
Antitumoral Activity of the MEK Inhibitor Trametinib (TMT212) Alone and in Combination with the CDK4/6 Inhibitor Ribociclib (LEE011) in Neuroendocrine Tumor Cells In Vitro


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines, Culture, Reagents, and Treatment
2.2. Cell Proliferation Assays
2.3. Flow Cytometric Cell Cycle Distribution Assay
2.4. Western Blot
2.5. Statistical Analysis
3. Results
3.1. The MEKi Trametinib and the ERKi SCH772984 Suppress NET Cell Proliferation
3.2. Synergistic Inhibitory Effects of the MEKi Trametinib (TMT212) or the ERKi SCH772984 in Combination with the CDK4/6i Ribociclib (LEE011) on NET Cell Proliferation
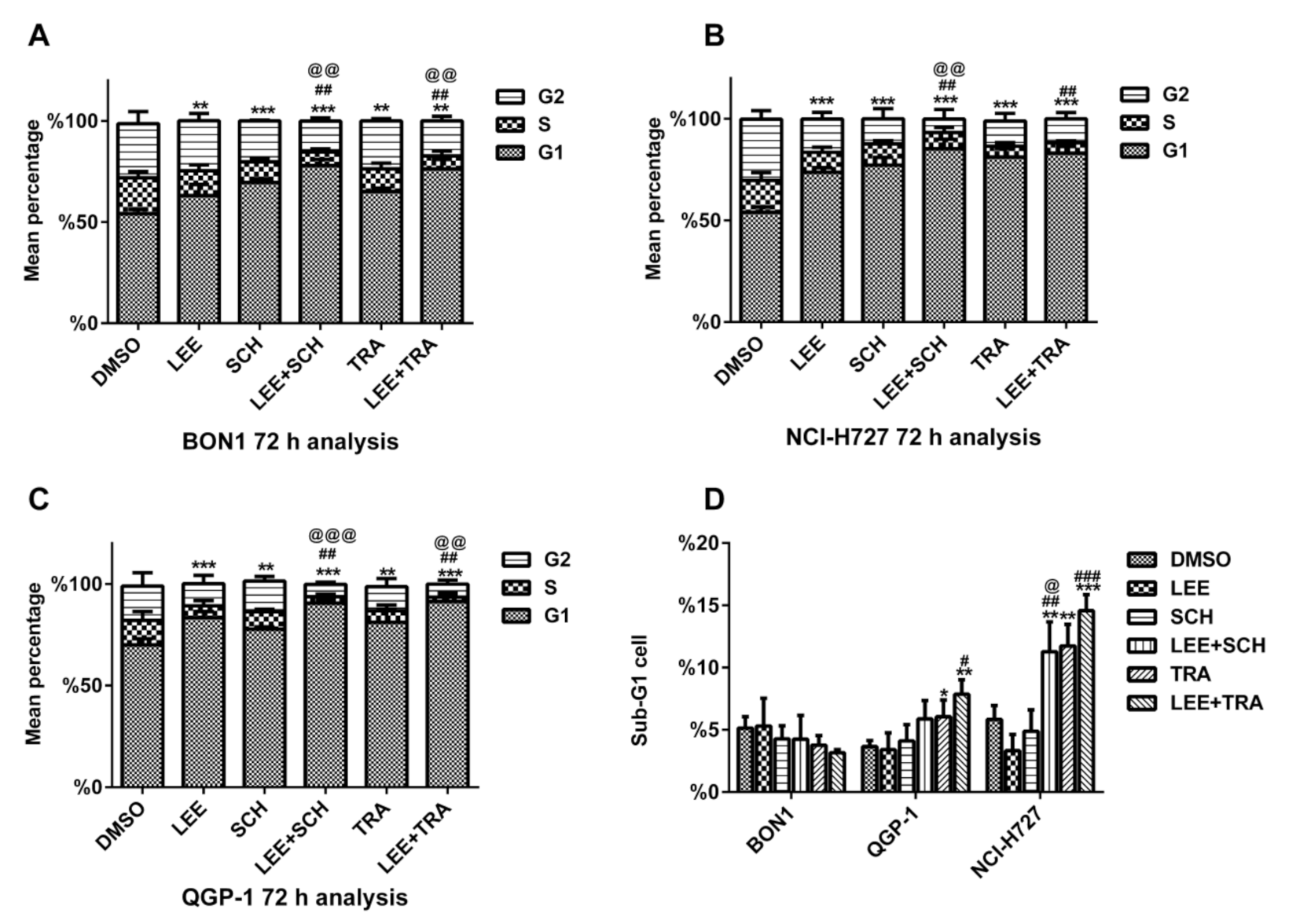
3.3. Synergistic Effects of the MEKi Trametinib (TMT212) or the ERKi SCH772984 in Combination with the CDK4/6i Ribociclib (LEE011) on NET Cell Cycle G1 Arrest, Sub G1 Events and Apoptosis
3.4. Synergistic Effects of the MEKi Trametinib (TMT212), the ERKi SCH772984, and the CDK4/6i Ribociclib (LEE011) on CDK4/6/Rb Signaling in NET Cell Lines
3.5. Synergistic Downregulation of the MEK/ERK and MYC Signaling after Treatment of NET Cells with Ribociclib (LEE011) and Trametinib or Ribociclib (LEE011) and SCH772984
3.6. Synergistic Downregulation of the p38 and JNK Signaling after Treatment of NET Cells with Ribociclib (LEE011) and Trametinib or Ribociclib (LEE011) and SCH772984
3.7. Variable Cell-Line-Dependent Effects of the MEKi Trametinib (TMT212), the ERKi SCH772984, and the CDK4/6i Ribociclib (LEE011) on PI3K/AKT Signaling
3.8. Regulation of the pEGFR and pIGFR Signaling after Combined Treatment of NET Cells with LEE011 and Trametinib or SCH772984
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients with Neuroendocrine Tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef]
- Rindi, G.; Klimstra, D.S.; Abedi-Ardekani, B.; Asa, S.L.; Bosman, F.T.; Brambilla, E.; Busam, K.J.; De Krijger, R.R.; Dietel, M.; El-Naggar, A.K.; et al. A common classification framework for neuroendocrine neoplasms: An International Agency for Research on Cancer (IARC) and World Health Organization (WHO) expert consensus proposal. Mod. Pathol. 2018, 31, 1770–1786. [Google Scholar] [CrossRef] [PubMed]
- Auernhammer, C.J.; Spitzweg, C.; Angele, M.K.; Boeck, S.; Grossman, A.; Nölting, S.; Ilhan, H.; Knösel, T.; Mayerle, J.; Reincke, M.; et al. Advanced neuroendocrine tumours of the small intestine and pancreas: Clinical developments, controversies, and future strategies. Lancet Diabetes Endocrinol. 2018, 6, 404–415. [Google Scholar] [CrossRef]
