
Clinical Features and Multiplatform Molecular Analysis Assist in Understanding Patient Response to Anti-PD-1/PD-L1 in Renal Cell Carcinoma

 , , , ,
, , , ,  , , , , and add
Show full author list
, , , , and add
Show full author list
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Population and Data Collection
2.2. DNA and RNA Extraction
2.3. mIF Immunohistochemistry
2.3.1. Immunofluorescence Staining
2.3.2. Sample Imaging
2.4. WES and Analysis
2.5. TCR Sequencing and Analysis
2.6. RNA-seq and Analysis
2.7. Statistical Analysis
3. Results
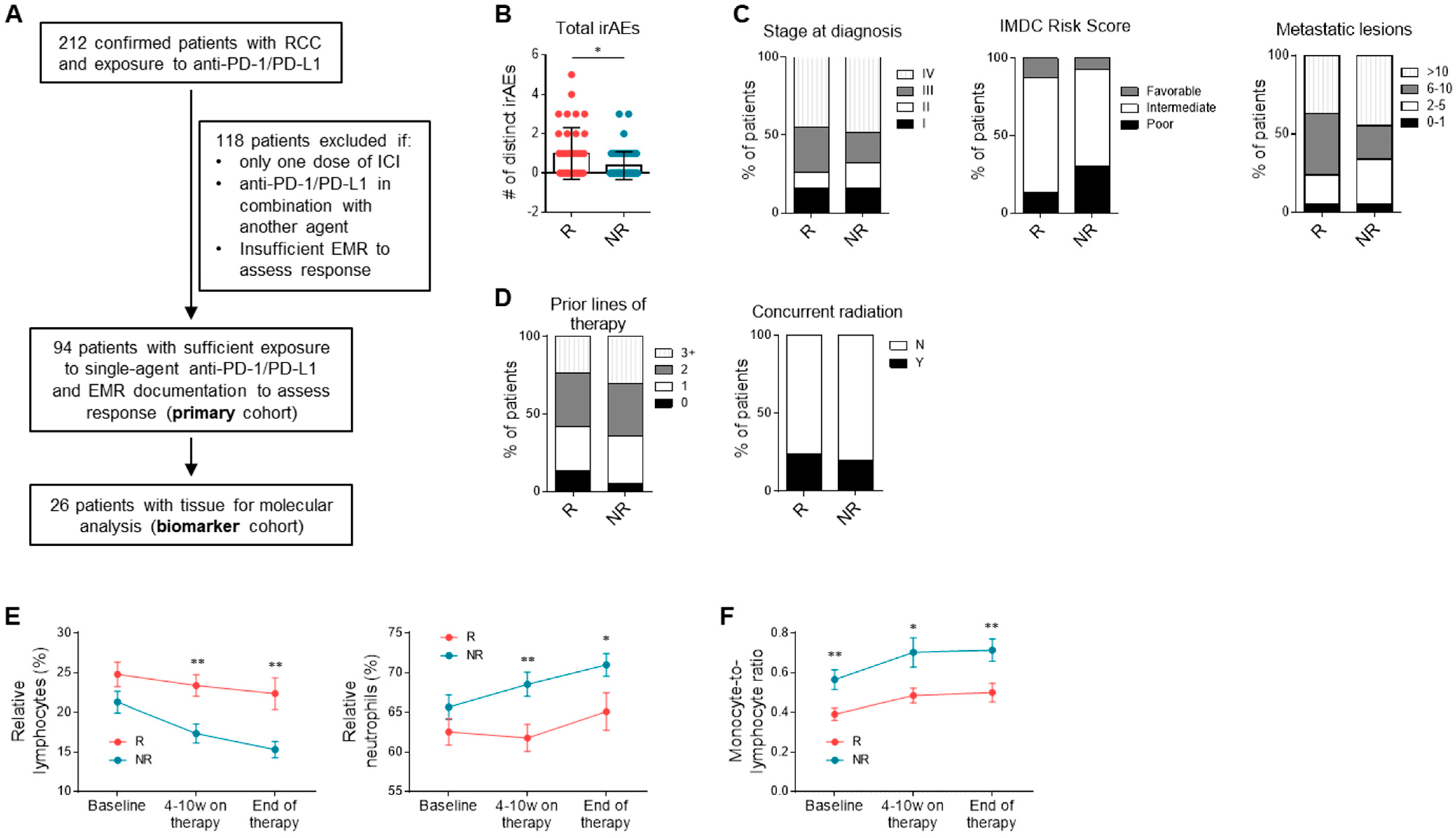
3.1. Study Population
3.2. Clinical Correlates and Response to Anti-PD-1/PD-L1
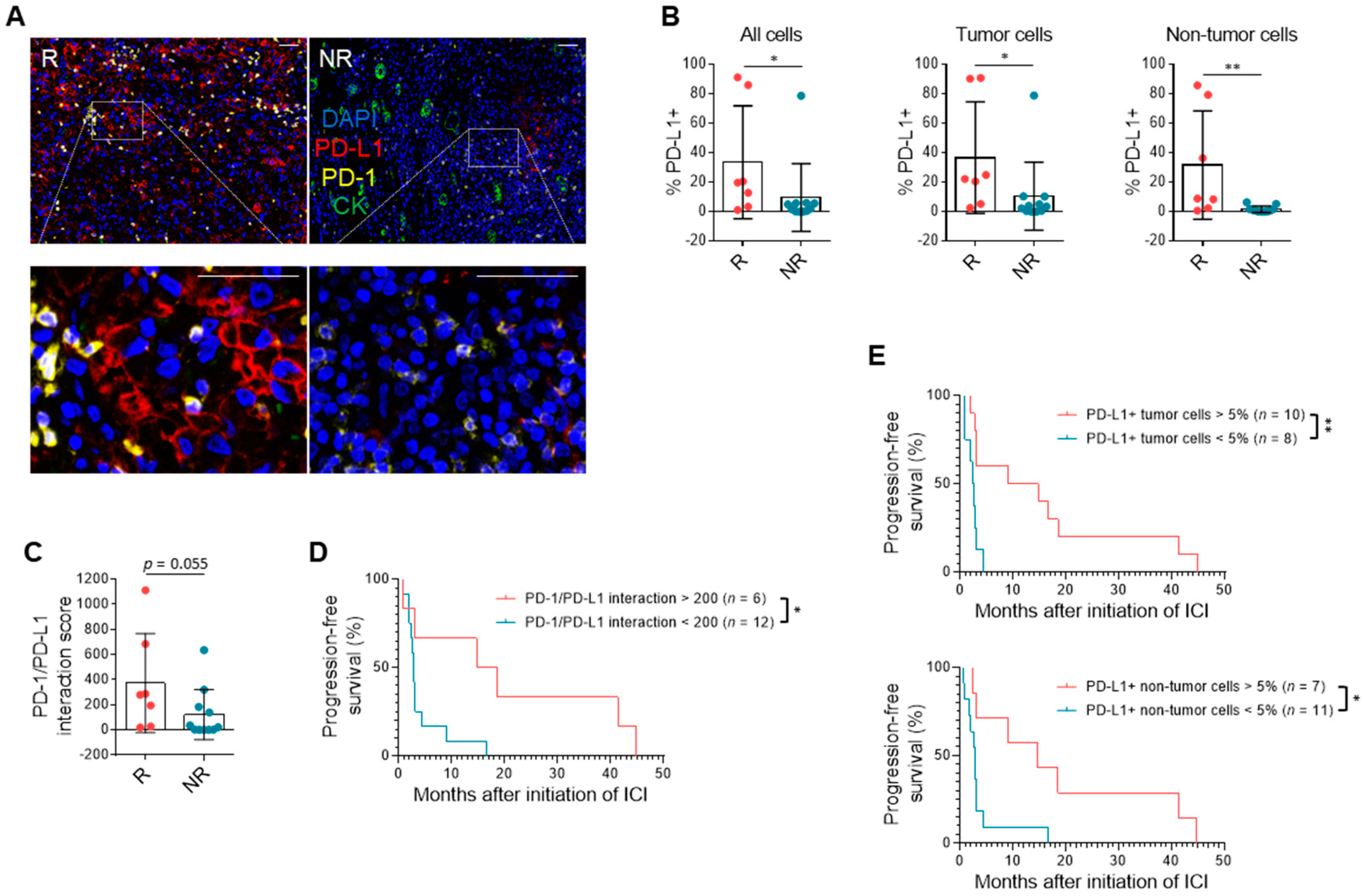
3.3. PD-L1 Expression and Immune Milieu
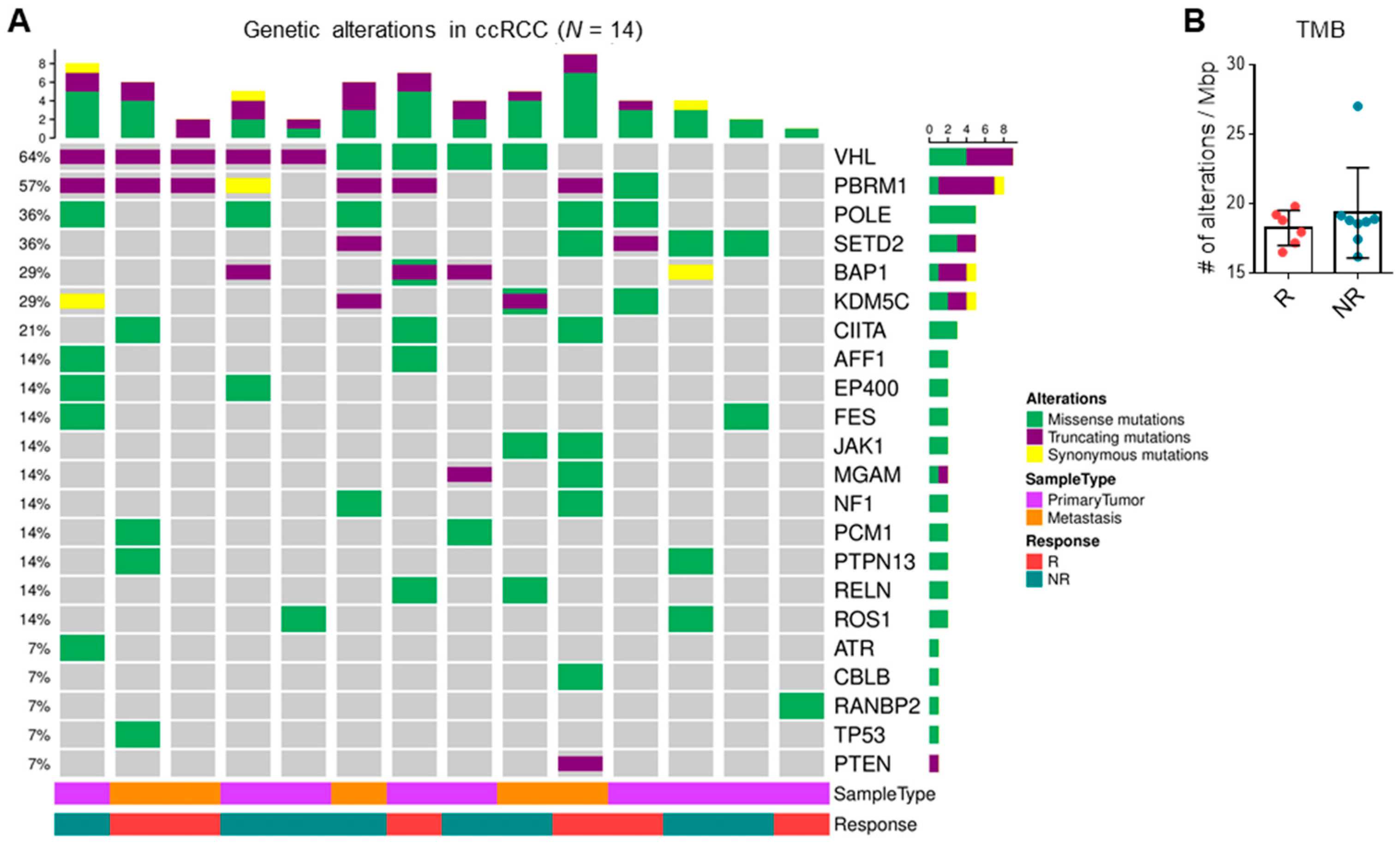
3.4. TMB and Driver Mutations Do Not Correlate with ICI Response
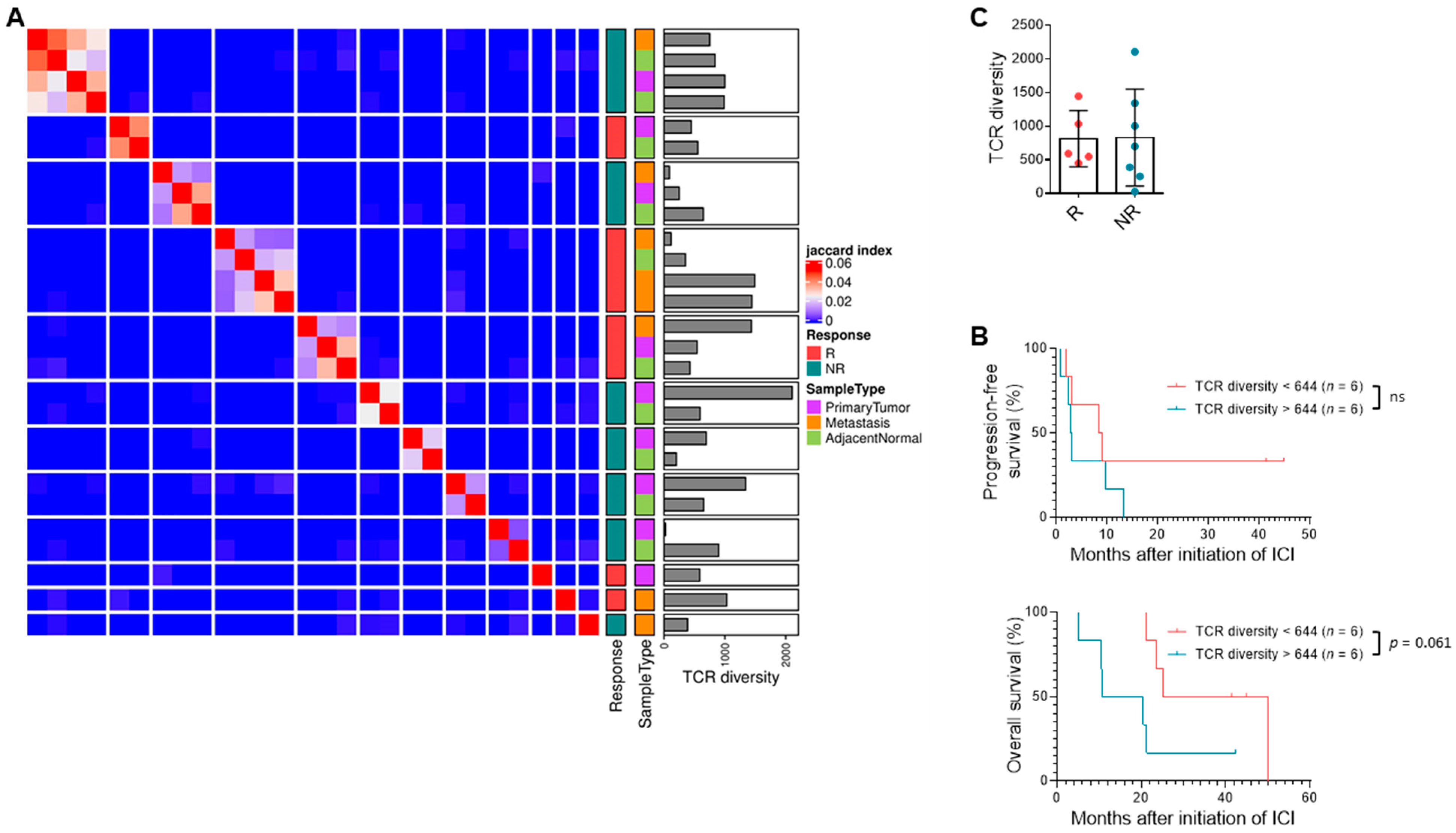
3.5. TCR Clonal Diversity Does Not Correlate with Response but May Impact Survival
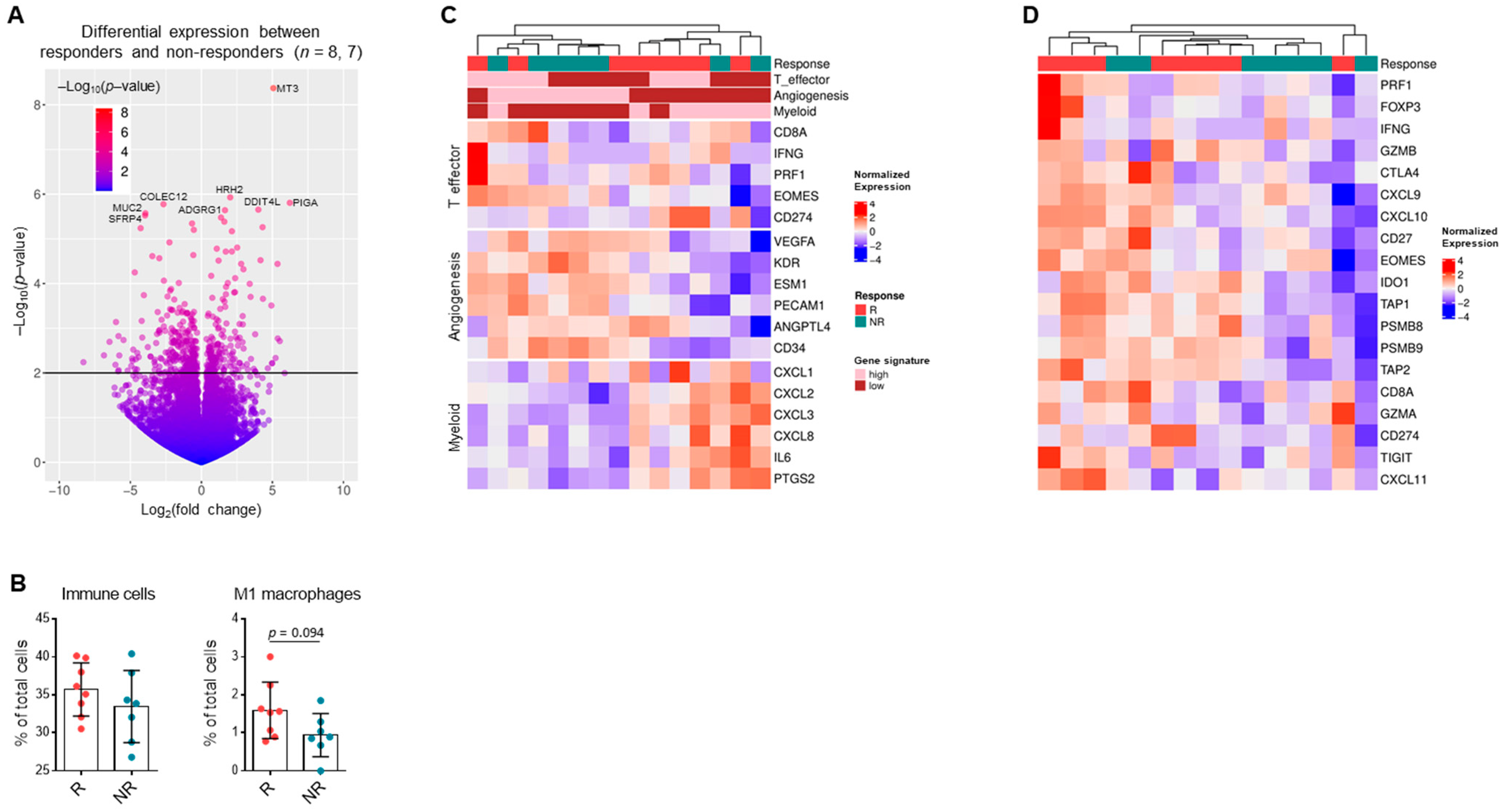
3.6. Gene Expression Patterns in RCC Suggest Response to Single-Agent Immunotherapy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Velasco, G.; Miao, D.; Voss, M.H.; Hakimi, A.A.; Hsieh, J.J.; Tannir, N.M.; Tamboli, P.; Appleman, L.J.; Rathmell, W.K.; Van Allen, E.M.; et al. Tumor Mutational Load and Immune Parameters across Metastatic Renal Cell Carcinoma Risk Groups. Cancer Immunol. Res. 2016, 4, 820–822. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.F.; Lee, J.-L.; Szczylik, C.; Donskov, F.; Malik, J.; Alekseev, B.Y.; Larkin, J.M.G.; Matveev, V.B.; Gafanov, R.A.; Tomczak, P.; et al. Pembrolizumab monotherapy as first-line therapy in advanced clear cell renal cell carcinoma (accRCC): Results from cohort A of KEYNOTE-427. J. Clin. Oncol. 2018, 36, 4500. [Google Scholar] [CrossRef]
