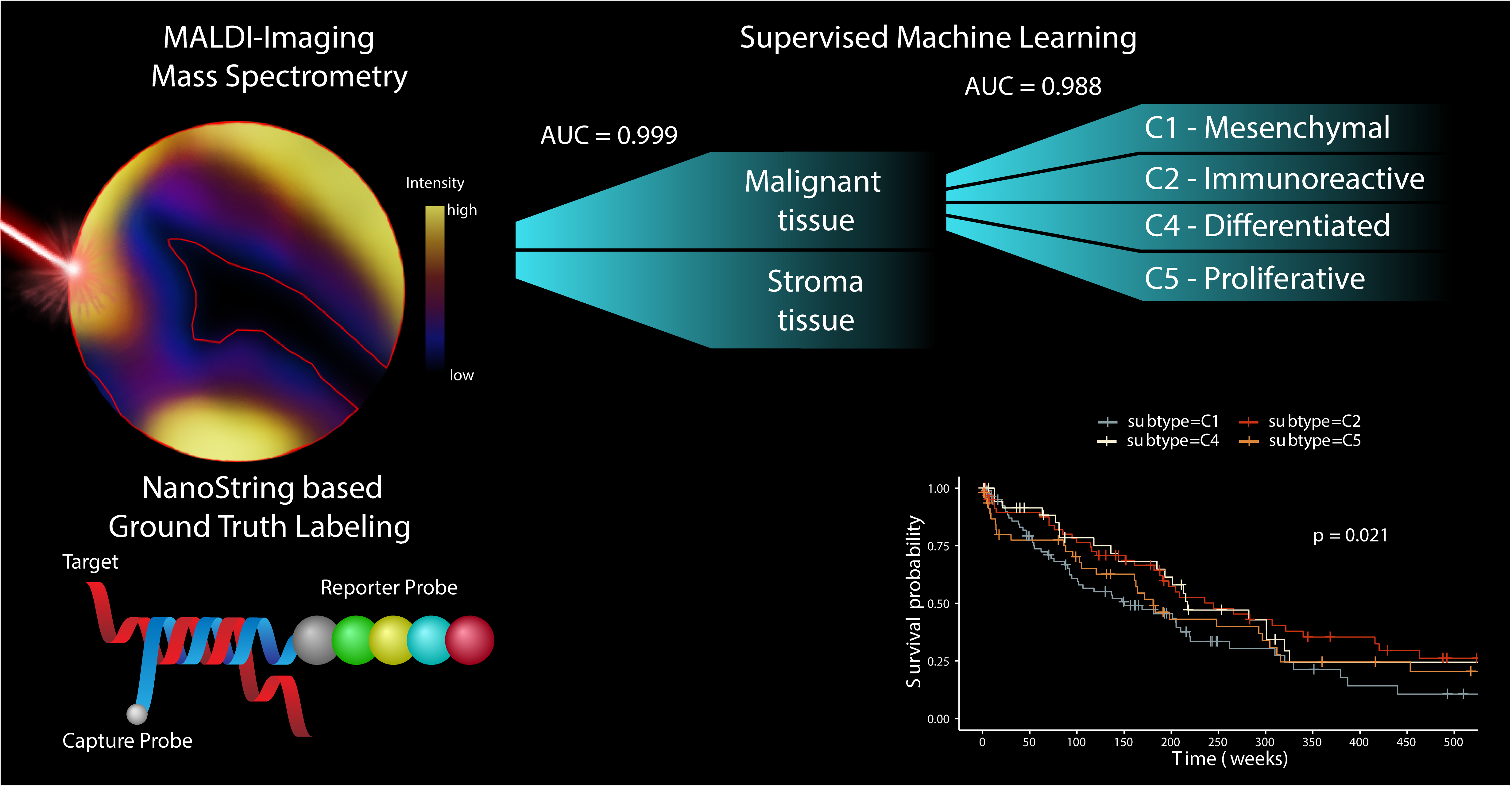
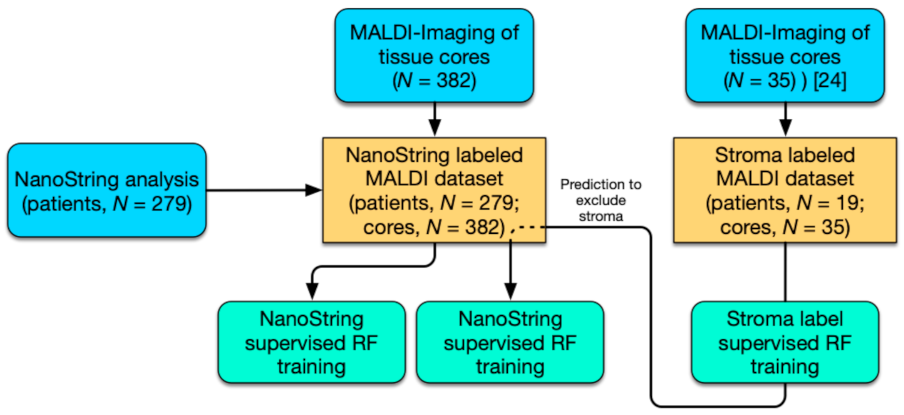
2.1. Two-Pronged Subtype Classification Workflow
2.2. Subtype Identification via NanoString Analysis
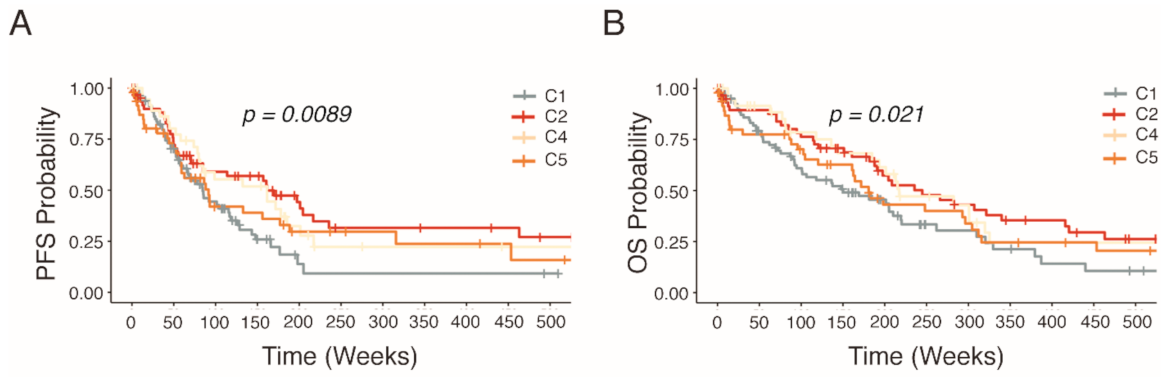
2.3. Distinct Survival Characteristics of Molecular Subtypes
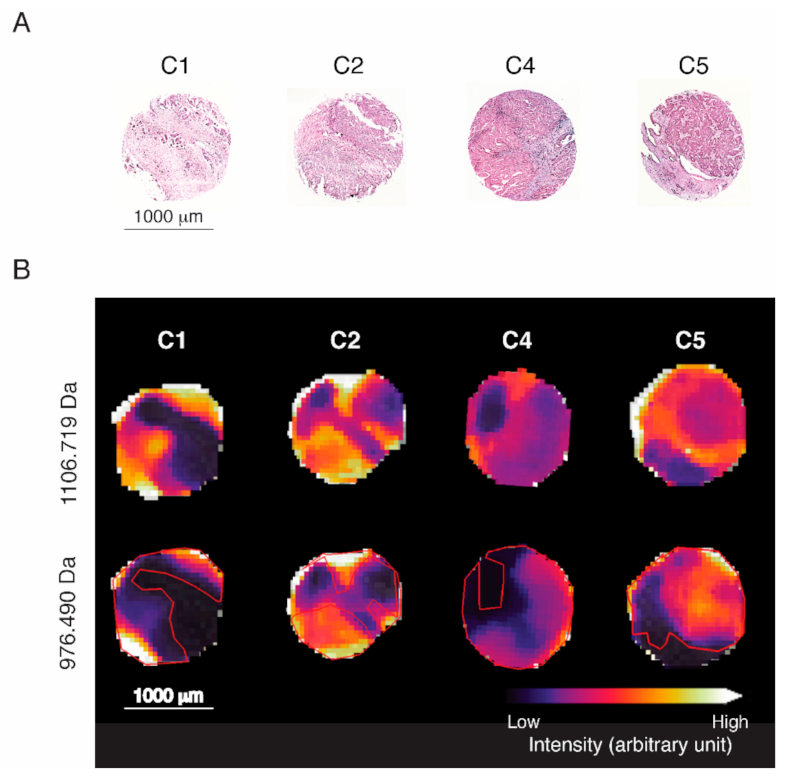
2.4. Accumulation of Proteomics Data by MALDI-IMS
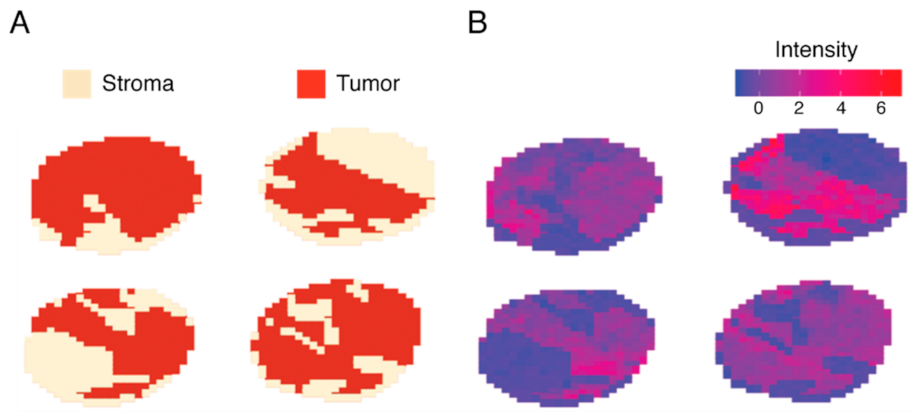
2.5. Classification of Stroma Compartments
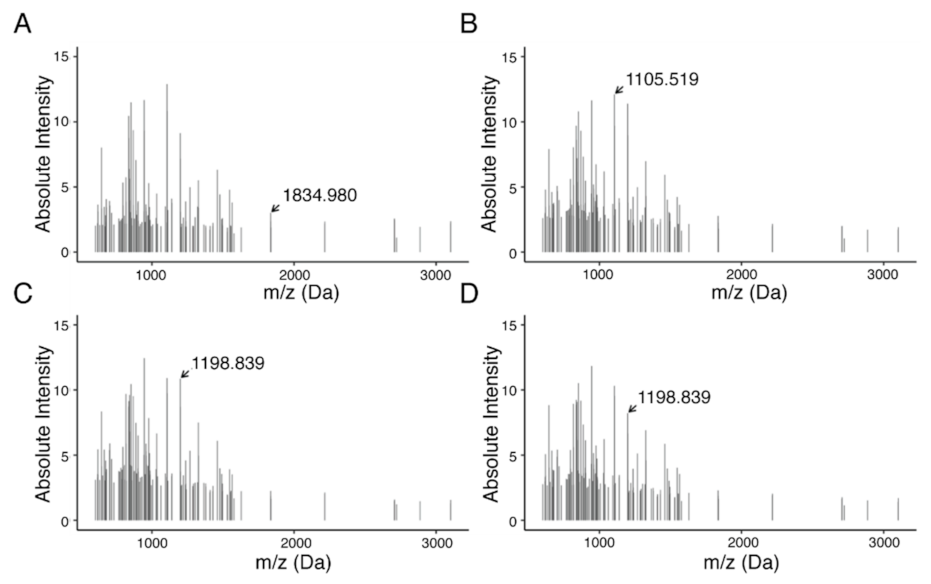
2.6. Discovery of Predictive Proteomic Signature of Tumor Subtypes
3. Discussion
4. Materials and Methods
4.1. HGSOC Patient Cohort
4.2. RNA Extraction and Classification by NanoString Technology
4.3. Statistical Analysis of Patient Outcome
4.4. Reference Dataset for Subtype Classification Based on Gene Expression Analysis
4.5. MALDI-Imaging and Peptide Identification by “Bottom-Up”-nHPLC Mass Spectrometry
4.6. Dataset Preparation
4.7. Exclusion of Spectra of Stromal Origin
4.8. Machine Learning and Model Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bowtell, D.D.; Bohm, S.; Ahmed, A.A.; Aspuria, P.J.; Bast, R.C., Jr.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef]
- Jiang, X.; Li, W.; Li, X.; Bai, H.; Zhang, Z. Current status and future prospects of PARP inhibitor clinical trials in ovarian cancer. Cancer Manag. Res. 2019, 11, 4371–4390. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef]
- Riester, M.; Wei, W.; Waldron, L.; Culhane, A.C.; Trippa, L.; Oliva, E.; Kim, S.H.; Michor, F.; Huttenhower, C.; Parmigiani, G.; et al. Risk prediction for late-stage ovarian cancer by meta-analysis of 1525 patient samples. J. Natl. Cancer Inst. 2014, 106, dju048. [Google Scholar] [CrossRef] [PubMed]