- Caplin, M.E.; Baudin, E.; Ferolla, P.; Filosso, P.; Garcia-Yuste, M.; Lim, E.; Oberg, K.; Pelosi, G.; Perren, A.; Rossi, R.E.; et al. Pulmonary neuroendocrine (carcinoid) tumors: European Neuroendocrine Tumor Society expert consensus and recommendations for best practice for typical and atypical pulmonary carcinoids. Ann. Oncol. 2015, 26, 1604–1620. [Google Scholar] [CrossRef] [PubMed]
- Baudin, E.; Hayes, A.R.; Scoazec, J.Y.; Filosso, P.L.; Lim, E.; Kaltsas, G.; Frilling, A.; Chen, J.; Kos-Kudla, B.; Gorbunova, V.; et al. Unmet Medical Needs in Pulmonary Neuroendocrine (Carcinoid) Neoplasms. Neuroendocrinology 2018, 108, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Grøndahl, V.; Binderup, T.; Langer, S.; Petersen, R.; Nielsen, K.; Kjaer, A.; Federspiel, B.; Knigge, U. Characteristics of 252 patients with bronchopulmonary neuroendocrine tumours treated at the Copenhagen NET Centre of Excellence. Lung Cancer 2019, 132, 141–149. [Google Scholar] [CrossRef]
- Robelin, P.; Hadoux, J.; Forestier, J.; Planchard, D.; Hervieu, V.; Berdelou, A.; Scoazec, J.-Y.; Valette, P.-J.; Leboulleux, S.; Ducreux, M.; et al. Characterization, Prognosis, and Treatment of Patients with Metastatic Lung Carcinoid Tumors. J. Thorac. Oncol. 2019, 14, 993–1002. [Google Scholar] [CrossRef]
- Rinke, A.; Auernhammer, C.J.; Bodei, L.; Kidd, M.; Krug, S.; Lawlor, R.; Marinoni, I.; Perren, A.; Scarpa, A.; Sorbye, H.; et al. Treatment of advanced gastroenteropancreatic neuroendocrine neoplasia, are we on the way to personalised medicine? Gut 2021. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.-M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017, 543, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, A.; Wiedmer, T.; Marinoni, I.; Perren, A. Genetic and epigenetic drivers of neuroendocrine tumours (NET). Endocr. Relat. Cancer 2017, 24, R315–R334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simbolo, M.; Barbi, S.; Fassan, M.; Mafficini, A.; Ali, G.; Vicentini, C.; Sperandio, N.; Corbo, V.; Rusev, B.; Mastracci, L.; et al. Gene Expression Profiling of Lung Atypical Carcinoids and Large Cell Neuroendocrine Carcinomas Identifies Three Transcriptomic Subtypes with Specific Genomic Alterations. J. Thorac. Oncol. 2019, 14, 1651–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobhani, N.; D’Angelo, A.; Pittacolo, M.; Roviello, G.; Miccoli, A.; Corona, S.P.; Bernocchi, O.; Generali, D.; Otto, T. Updates on the CDK4/6 Inhibitory Strategy and Combinations in Breast Cancer. Cells 2019, 8, 321. [Google Scholar] [CrossRef] [Green Version]