- Atkins, M.B.; Jegede, O.; Haas, N.B.; McDermott, D.F.; Bilen, M.A.; Drake, C.G.; Sosman, J.A.; Alter, R.S.; Plimack, E.R.; Rini, B.I.; et al. Phase II study of nivolumab and salvage nivolumab + ipilimumab in treatment-naïve patients (pts) with advanced renal cell carcinoma (RCC) (HCRN GU16-260). J. Clin. Oncol. 2020, 38, 5006. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Frontera, O.A.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Rini, B.I.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, W.M.; Donskov, F.; Lee, J.L.; et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): A multicentre, open-label, phase 3, randomised controlled trial. Lancet 2019, 393, 2404–2415. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef]
- Motzer, R.J.; Robbins, P.B.; Powles, T.; Albiges, L.; Haanen, J.B.; Larkin, J.; Mu, X.J.; Ching, K.A.; Uemura, M.; Pal, S.K.; et al. Avelumab plus axitinib versus sunitinib in advanced renal cell carcinoma: Biomarker analysis of the phase 3 JAVELIN Renal 101 trial. Nat. Med. 2020, 26, 1733–1741. [Google Scholar] [CrossRef]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 2018, 24, 749–757. [Google Scholar] [CrossRef]
- Braun, D.A.; Hou, Y.; Bakouny, Z.; Ficial, M.; Angelo, M.S.; Forman, J.; Ross-Macdonald, P.; Berger, A.C.; Jegede, O.A.; Elagina, L.; et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat. Med. 2020, 26, 909–918. [Google Scholar] [CrossRef]
- Huszno, J.; Kolosza, Z.; Mrochem-Kwarciak, J.; Rutkowski, T.; Skladowski, K. The Role of Neutrophil-Lymphocyte Ratio, Platelet-Lymphocyte Ratio, and Platelets in the Prognosis of Metastatic Renal Cell Carcinoma. Oncology 2019, 97, 7–17. [Google Scholar] [CrossRef]