- Waldron, L.; Riester, M.; Birrer, M. Molecular subtypes of high-grade serous ovarian cancer: The holy grail? J. Natl. Cancer Inst. 2014, 106, dju297. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.Z.; Miow, Q.H.; Huang, R.Y.; Wong, M.K.; Ye, J.; Lau, J.A.; Wu, M.C.; Bin Abdul Hadi, L.H.; Soong, R.; Choolani, M.; et al. Functional genomics identifies five distinct molecular subtypes with clinical relevance and pathways for growth control in epithelial ovarian cancer. EMBO Mol. Med. 2013, 5, 1051–1066. [Google Scholar] [CrossRef] [PubMed]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Helland, A.; Anglesio, M.S.; George, J.; Cowin, P.A.; Johnstone, C.N.; House, C.M.; Sheppard, K.E.; Etemadmoghadam, D.; Melnyk, N.; Rustgi, A.K.; et al. Deregulation of MYCN, LIN28B and LET7 in a molecular subtype of aggressive high-grade serous ovarian cancers. PLoS ONE 2011, 6, e18064. [Google Scholar] [CrossRef]
- Zhang, S.; Jing, Y.; Zhang, M.; Zhang, Z.; Ma, P.; Peng, H.; Shi, K.; Gao, W.Q.; Zhuang, G. Stroma-associated master regulators of molecular subtypes predict patient prognosis in ovarian cancer. Sci. Rep. 2015, 5, 16066. [Google Scholar] [CrossRef]
- Rankin, E.B.; Fuh, K.C.; Taylor, T.E.; Krieg, A.J.; Musser, M.; Yuan, J.; Wei, K.; Kuo, C.J.; Longacre, T.A.; Giaccia, A.J. AXL is an essential factor and therapeutic target for metastatic ovarian cancer. Cancer Res. 2010, 70, 7570–7579. [Google Scholar] [CrossRef] [PubMed]
- Antony, J.; Tan, T.Z.; Kelly, Z.; Low, J.; Choolani, M.; Recchi, C.; Gabra, H.; Thiery, J.P.; Huang, R.Y. The GAS6-AXL signaling network is a mesenchymal (Mes) molecular subtype-specific therapeutic target for ovarian cancer. Sci. Signal. 2016, 9, ra97. [Google Scholar] [CrossRef] [PubMed]
- Konecny, G.E.; Wang, C.; Hamidi, H.; Winterhoff, B.; Kalli, K.R.; Dering, J.; Ginther, C.; Chen, H.W.; Dowdy, S.; Cliby, W.; et al. Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. J. Natl. Cancer Inst. 2014, 106, dju249. [Google Scholar] [CrossRef]
- Chen, G.M.; Kannan, L.; Geistlinger, L.; Kofia, V.; Safikhani, Z.; Gendoo, D.M.A.; Parmigiani, G.; Birrer, M.; Haibe-Kains, B.; Waldron, L. Consensus on Molecular Subtypes of High-Grade Serous Ovarian Carcinoma. Clin. Cancer Res. 2018, 24, 5037–5047. [Google Scholar] [CrossRef]