- Schettini, F.; De Santo, I.; Rea, C.G.; De Placido, P.; Formisano, L.; Giuliano, M.; Arpino, G.; De Laurentiis, M.; Puglisi, F.; De Placido, S.; et al. CDK 4/6 Inhibitors as Single Agent in Advanced Solid Tumors. Front. Oncol. 2018, 8, 608. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.H.; Contractor, T.; Clausen, R.; Klimstra, D.S.; Du, Y.-C.N.; Allen, P.J.; Brennan, M.F.; Levine, A.J.; Harris, C.R. Attenuation of the Retinoblastoma Pathway in Pancreatic Neuroendocrine Tumors Due to Increased Cdk4/Cdk6. Clin. Cancer Res. 2012, 18, 4612–4620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grande, E.; Teulé, A.; Alonso-Gordoa, T.; Jiménez-Fonseca, P.; Benavent, M.; Capdevila, J.; Custodio, A.; Vera, R.; Munarriz, J.; La Casta, A.; et al. The PALBONET Trial: A Phase II Study of Palbociclib in Metastatic Grade 1 and 2 Pancreatic Neuroendocrine Tumors (GETNE-1407). Oncologist 2020, 25, e745–e1265. [Google Scholar] [CrossRef] [Green Version]
- Prada, E.T.A.; Nölting, S.; Spoettl, G.; Maurer, J.; Auernhammer, C.J. The Novel Cyclin-Dependent Kinase 4/6 Inhibitor Ribociclib (LEE011) Alone and in Dual-Targeting Approaches Demonstrates Antitumoral Efficacy in Neuroendocrine Tumors in vitro. Neuroendocrinology 2018, 106, 58–73. [Google Scholar] [CrossRef]
- Dasari, A.; Halperin, D.; Coya, T.; Mahvash, A.; Zorilla, I.; Meric-Bernstam, F. A Pilot Study of the Cyclin Dependent Kinases 4, 6 Inhibitor Ribociclib in Patients with Foregut Neuroendocrine Tumors. Neuroendocrinology 2018, 106, 1–301. [Google Scholar]
- Álvarez-Fernández, M.; Malumbres, M. Mechanisms of Sensitivity and Resistance to CDK4/6 Inhibition. Cancer Cell 2020, 37, 514–529. [Google Scholar] [CrossRef]
- Wagner, V.; Gil, J. Senescence as a therapeutically relevant response to CDK4/6 inhibitors. Oncogene 2020, 39, 5165–5176. [Google Scholar] [CrossRef]
- Takkenkamp, T.J.; Jalving, M.; Hoogwater, F.J.H.; Walenkamp, A.M.E. The immune tumour microenvironment of neuroendocrine tumours and its implications for immune checkpoint inhibitors. Endocr. Relat. Cancer 2020, 27, R329–R343. [Google Scholar] [CrossRef] [PubMed]
- Varjosalo, M.; Keskitalo, S.; Van Drogen, A.; Nurkkala, H.; Vichalkovski, A.; Aebersold, R.; Gstaiger, M. The Protein Interaction Landscape of the Human CMGC Kinase Group. Cell Rep. 2013, 3, 1306–1320. [Google Scholar] [CrossRef] [Green Version]
- Hart, L.S.; Rader, J.; Raman, P.; Batra, V.; Russell, M.R.; Tsang, M.; Gagliardi, M.; Chen, L.; Martinez, D.; Li, Y.; et al. Preclinical Therapeutic Synergy of MEK1/2 and CDK4/6 Inhibition in Neuroblastoma. Clin. Cancer Res. 2017, 23, 1785–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.S.; Helms, T.L.; Feng, N.; Gay, J.; Chang, Q.E.; Tian, F.; Wu, J.Y.; Toniatti, C.; Heffernan, T.P.; Powis, G.; et al. Efficacy of the combination of MEK and CDK4/6 inhibitors in vitro and in vivo in KRAS mutant colorectal cancer models. Oncotarget 2016, 7, 39595–39608. [Google Scholar] [CrossRef] [Green Version]