- Capone, M.; Giannarelli, D.; Mallardo, D.; Madonna, G.; Festino, L.; Grimaldi, A.M.; Vanella, V.; Simeone, E.; Paone, M.; Palmieri, G.; et al. Baseline neutrophil-to-lymphocyte ratio (NLR) and derived NLR could predict overall survival in patients with advanced melanoma treated with nivolumab. J. Immunother. Cancer 2018, 6, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Giorgi, U.; Procopio, G.; Giannarelli, D.; Sabbatini, R.; Bearz, A.; Buti, S.; Basso, U.; Mitterer, M.; Ortega, C.; Bidoli, P.; et al. Association of Systemic Inflammation Index and Body Mass Index with Survival in Patients with Renal Cell Cancer Treated with Nivolumab. Clin. Cancer Res. 2019, 25, 3839–3846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martini, D.J.; Liu, Y.; Shabto, J.M.; Carthon, B.C.; Hitron, E.E.; Russler, G.A.; Caulfield, S.; Kissick, H.T.; Harris, W.B.; Kucuk, O.; et al. Novel Risk Scoring System for Patients with Metastatic Renal Cell Carcinoma Treated with Immune Checkpoint Inhibitors. Oncology 2019, 25, e484–e491. [Google Scholar] [CrossRef] [Green Version]
- Cortellini, A.; Bersanelli, M.; Buti, S.; Cannita, K.; Santini, D.; Perrone, F.; Giusti, R.; Tiseo, M.; Michiara, M.; Di Marino, P.; et al. A multicenter study of body mass index in cancer patients treated with anti-PD-1/PD-L1 immune checkpoint inhibitors: When overweight becomes favorable. J. Immunother. Cancer 2019, 7, 57. [Google Scholar] [CrossRef] [PubMed]
- Labadie, B.W.; Liu, P.; Bao, R.; Crist, M.; Fernandes, R.; Ferreira, L.; Graupner, S.; Poklepovic, A.S.; Duran, I.; Vareki, S.M.; et al. BMI, irAE, and gene expression signatures associate with resistance to immune-checkpoint inhibition and outcomes in renal cell carcinoma. J. Transl. Med. 2019, 17, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Boi, S.K.; Orlandella, R.M.; Gibson, J.T.; Turbitt, W.J.; Wald, G.; Thomas, L.; Rosean, C.B.; Norris, K.E.; Bing, M.; Bertrand, L.; et al. Obesity diminishes response to PD-1-based immunotherapies in renal cancer. J. Immunother. Cancer 2020, 8, e000725. [Google Scholar] [CrossRef]
