
Osteoblast-Derived Paracrine and Juxtacrine Signals Protect Disseminated Breast Cancer Cells from Stress

 ,
,  , , , and
, , , and
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
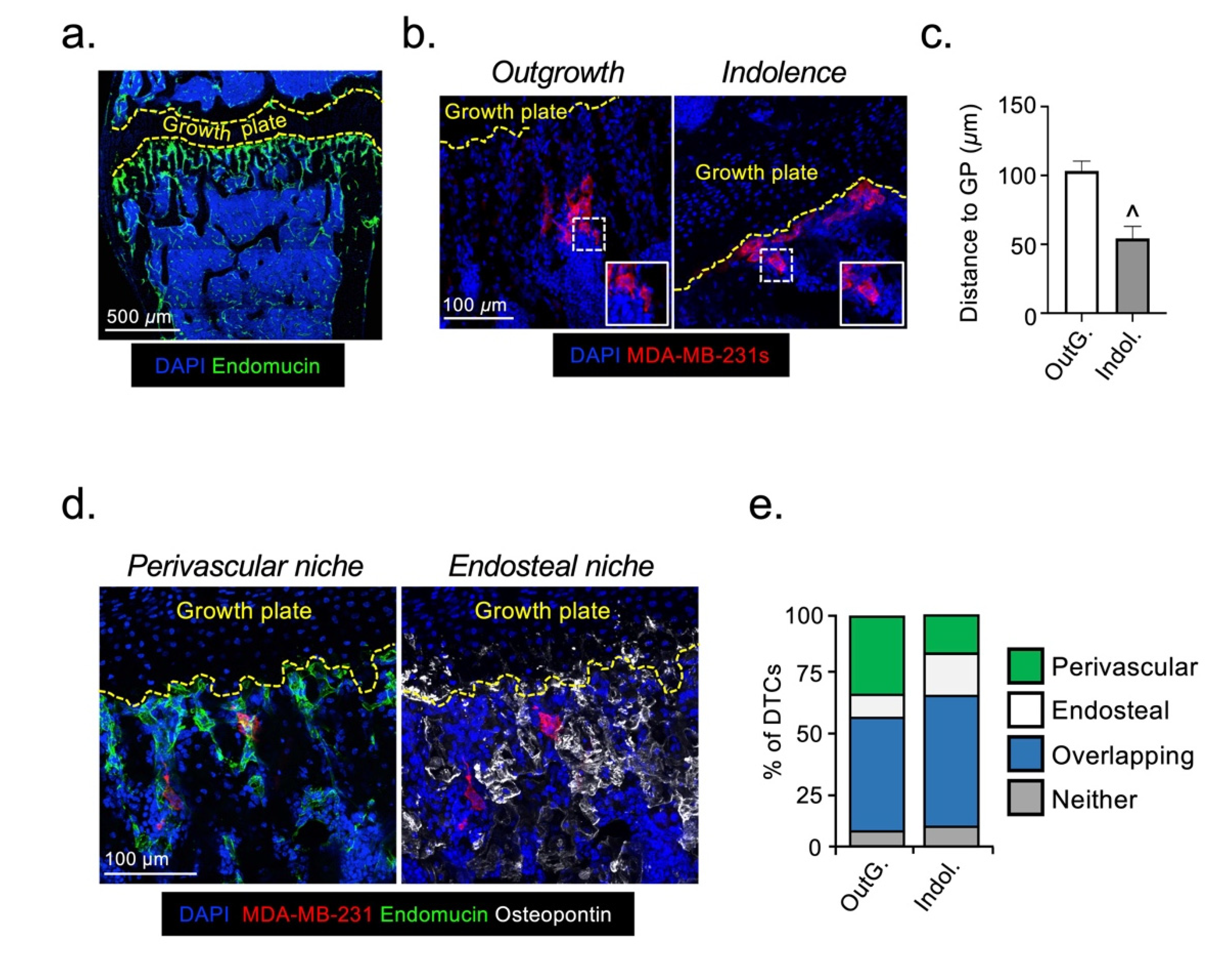
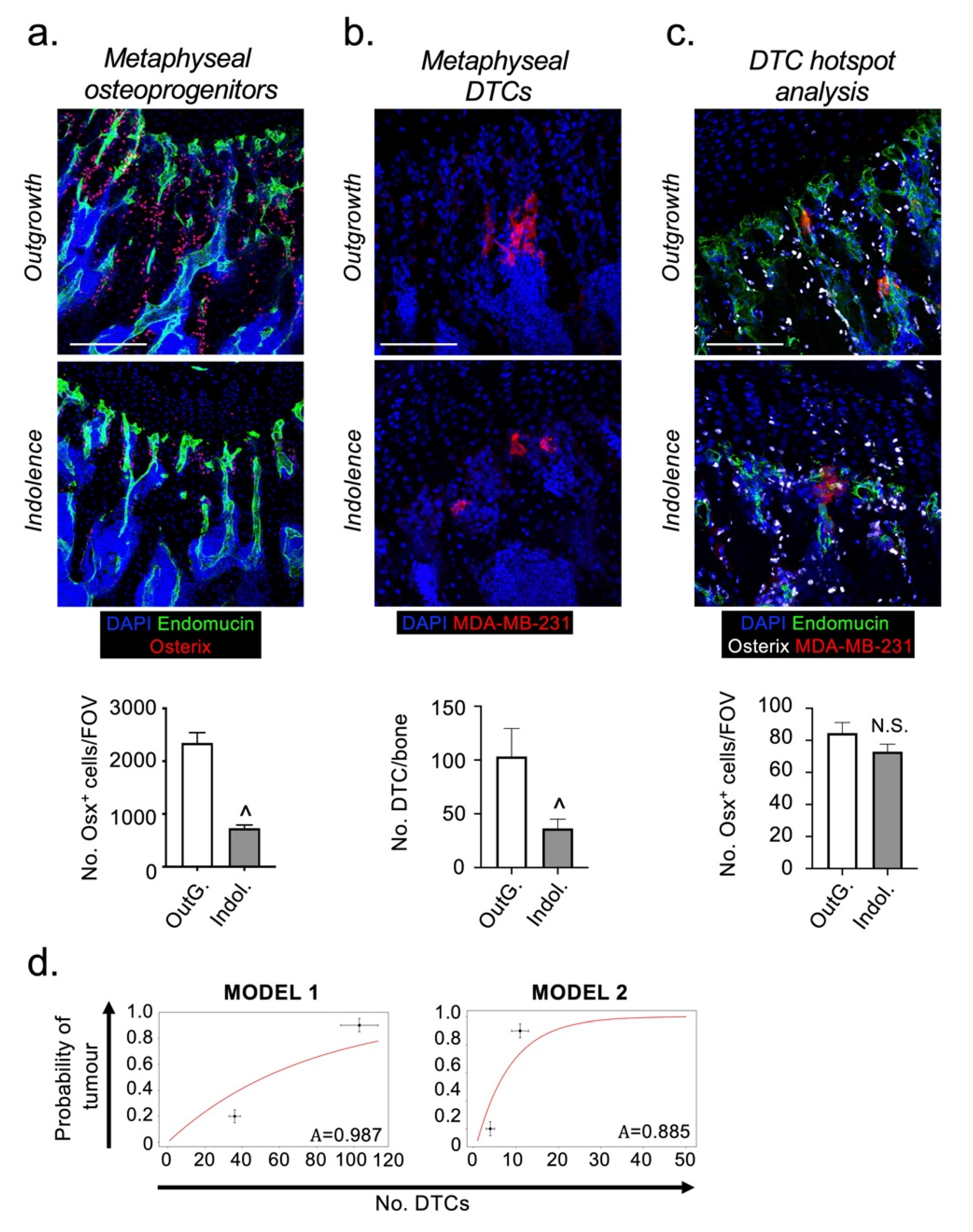
2.1. Disseminated Breast Cancer Cells Occupy Distinct Micro-Anatomical Niches during Metastatic Dormancy Compared to Outgrowth
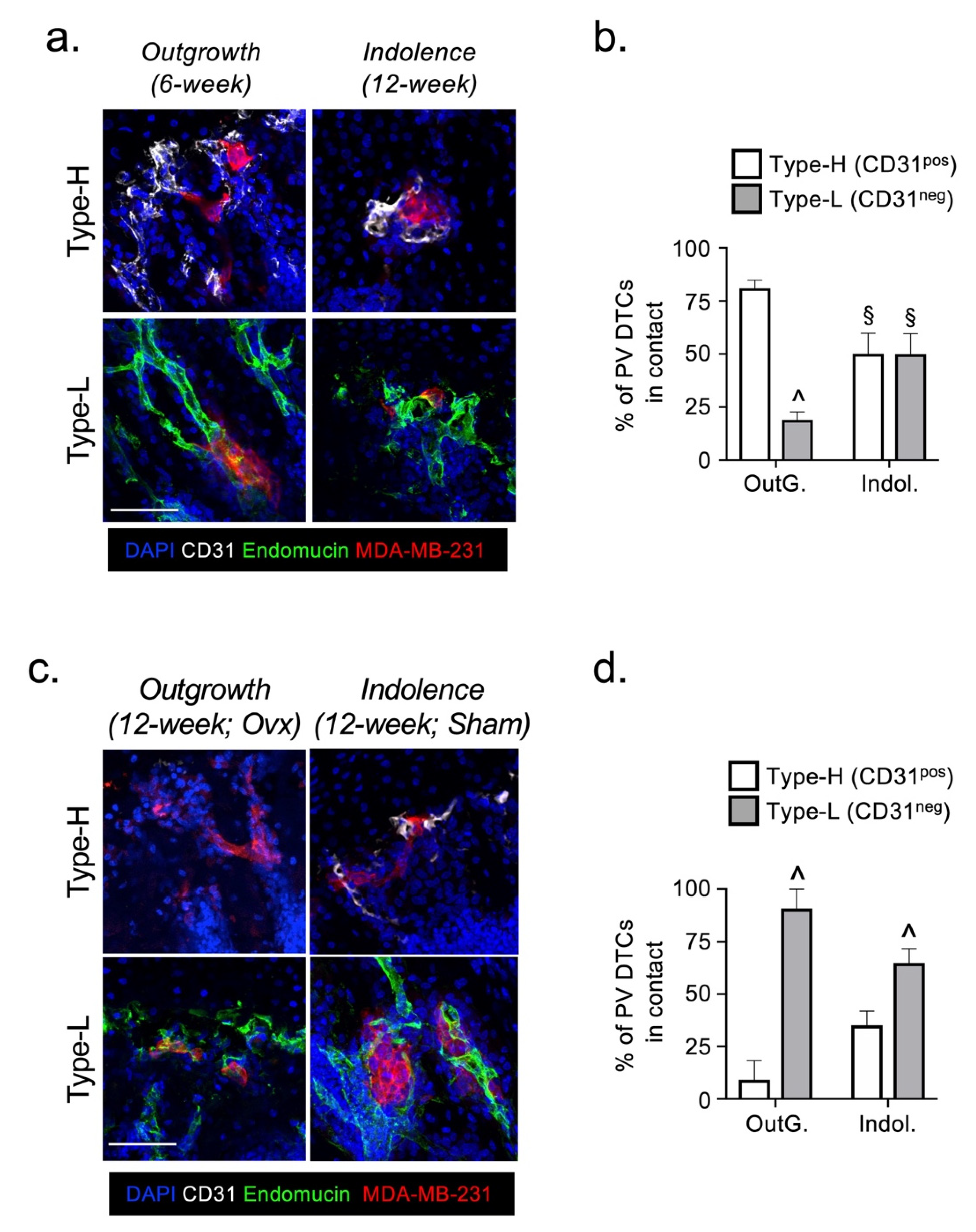
2.2. The Fate of DTCs in Bone Is Not Determined by Their Interaction with Specific Vessel Subtypes
2.3. Osteoprogenitors Are Potential Accessory Cells for DTCs
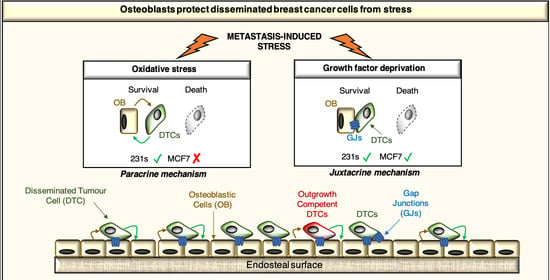
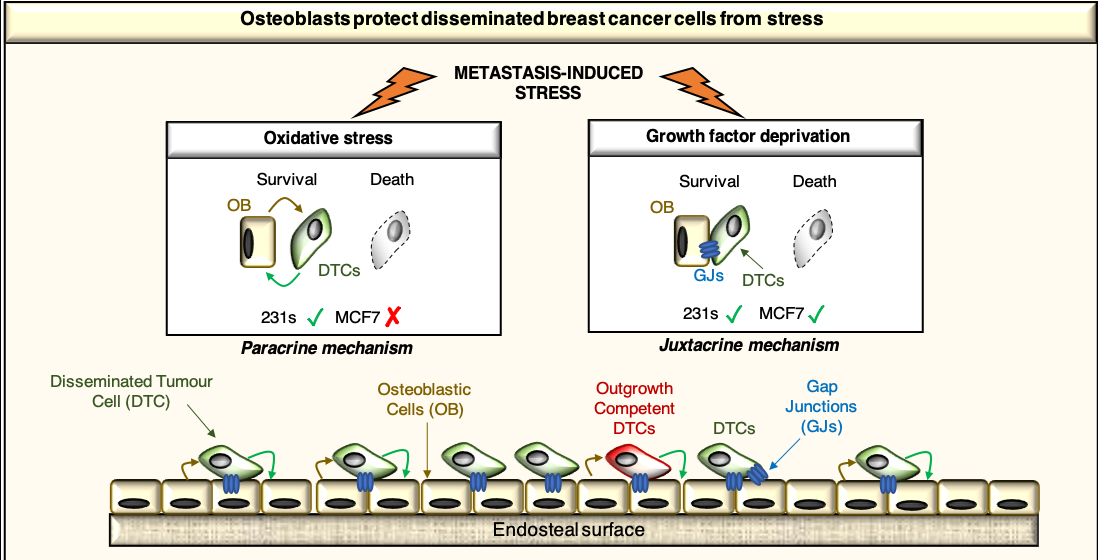