- Ziemke, E.K.; Dosch, J.S.; Maust, J.D.; Shettigar, A.; Sen, A.; Welling, T.H.; Hardiman, K.M.; Sebolt-Leopold, J.S. Sensitivity of KRAS-Mutant Colorectal Cancers to Combination Therapy That Cotargets MEK and CDK4/6. Clin. Cancer Res. 2016, 22, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Tao, Z.; Le Blanc, J.M.; Wang, C.; Zhan, T.; Zhuang, H.; Wang, P.; Yuan, Z.; Lu, B. Coadministration of Trametinib and Palbociclib Radiosensitizes KRAS-Mutant Non–Small Cell Lung Cancers In Vitro and In Vivo. Clin. Cancer Res. 2016, 22, 122–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teh, J.L.; Purwin, T.J.; Greenawalt, E.J.; Chervoneva, I.; Goldberg, A.; Davies, M.A.; Aplin, A.E. An In Vivo Reporter to Quantitatively and Temporally Analyze the Effects of CDK4/6 Inhibitor-Based Therapies in Melanoma. Cancer Res. 2016, 76, 5455–5466. [Google Scholar] [CrossRef] [Green Version]
- Martin, C.A.; Cullinane, C.; Kirby, L.; Abuhammad, S.; Lelliott, E.J.; Waldeck, K.; Young, R.J.; Brajanovski, N.; Cameron, D.P.; Walker, R.; et al. Palbociclib synergizes with BRAF and MEK inhibitors in treatment naïve melanoma but not after the development of BRAF inhibitor resistance. Int. J. Cancer 2018, 142, 2139–2152. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, R.J.; Amaria, R.N.; Lawrence, D.P.; Brennan, J.; Leister, C.; Singh, R. Phase 1b dose-escalation study of trametinib (MEKi) plus palbociclib (CDK4/6i) in patients with advanced solid tumors. Mol. Cancer Ther. 2015, 14 (Suppl. 2). [Google Scholar] [CrossRef]
- Abe, H.; Kikuchi, S.; Hayakawa, K.; Iida, T.; Nagahashi, N.; Maeda, K.; Sakamoto, J.; Matsumoto, N.; Miura, T.; Matsumura, K.; et al. Discovery of a Highly Potent and Selective MEK Inhibitor: GSK1120212 (JTP-74057 DMSO Solvate). ACS Med. Chem. Lett. 2011, 2, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Kakadia, S.; Yarlagadda, N.; Awad, R.; Kundranda, M.; Niu, J.; Naraev, B.; Mina, L.; Dragovich, T.; Gimbel, M.; Mahmoud, F. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. OncoTargets Ther. 2018, 11, 7095–7107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weart, T.C.; Miller, K.D.; Simone, C.B., 2nd. Spotlight on dabrafenib/trametinib in the treatment of non-small-cell lung cancer: Place in therapy. Cancer Manag. Res. 2018, 10, 647–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Schellens, J.H.; Soria, J.C.; Wen, P.Y.; Zielinski, C.; Cabanillas, M.E.; Urbanowitz, G.; et al. Dabrafenib and Trametinib Treatment in Patients with Locally Advanced or Metastatic BRAF V600–Mutant Anaplastic Thyroid Cancer. J. Clin. Oncol. 2018, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Prada, E.T.A.; Auernhammer, C.J. Targeted therapy of gastroenteropancreatic neuroendocrine tumours: Preclinical strategies and future targets. Endocr. Connect. 2018, 7, R1–R25. [Google Scholar] [CrossRef] [Green Version]