- Panda, A.; De Cubas, A.A.; Stein, M.; Riedlinger, G.; Kra, J.; Mayer, T.; Smith, C.C.; Vincent, B.G.; Serody, J.S.; Beckermann, K.E.; et al. Endogenous retrovirus expression is associated with response to immune checkpoint pathway in clear cell renal cell carcinoma. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Beckermann, K.E.; Bortone, D.S.; De Cubas, A.A.; Bixby, L.M.; Lee, S.J.; Panda, A.; Ganesan, S.; Bhanot, G.; Wallen, E.M.; et al. Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J. Clin. Investig. 2018, 128, 4804–4820. [Google Scholar] [CrossRef] [Green Version]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Ascierto, M.L.; McMiller, T.L.; Berger, A.E.; Danilova, L.; Anders, R.A.; Netto, G.J.; Xu, H.; Pritchard, T.S.; Fan, J.; Cheadle, C.; et al. The Intratumoral Balance between Metabolic and Immunologic Gene Expression Is Associated with Anti–PD-1 Response in Patients with Renal Cell Carcinoma. Cancer Immunol. Res. 2016, 4, 726–733. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Banchereau, R.; Hamidi, H.; Powles, T.; McDermott, D.; Atkins, M.B.; Escudier, B.; Liu, L.-F.; Leng, N.; Abbas, A.R.; et al. Molecular Subsets in Renal Cancer Determine Outcome to Checkpoint and Angiogenesis Blockade. Cancer Cell 2020, 38, 803–817.e4. [Google Scholar] [CrossRef] [PubMed]
- Miao, D.; Margolis, C.A.; Gao, W.; Voss, M.H.; Li, W.; Martini, D.J.; Norton, C.; Bossé, D.; Wankowicz, S.M.; Cullen, D.; et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018, 359, 801–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, D.A.; Ishii, Y.; Walsh, A.M.; Van Allen, E.M.; Wu, C.J.; Shukla, S.A.; Choueiri, T.K. Clinical Validation of PBRM1 Alterations as a Marker of Immune Checkpoint Inhibitor Response in Renal Cell Carcinoma. JAMA Oncol. 