- Smits, A.J.; Kummer, J.A.; de Bruin, P.C.; Bol, M.; van den Tweel, J.G.; Seldenrijk, K.A.; Willems, S.M.; Offerhaus, G.J.; de Weger, R.A.; van Diest, P.J.; et al. The estimation of tumor cell percentage for molecular testing by pathologists is not accurate. Mod. Pathol. 2014, 27, 168–174. [Google Scholar] [CrossRef]
- Schwede, M.; Waldron, L.; Mok, S.C.; Wei, W.; Basunia, A.; Merritt, M.A.; Mitsiades, C.S.; Parmigiani, G.; Harrington, D.P.; Quackenbush, J.; et al. The Impact of Stroma Admixture on Molecular Subtypes and Prognostic Gene Signatures in Serous Ovarian Cancer. Cancer Epidemiol. Biomark. Prev. 2020, 29, 509–519. [Google Scholar] [CrossRef]
- Tothill, R.W.; Shi, F.; Paiman, L.; Bedo, J.; Kowalczyk, A.; Mileshkin, L.; Buela, E.; Klupacs, R.; Bowtell, D.; Byron, K. Development and validation of a gene expression tumour classifier for cancer of unknown primary. Pathology 2015, 47, 7–12. [Google Scholar] [CrossRef]
- Leong, H.S.; Galletta, L.; Etemadmoghadam, D.; George, J.; Australian Ovarian Cancer, S.; Kobel, M.; Ramus, S.J.; Bowtell, D. Efficient molecular subtype classification of high-grade serous ovarian cancer. J. Pathol. 2015, 236, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Talhouk, A.; George, J.; Wang, C.; Budden, T.; Tan, T.Z.; Chiu, D.S.; Kommoss, S.; Leong, H.S.; Chen, S.; Intermaggio, M.P.; et al. Development and Validation of the Gene Expression Predictor of High-grade Serous Ovarian Carcinoma Molecular SubTYPE (PrOTYPE). Clin. Cancer Res. 2020, 26, 5411–5423. [Google Scholar] [CrossRef]
- Walch, A.; Rauser, S.; Deininger, S.O.; Hofler, H. MALDI imaging mass spectrometry for direct tissue analysis: A new frontier for molecular histology. Histochem. Cell Biol. 2008, 130, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Casadonte, R.; Caprioli, R.M. Proteomic analysis of formalin-fixed paraffin-embedded tissue by MALDI imaging mass spectrometry. Nat. Protoc. 2011, 6, 1695–1709. [Google Scholar] [CrossRef]