- Valentino, J.D.; Li, J.; Zaytseva, Y.Y.; Mustain, W.C.; Elliott, V.A.; Kim, J.T.; Harris, J.W.; Campbell, K.; Weiss, H.; Wang, C.; et al. Cotargeting the PI3K and RAS Pathways for the Treatment of Neuroendocrine Tumors. Clin. Cancer Res. 2014, 20, 1212–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iida, S.; Miki, Y.; Ono, K.; Akahira, J.-I.; Nakamura, Y.; Suzuki, T.; Sasano, H. Synergistic anti-tumor effects of RAD001 with MEK inhibitors in neuroendocrine tumors: A potential mechanism of therapeutic limitation of mTOR inhibitor. Mol. Cell. Endocrinol. 2012, 350, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Hofving, T.; Arvidsson, Y.; Almobarak, B.; Inge, L.; Pfragner, R.; Persson, M.; Stenman, G.; Kristiansson, E.; Johanson, V.; Nilsson, O. The neuroendocrine phenotype, genomic profile and therapeutic sensitivity of GEPNET cell lines. Endocr. Relat. Cancer 2018, 25, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Vandamme, T.; Peeters, M.; Dogan, F.; Pauwels, P.; Van Assche, E.; Beyens, M.; Mortier, G.; Vandeweyer, G.; De Herder, W.; Van Camp, G.; et al. Whole-exome characterization of pancreatic neuroendocrine tumor cell lines BON-1 and QGP-1. J. Mol. Endocrinol. 2015, 54, 137–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boora, G.K.; Kanwar, R.; Kulkarni, A.A.; Pleticha, J.; Ames, M.M.; Schroth, G.P.; Beutler, A.S.; Banck, M.S. Exome-level comparison of primary well-differentiated neuroendocrine tumors and their cell lines. Cancer Genet. 2015, 208, 374–381. [Google Scholar] [CrossRef]
- Jin, X.-F.; Spoettl, G.; Maurer, J.; Nölting, S.; Auernhammer, C.J. Inhibition of Wnt/β-Catenin Signaling in Neuroendocrine Tumors In Vitro: Antitumoral Effects. Cancers 2020, 12, 345. [Google Scholar] [CrossRef] [Green Version]
- Foucquier, J.; Guedj, M. Analysis of drug combinations: Current methodological landscape. Pharmacol. Res. Perspect. 2015, 3, e00149. [Google Scholar] [CrossRef]
- Vakil, V.; Trappe, W. Drug Combinations: Mathematical Modeling and Networking Methods. Pharmaceutics 2019, 11, 208. [Google Scholar] [CrossRef] [Green Version]