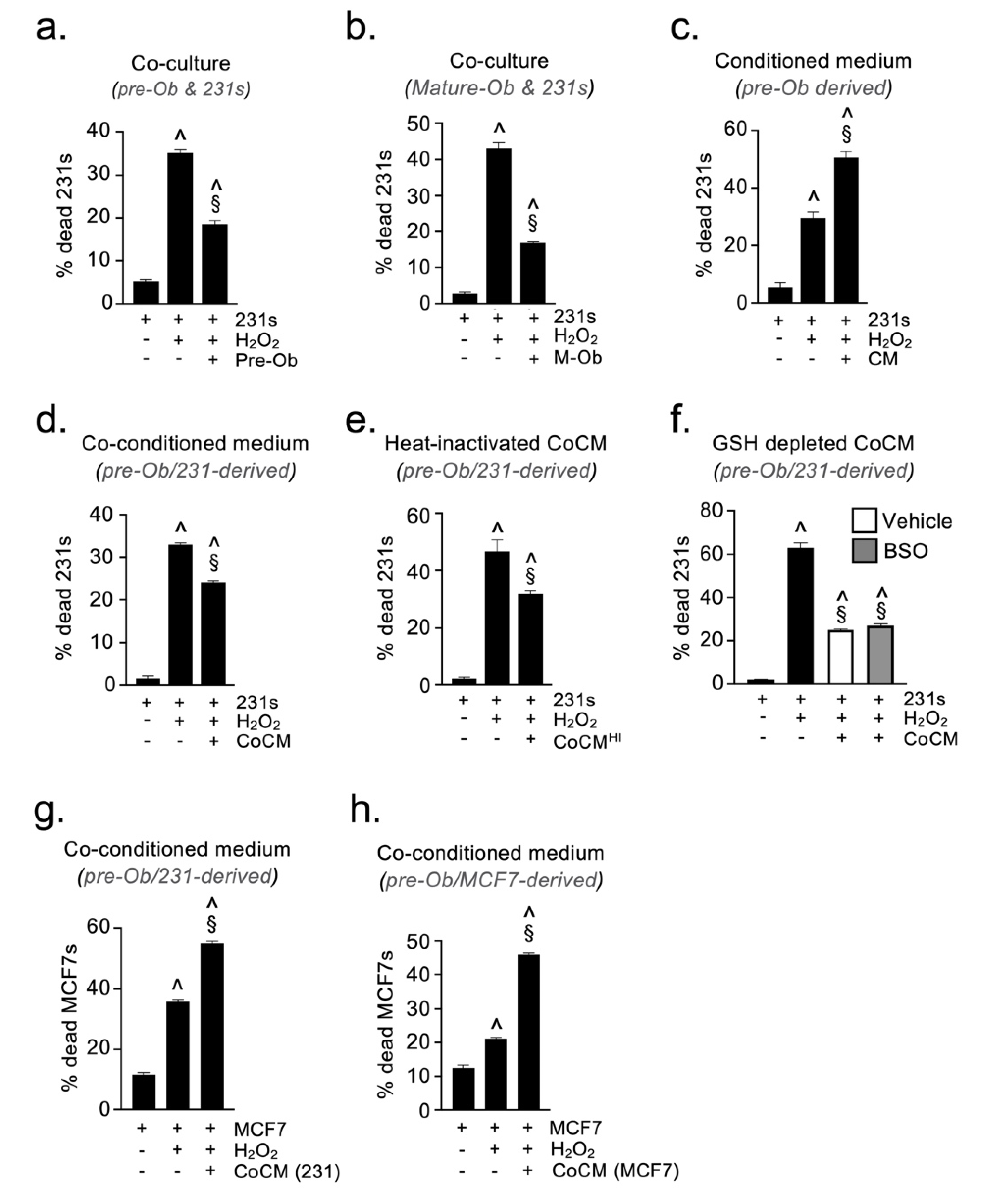
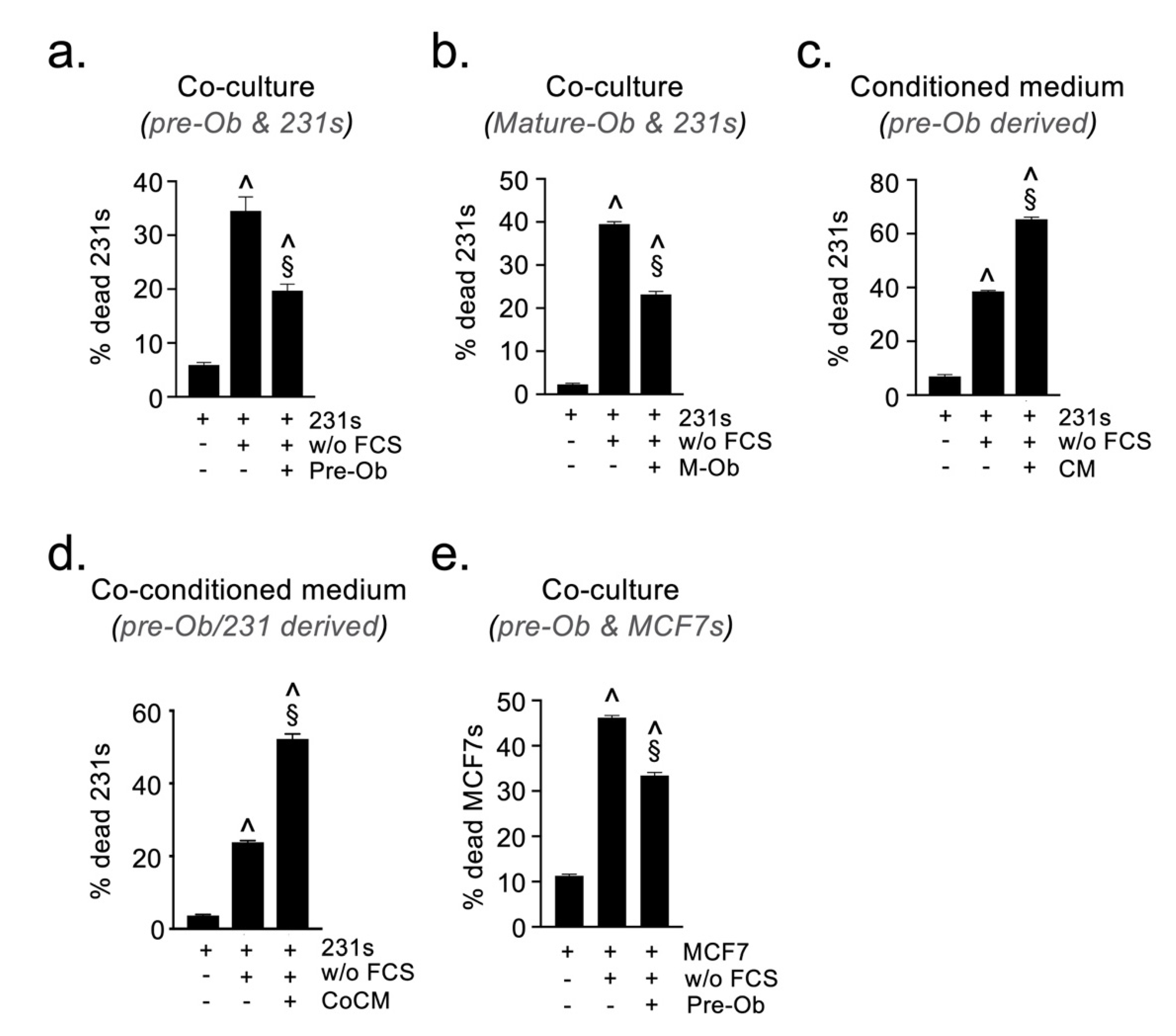
2.4. Osteoblast-Derived Paracrine Signals Protect Breast Cancer Cells from Oxidative Stress
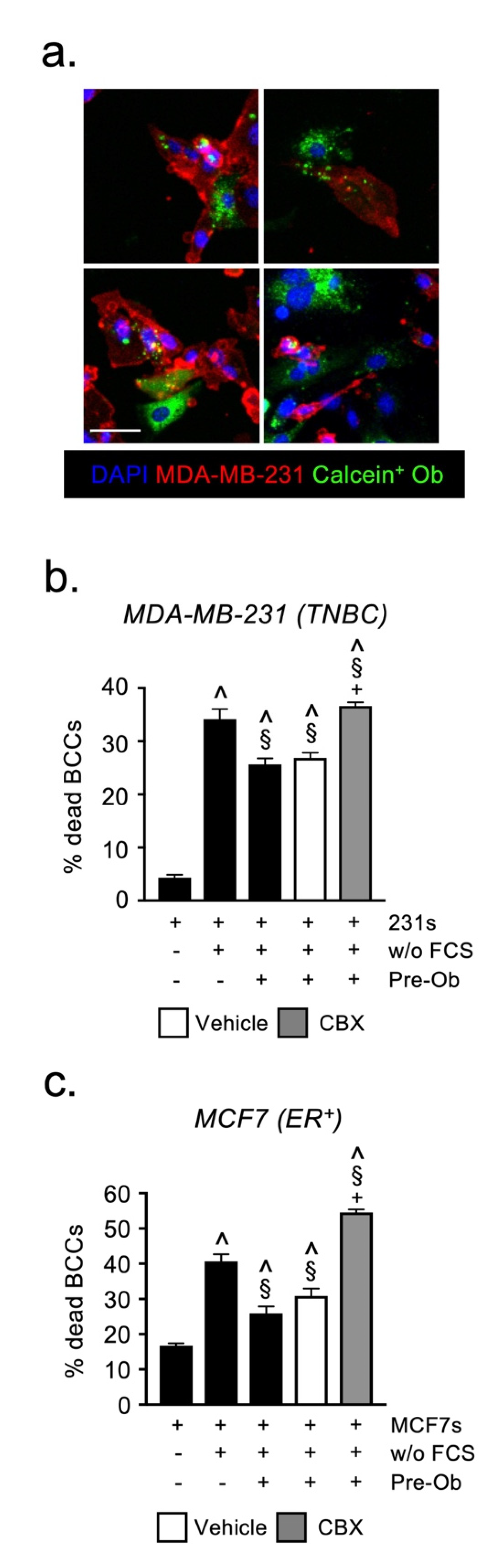
2.5. Osteoblast-Derived Juxtacrine Signaling Protects Breast Cancer Cells from Cell Stress
2.6. Candidate Growth Factors, Cytokines or Angiogenic Mediator Expression Does Not Explain Differences in the Fate of DTCs
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.2. Cell Lines and Tissue Culture, Reagents
4.3. Immunofluorescent Analysis
4.4. Oxidative Stress and Serum Deprivation Assays
4.5. Protein Array and ELISA
4.6. Gap Junction-Mediated Transfer of Calcein