2019, 5, 1631–1633. [Google Scholar] [CrossRef] [PubMed]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Chambers, T.; Lopez, J.I.; Nicol, D.; O’Brien, T.; Larkin, J.; Horswell, S.; et al. Tracking Cancer Evolution Reveals Constrained Routes to Metastases: TRACERx Renal. Cell 2018, 173, 581–594.e12. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.-D.; Kong, W.; Peterson, C.B.; McGrail, D.J.; Hoang, A.; Zhang, X.; Lam, T.; Pilie, P.G.; Zhu, H.; Beckermann, K.E.; et al. PBRM1 loss defines a nonimmunogenic tumor phenotype associated with checkpoint inhibitor resistance in renal carcinoma. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Johnson, D.B.; Bordeaux, J.M.; Kim, J.-Y.; Vaupel, C.A.; Rimm, D.L.; Ho, T.H.; Joseph, R.W.; Daud, A.I.; Conry, R.M.; Gaughan, E.M.; et al. Quantitative Spatial Profiling of PD-1/PD-L1 Interaction and HLA-DR/IDO-1 Predicts Improved Outcomes of anti-PD-1 Therapies in Metastatic Melanoma. Clin. Cancer Res. 2018, 24, 5250–5260. [Google Scholar] [CrossRef] [Green Version]
- Depristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. BioRxiv 2017. [Google Scholar] [CrossRef] [Green Version]
- Voss, K.; Van der Auwera, G.; Gentry, J. Full-stack genomics pipelining with GATK4 + WDL + Cromwell. F1000Research 2017, 6. [Google Scholar] [CrossRef]
- Yang, H.; Wang, K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 2015, 10, 1556–1566. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Minikel, E.V.; Karczewski, K.J.; Martin, H.C.; Cummings, B.B.; Whiffin, N.; Alföldi, J.; MacArthur, D.G.; Genome Aggregation Database (gnomAD) Production Team; Genome Aggregation Database (gnomAD) Consortium; Schreiber, S.L.; et al. Evaluating potential drug targets through human loss-of-function genetic variation. BioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- The 