- De Leeuw, R.; McNair, C.; Schiewer, M.J.; Neupane, N.P.; Brand, L.J.; Augello, M.A.; Li, Z.; Cheng, L.C.; Yoshida, A.; Courtney, S.M.; et al. MAPK Reliance via Acquired CDK4/6 Inhibitor Resistance in Cancer. Clin. Cancer Res. 2018, 24, 4201–4214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, A.L.; Lee, S.E.; Dawson, L.K.; Marlow, L.A.; Edenfield, B.H.; Durham, W.F.; Flotte, T.J.; Thompson, M.; Small, D.L.; Synnott, A.J.; et al. Targeting the cyclin dependent kinase and retinoblastoma axis overcomes standard of care resistance in BRAFV600E-mutant melanoma. Oncotarget 2018, 9, 10905–10919. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Jung, M.; Kang, H.N.; Kim, H.; Park, C.W.; Kim, S.M.; Shin, S.J.; Kim, S.H.; Kim, S.G.; Kim, E.K.; et al. Oncogenic BRAF fusions in mucosal melanomas activate the MAPK pathway and are sensitive to MEK/PI3K inhibition or MEK/CDK4/6 inhibition. Oncogene 2017, 36, 3334–3345. [Google Scholar] [CrossRef]
- Pek, M.; Yatim, S.M.J.M.; Chen, Y.; Li, J.; Gong, M.; Jiang, X.; Zhang, F.; Zheng, J.; Wu, X.; Yu, Q. Oncogenic KRAS-associated gene signature defines co-targeting of CDK4/6 and MEK as a viable therapeutic strategy in colorectal cancer. Oncogene 2017, 36, 4975–4986. [Google Scholar] [CrossRef]
- Franco, J.; Witkiewicz, A.K.; Knudsen, E.S. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget 2014, 5, 6512–6525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante, J.R.; Fecher, L.A.; Falchook, G.S.; Nallapareddy, S.; Gordon, M.S.; Becerra, C.; DeMarini, D.J.; Cox, D.S.; Xu, Y.; Morris, S.R.; et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 773–781. [Google Scholar] [CrossRef]
- Braal, C.L.; Jongbloed, E.M.; Wilting, S.M.; Mathijssen, R.H.J.; Koolen, S.L.W.; Jager, A. Inhibiting CDK4/6 in Breast Cancer with Palbociclib, Ribociclib, and Abemaciclib: Similarities and Differences. Drugs 2020. [Google Scholar] [CrossRef]
- Girardi, D.M.; Silva, A.C.; Rêgo, J.F.M.; Coudry, R.A.; Riechelmann, R.P. Unraveling molecular pathways of poorly differentiated neuroendocrine carcinomas of the gastroenteropancreatic system: A systematic review. Cancer Treat. Rev. 2017, 56, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Klempner, S.J.; Gershenhorn, B.; Tran, P.; Lee, T.K.; Erlander, M.G.; Gowen, K.; Schrock, A.B.; Morosini, D.; Ross, J.S.; Miller, V.A.; et al. BRAFV600E Mutations in High-Grade Colorectal Neuroendocrine Tumors May Predict Responsiveness to BRAF–MEK Combination Therapy. Cancer Discov. 2016, 6, 594–600. [Google Scholar] [CrossRef] [Green Version]
- Dizdar, L.; Werner, T.A.; Drusenheimer, J.C.; Möhlendick, B.; Raba, K.; Boeck, I.; Anlauf, M.; Schott, M.; Göring, W.; Esposito, I.; et al. BRAFV600E mutation: A promising target in colorectal neuroendocrine carcinoma. Int. J. Cancer 2019, 144, 1379–1390. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.; DeCristo, M.J.; McAllister, S.S.; Zhao, J.J. CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest. Trends Cell Biol. 2018, 28, 911–925. [Google Scholar] [CrossRef]