4.7. Real-Time Quantitative PCR
4.8. Mathematical Modelling of Outgrowth Probability
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braun, S.; Pantel, K.; Muller, P.; Janni, W.; Hepp, F.; Kentenich, C.R.; Gastroph, S.; Wischnik, A.; Dimpfl, T.; Kindermann, G.; et al. Cytokeratin-positive cells in the bone marrow and survival of patients with stage I, II, or III breast cancer. N. Engl. J. Med. 2000, 342, 525–533. [Google Scholar] [CrossRef] [Green Version]
- Braun, S.; Vogl, F.D.; Naume, B.; Janni, W.; Osborne, M.P.; Coombes, R.C.; Schlimok, G.; Diel, I.J.; Gerber, B.; Gebauer, G.; et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N. Engl. J. Med. 2005, 353, 793–802. [Google Scholar] [CrossRef]
- Price, T.T.; Burness, M.L.; Sivan, A.; Warner, M.J.; Cheng, R.; Lee, C.H.; Olivere, L.; Comatas, K.; Magnani, J.; Kim Lyerly, H.; et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci. Transl. Med. 2016, 8, 340ra373. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pedersen, E.A.; Havens, A.M.; Jung, Y.; Mishra, A.; Joseph, J.; Kim, J.K.; Patel, L.R.; Ying, C.; Ziegler, A.M.; et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J. Clin. Investig. 2011, 121, 1298–1312. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yu, C.; Gao, X.; Welte, T.; Muscarella, A.M.; Tian, L.; Zhao, H.; Zhao, Z.; Du, S.; Tao, J.; et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell 2015, 27, 193–210. [Google Scholar] [CrossRef] [Green Version]