- Addie, R.D.; Balluff, B.; Bovee, J.V.; Morreau, H.; McDonnell, L.A. Current State and Future Challenges of Mass Spectrometry Imaging for Clinical Research. Anal. Chem. 2015, 87, 6426–6433. [Google Scholar] [CrossRef]
- Schwamborn, K.; Caprioli, R.M. Molecular imaging by mass spectrometry--looking beyond classical histology. Nat. Rev. Cancer 2010, 10, 639–646. [Google Scholar] [CrossRef]
- Klein, O.; Kanter, F.; Kulbe, H.; Jank, P.; Denkert, C.; Nebrich, G.; Schmitt, W.D.; Wu, Z.; Kunze, C.A.; Sehouli, J.; et al. MALDI-Imaging for Classification of Epithelial Ovarian Cancer Histotypes from a Tissue Microarray Using Machine Learning Methods. Proteom. Clin. Appl. 2019, 13, e1700181. [Google Scholar] [CrossRef]
- Millstein, J.; Budden, T.; Goode, E.L.; Anglesio, M.S.; Talhouk, A.; Intermaggio, M.P.; Leong, H.S.; Chen, S.; Elatre, W.; Gilks, B.; et al. Prognostic gene expression signature for high-grade serous ovarian cancer. Ann. Oncol. 2020, 31, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- Trusheim, M.R.; Berndt, E.R.; Douglas, F.L. Stratified medicine: Strategic and economic implications of combining drugs and clinical biomarkers. Nat. Rev. Drug Discov. 2007, 6, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Kulbe, H.; Klein, O.; Wu, Z.; Taube, E.T.; Kassuhn, W.; Horst, D.; Darb-Esfahani, S.; Jank, P.; Abobaker, S.; Ringel, F.; et al. Discovery of Prognostic Markers for Early-Stage High-Grade Serous Ovarian Cancer by Maldi-Imaging. Cancers 2020, 12, 2000. [Google Scholar] [CrossRef] [PubMed]
- Geessink, O.G.F.; Baidoshvili, A.; Klaase, J.M.; Ehteshami Bejnordi, B.; Litjens, G.J.S.; van Pelt, G.W.; Mesker, W.E.; Nagtegaal, I.D.; Ciompi, F.; van der Laak, J. Computer aided quantification of intratumoral stroma yields an independent prognosticator in rectal cancer. Cell. Oncol. 2019, 42, 331–341. [Google Scholar] [CrossRef]