- Carragher, L.A.S.; Snell, K.R.; Giblett, S.M.; Aldridge, V.S.S.; Patel, B.; Cook, S.J.; Winton, D.J.; Marais, R.; Pritchard, C.A. V600EBraf induces gastrointestinal crypt senescence and promotes tumour progression through enhanced CpG methylation of p16 INK4a. EMBO Mol. Med. 2010, 2, 458–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.P.; VanBrocklin, M.W.; Lastwika, K.J.; McKinney, A.J.; Brandner, S.; Holmen, S.L. Activated MEK cooperates with Ink4a/Arf loss or Akt activation to induce gliomas in vivo. Oncogene 2011, 30, 1341–1350. [Google Scholar] [CrossRef] [Green Version]
- Pratilas, C.A.; Taylor, B.S.; Ye, Q.; Viale, A.; Sander, C.; Solit, D.B.; Rosen, N. V600EBRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc. Natl. Acad. Sci. USA 2009, 106, 4519–4524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonelli, M.A.; Digiacomo, G.; Fumarola, C.; Alfieri, R.; Quaini, F.; Falco, A.; Madeddu, D.; La Monica, S.; Cretella, D.; Ravelli, A.; et al. Combined Inhibition of CDK4/6 and PI3K/AKT/mTOR Pathways Induces a Synergistic Anti-Tumor Effect in Malignant Pleural Mesothelioma Cells. Neoplasia 2017, 19, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Basken, J.; Stuart, S.A.; Kavran, A.J.; Lee, T.; Ebmeier, C.C.; Old, W.M.; Ahn, N.G. Specificity of Phosphorylation Responses to Mitogen Activated Protein (MAP) Kinase Pathway Inhibitors in Melanoma Cells. Mol. Cell. Proteom. 2018, 17, 550–564. [Google Scholar] [CrossRef] [Green Version]
- Luley, K.B.; Biedermann, S.B.; Künstner, A.; Busch, H.; Franzenburg, S.; Schrader, J.; Grabowski, P.; Wellner, U.F.; Keck, T.; Brabant, G.; et al. A Comprehensive Molecular Characterization of the Pancreatic Neuroendocrine Tumor Cell Lines BON-1 and QGP-1. Cancers 2020, 12, 691. [Google Scholar] [CrossRef] [Green Version]
- Ear, P.H.; Li, G.; Wu, M.; Abusada, E.; Bellizzi, A.M.; Howe, J.R. Establishment and Characterization of Small Bowel Neuroendocrine Tumor Spheroids. J. Vis. Exp. 2019, e60303. [Google Scholar] [CrossRef] [PubMed]
- April-Monn, S.L.; Wiedmer, T.; Skowronska, M.; Maire, R.; Lena, M.S.; Trippel, M.; Di Domenico, A.; Muffatti, F.; Andreasi, V.; Capurso, G.; et al. 3D Primary Cell Culture: A Novel Preclinical Model For Pancreatic Neuroendocrine Tumors. Neuroendocrinology 2021, 111, 273–287. [Google Scholar] [CrossRef]
- Kawasaki, K.; Toshimitsu, K.; Matano, M.; Fujita, M.; Fujii, M.; Togasaki, K.; Ebisudani, T.; Shimokawa, M.; Takano, A.; Takahashi, S.; et al. An Organoid Biobank of Neuroendocrine Neoplasms Enables Genotype-Phenotype Mapping. Cell 2020, 183, 1420–1435.e21. [Google Scholar] [CrossRef]