1000 Genomes Project Consortium. An integrated map of genetic variation from 1092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; O’Connor, T.D.; Jun, G.; Kang, H.M.; Abecasis, G.; Leal, S.M.; Gabriel, S.; Rieder, M.J.; Altshuler, D.; Shendure, J.; et al. Analysis of 6515 exomes reveals the recent origin of most human protein-coding variants. Nature 2013, 493, 216–220. [Google Scholar] [CrossRef] [Green Version]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep. 2018, 23, 313–326.e5. [Google Scholar] [CrossRef] [Green Version]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Shugay, M.; Bagaev, D.V.; Turchaninova, M.A.; Bolotin, D.A.; Britanova, O.V.; Putintseva, E.V.; Pogorelyy, M.V.; Nazarov, V.I.; Zvyagin, I.V.; Kirgizova, V.I.; et al. VDJtools: Unifying Post-analysis of T Cell Receptor Repertoires. PLoS Comput. Biol. 2015, 11, e1004503. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Finotello, F.; Mayer, C.; Plattner, C.; Laschober, G.; Rieder, D.; Hackl, H.; Krogsdam, A.; Loncova, Z.; Posch, W.; Wilflingseder, D.; et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 2019, 11, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brannon, A.R.; Reddy, A.; Seiler, M.; Arreola, A.; Moore, D.T.; Pruthi, R.S.; Wallen, E.M.; Nielsen, M.E.; Liu, H.; Nathanson, K.L.; et al. Molecular stratification of clear cell renal cell carci-noma by consensus clustering reveals distinct subtypes and survival patterns. Genes Cancer 2010, 1, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, N.A.; Becht, E.; Vano, Y.; Petitprez, F.; Lacroix, L.; Validire, P.; Sanchez-Salas, R.; Ingels, A.; Oudard, S.; Moatti, A.; et al. Tumor-Infiltrating and Peripheral Blood T-cell Immunophenotypes Predict Early Relapse in Localized Clear Cell Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 4416–4428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singla, N.; Xie, Z.; Zhang, Z.; Gao, M.; Yousuf, Q.; Onabolu, O.; McKenzie, T.; Tcheuyap, V.T.; Ma, Y.; Choi, J.; et al. Pancreatic tropism of metastatic renal cell carcinoma. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, J.; Liu, B. Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front. Immunol. 2018, 9, 978. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Characteristic | Primary Cohort, n = 94 | Responders (CR, PR, Mixed), n = 38 | Non-Responders (PD, SD), n = 56 |
|---|---|---|---|
| Best response to ICI therapy (%) | |||
| CR | 2 (2.1) | 2 (5.3) | 0 (0.0) |
| PR | 23 (24.5) | 23 (60.5) | 0 (0.0) |
| SD | 18 (19.1) | 0 (0.0) | 18 (32.1) |
| PD | 38 (40.4) | 0 (0.0) | 38 (67.9) |
| Mixed | 13 (13.8) | 13 (34.2) | 0 (0.0) |
| Median age at initiation of ICI (range), year | 63 (27–82) | 62 (27–79) | 63 (31–82) |
| Sex (%) | |||
| Male | 71 (75.5) | 30 (78.9) | 41 (73.2) |
| Female | 23 (24.5) | 8 (21.1) | 15 (26.8) |
| Stage at diagnosis (%) | |||
| I | 15 (16.0) | 6 (15.8) | 9 (16.1) |
| II | 13 (13.8) | 4 (10.5) | 9 (16.1) |
| III | 22 (23.4) | 11 (28.9) | 11 (19.6) |
| IV | 44 (46.8) | 17 (44.7) | 27 (48.2) |
| Histology | |||
| Clear cell | 79 (84.0) | 32 (84.2) | 47 (83.9) |
| Papillary | 4 (4.3) | 1 (2.6) | 3 (5.4) |
| Sarcomatoid | 2 (2.1) | 1 (2.6) | 1 (1.8) |
| Chromophobe | 2 (2.1) | 0 (0.0) | 2 (3.6) |
| Undifferentiated | 7 (7.4) | 4 (10.5) | 3 (5.4) |
| IMDC risk group (%) | |||
| Favorable | 9 (9.6) | 5 (13.2) | 4 (7.1) |
| Intermediate | 63 (67.0) | 28 (73.7) | 35 (62.5) |
| Poor | 22 (23.4) | 5 (13.2) | 17 (30.4) |
| Previous therapies (%) | |||
| Nephrectomy | 90 (95.7) | 35 (92.1) | 55 (98.2) |
| Radiation | 32 (34.0) | 13 (34.2) | 19 (33.9) |
| Anti-angiogenic agent | 81 (86.2) | 30 (78.9) | 51 (91.1) |