- Kim, J.M.; Kim, K.; Punj, V.; Liang, G.; Ulmer, T.S.; Lu, W.; An, W. Linker histone H1.2 establishes chromatin compaction and gene silencing through recognition of H3K27me3. Sci. Rep. 2015, 5, 16714. [Google Scholar] [CrossRef]
- Izar, B.; Tirosh, I.; Stover, E.H.; Wakiro, I.; Cuoco, M.S.; Alter, I.; Rodman, C.; Leeson, R.; Su, M.J.; Shah, P.; et al. A single-cell landscape of high-grade serous ovarian cancer. Nat. Med. 2020, 26, 1271–1279. [Google Scholar] [CrossRef]
- Kommoss, S.; Winterhoff, B.; Oberg, A.L.; Konecny, G.E.; Wang, C.; Riska, S.M.; Fan, J.B.; Maurer, M.J.; April, C.; Shridhar, V.; et al. Bevacizumab May Differentially Improve Ovarian Cancer Outcome in Patients with Proliferative and Mesenchymal Molecular Subtypes. Clin. Cancer Res. 2017, 23, 3794–3801. [Google Scholar] [CrossRef]
- Yu, M.; Guo, G.; Huang, L.; Deng, L.; Chang, C.S.; Achyut, B.R.; Canning, M.; Xu, N.; Arbab, A.S.; Bollag, R.J.; et al. CD73 on cancer-associated fibroblasts enhanced by the A2B-mediated feedforward circuit enforces an immune checkpoint. Nat. Commun. 2020, 11, 515. [Google Scholar] [CrossRef] [PubMed]
- Aichler, M.; Walch, A. MALDI Imaging mass spectrometry: Current frontiers and perspectives in pathology research and practice. Lab. Investig. 2015, 95, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Klein, O.; Strohschein, K.; Nebrich, G.; Oetjen, J.; Trede, D.; Thiele, H.; Alexandrov, T.; Giavalisco, P.; Duda, G.N.; von Roth, P.; et al. MALDI imaging mass spectrometry: Discrimination of pathophysiological regions in traumatized skeletal muscle by characteristic peptide signatures. Proteomics 2014, 14, 2249–2260. [Google Scholar] [CrossRef]