- Benten, D.; Behrang, Y.; Unrau, L.; Weissmann, V.; Wolters-Eisfeld, G.; Burdak-Rothkamm, S.; Stahl, F.R.; Anlauf, M.; Grabowski, P.; Möbs, M.; et al. Establishment of the First Well-differentiated Human Pancreatic Neuroendocrine Tumor Model. Mol. Cancer Res. 2018, 16, 496–507. [Google Scholar] [CrossRef] [Green Version]
- Bresciani, G.; Hofland, L.J.; Dogan, F.; Giamas, G.; Gagliano, T.; Zatelli, M.C. Evaluation of Spheroid 3D Culture Methods to Study a Pancreatic Neuroendocrine Neoplasm Cell Line. Front. Endocrinol. 2019, 10, 682. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Martínez, A.D.; van den Dungen, R.; Dogan-Oruc, F.; Van Koetsveld, P.M.; Culler, M.D.; De Herder, W.W.; Luque, R.M.; Feelders, R.A.; Hofland, L.J. Effects of novel somatostatin-dopamine chimeric drugs in 2D and 3D cell culture models of neuroendocrine tumors. Endocr. Relat. Cancer 2019, 26, 585–599. [Google Scholar] [CrossRef] [PubMed]
- Yu, R. Animal models of spontaneous pancreatic neuroendocrine tumors. Mol. Cell. Endocrinol. 2016, 421, 60–67. [Google Scholar] [CrossRef]
- Wong, C.; Tang, L.H.; Davidson, C.; Vosburgh, E.; Chen, W.; Foran, D.J.; Notterman, D.A.; Levine, A.J.; Xu, E.Y. Two well-differentiated pancreatic neuroendocrine tumor mouse models. Cell Death Differ. 2020, 27, 269–283. [Google Scholar] [CrossRef] [Green Version]
- Gaudenzi, G.; Carra, S.; Dicitore, A.; Cantone, M.C.; Persani, L.; Vitale, G. Fishing for neuroendocrine tumors. Endocr. Relat. Cancer 2020, 27, R163–R176. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Adashek, J.J.; Shaya, J.; Okamura, R.; Jimenez, R.E.; Lee, S.; Sicklick, J.K.; Kurzrock, R. Concomitant MEK and Cyclin Gene Alterations: Implications for Response to Targeted Therapeutics. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, X.-F.; Spöttl, G.; Maurer, J.; Nölting, S.; Auernhammer, C.J. Antitumoral Activity of the MEK Inhibitor Trametinib (TMT212) Alone and in Combination with the CDK4/6 Inhibitor Ribociclib (LEE011) in Neuroendocrine Tumor Cells In Vitro. Cancers 2021, 13, 1485. https://doi.org/10.3390/cancers13061485
Jin X-F, Spöttl G, Maurer J, Nölting S, Auernhammer CJ. Antitumoral Activity of the MEK Inhibitor Trametinib (TMT212) Alone and in Combination with the CDK4/6 Inhibitor Ribociclib (LEE011) in Neuroendocrine Tumor Cells In Vitro. Cancers. 2021; 13(6):1485. https://doi.org/10.3390/cancers13061485
Chicago/Turabian StyleJin, Xi-Feng, Gerald Spöttl, Julian Maurer, Svenja Nölting, and Christoph Josef Auernhammer. 2021. "Antitumoral Activity of the MEK Inhibitor Trametinib (TMT212) Alone and in Combination with the CDK4/6 Inhibitor Ribociclib (LEE011) in Neuroendocrine Tumor Cells In Vitro" Cancers 13, no. 6: 1485. https://doi.org/10.3390/cancers13061485