| mTOR inhibitor | 25 (26.6) | 10 (26.3) | 15 (26.8) |
| High-dose IL-2 | 22 (23.4) | 11 (28.9) | 11 (19.6) |
| ICI agent (%) | |||
| Nivolumab | 79 (84.0) | 28 (73.7) | 51 (91.1) |
| Atezolizumab | 15 (16.0) | 10 (26.3) | 5 (8.9) |
| ICI line of therapy (%) | |||
| First-line | 8 (8.5) | 5 (13.2) | 3 (5.4) |
| Second-line | 28 (29.8) | 11 (28.9) | 17 (30.4) |
| Third-line | 32 (34.0) | 13 (34.2) | 19 (33.9) |
| Fourth-line+ | 26 (27.7) | 9 (23.7) | 17 (30.4) |
| Median duration of ICI therapy (range), days | 189 (12–1637) | 329 (28–1637) **** | 98 (12–769) **** |
| Median survival (95% CI), months | |||
| PFS | 6.6 (4.4–8.7) | 11.1 (9.0–23.6) #### | 3.1 (2.7–5.7) #### |
| OS | 23.5 (20.4–34.1) | 43.6 (29.4–not reached) #### | 16.4 (10.6–23.0) #### |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shiuan, E.; Reddy, A.; Dudzinski, S.O.; Lim, A.R.; Sugiura, A.; Hongo, R.; Young, K.; Liu, X.-D.; Smith, C.C.; O’Neal, J.; et al. Clinical Features and Multiplatform Molecular Analysis Assist in Understanding Patient Response to Anti-PD-1/PD-L1 in Renal Cell Carcinoma. Cancers 2021, 13, 1475. https://doi.org/10.3390/cancers13061475
Shiuan E, Reddy A, Dudzinski SO, Lim AR, Sugiura A, Hongo R, Young K, Liu X-D, Smith CC, O’Neal J, et al. Clinical Features and Multiplatform Molecular Analysis Assist in Understanding Patient Response to Anti-PD-1/PD-L1 in Renal Cell Carcinoma. Cancers. 2021; 13(6):1475. https://doi.org/10.3390/cancers13061475
Chicago/Turabian StyleShiuan, Eileen, Anupama Reddy, Stephanie O. Dudzinski, Aaron R. Lim, Ayaka Sugiura, Rachel Hongo, Kirsten Young, Xian-De Liu, Christof C. Smith, Jamye O’Neal, and et al. 2021. "Clinical Features and Multiplatform Molecular Analysis Assist in Understanding Patient Response to Anti-PD-1/PD-L1 in Renal Cell Carcinoma" Cancers 13, no. 6: 1475. https://doi.org/10.3390/cancers13061475