- Cillero-Pastor, B.; Heeren, R.M. Matrix-assisted laser desorption ionization mass spectrometry imaging for peptide and protein analyses: A critical review of on-tissue digestion. J. Proteome Res. 2014, 13, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Obermeyer, Z.; Emanuel, E.J. Predicting the Future—Big Data, Machine Learning, and Clinical Medicine. N. Engl. J. Med. 2016, 375, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Menze, B.H.; Kelm, B.M.; Masuch, R.; Himmelreich, U.; Bachert, P.; Petrich, W.; Hamprecht, F.A. A comparison of random forest and its Gini importance with standard chemometric methods for the feature selection and classification of spectral data. BMC Bioinform. 2009, 10, 213. [Google Scholar] [CrossRef]
- Lang, M.; Binder, M.; Richter, J.; Schratz, P.; Pfisterer, F.; Coors, S.; Au, Q.; Casalicchio, G.; Kotthoff, L.; Bischl, B. mlr3: A modern object-oriented machine learningframework in R. J. Open Source Softw. 2019, 4, 1903. [Google Scholar] [CrossRef]
- Marvin, N.; Wright, A.Z. Ranger: A Fast Implementation of Random Forests for High Dimensional Data in C++ and R. J. Stat. Softw. 2017, 77. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 77. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kassuhn, W.; Klein, O.; Darb-Esfahani, S.; Lammert, H.; Handzik, S.; Taube, E.T.; Schmitt, W.D.; Keunecke, C.; Horst, D.; Dreher, F.; et al. Classification of Molecular Subtypes of High-Grade Serous Ovarian Cancer by MALDI-Imaging. Cancers 2021, 13, 1512. https://doi.org/10.3390/cancers13071512
Kassuhn W, Klein O, Darb-Esfahani S, Lammert H, Handzik S, Taube ET, Schmitt WD, Keunecke C, Horst D, Dreher F, et al. Classification of Molecular Subtypes of High-Grade Serous Ovarian Cancer by MALDI-Imaging. Cancers. 2021; 13(7):1512. https://doi.org/10.3390/cancers13071512
Chicago/Turabian StyleKassuhn, Wanja, Oliver Klein, Silvia Darb-Esfahani, Hedwig Lammert, Sylwia Handzik, Eliane T. Taube, Wolfgang D. Schmitt, Carlotta Keunecke, David Horst, Felix Dreher, and et al. 2021. "Classification of Molecular Subtypes of High-Grade Serous Ovarian Cancer by MALDI-Imaging" Cancers 13, no. 7: 1512. https://doi.org/10.3390/cancers13071512