- Lo Celso, C.; Fleming, H.E.; Wu, J.W.; Zhao, C.X.; Miake-Lye, S.; Fujisaki, J.; Cote, D.; Rowe, D.W.; Lin, C.P.; Scadden, D.T. Live-animal tracking of individual haematopoietic stem/progenitor cells in their niche. Nature 2009, 457, 92–96. [Google Scholar] [CrossRef] [Green Version]
- Carlson, P.; Dasgupta, A.; Grzelak, C.A.; Kim, J.; Barrett, A.; Coleman, I.M.; Shor, R.E.; Goddard, E.T.; Dai, J.; Schweitzer, E.M.; et al. Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat. Cell Biol. 2019, 21, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Esposito, M.; Mondal, N.; Greco, T.M.; Wei, Y.; Spadazzi, C.; Lin, S.C.; Zheng, H.; Cheung, C.; Magnani, J.L.; Lin, S.H.; et al. Bone vascular niche E-selectin induces mesenchymal-epithelial transition and Wnt activation in cancer cells to promote bone metastasis. Nat. Cell Biol. 2019, 21, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Kusumbe, A.P.; Ramasamy, S.K.; Adams, R.H. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 2014, 507, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Bowers, M.; Zhang, B.; Ho, Y.; Agarwal, P.; Chen, C.C.; Bhatia, R. Osteoblast ablation reduces normal long-term hematopoietic stem cell self-renewal but accelerates leukemia development. Blood 2015, 125, 2678–2688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tian, L.; Liu, J.; Goldstein, A.; Bado, I.; Zhang, W.; Arenkiel, B.R.; Li, Z.; Yang, M.; Du, S.; et al. The Osteogenic Niche Is a Calcium Reservoir of Bone Micrometastases and Confers Unexpected Therapeutic Vulnerability. Cancer Cell 2018, 34, 823–839.e827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, M.D.; Schmidt, E.E.; Kerkvliet, N.; Nadkarni, K.V.; Morris, V.L.; Groom, A.C.; Chambers, A.F.; MacDonald, I.C. Temporal progression of metastasis in lung: Cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Res. 2000, 60, 2541–2546. [Google Scholar]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Luzzi, K.J.; MacDonald, I.C.; Schmidt, E.E.; Kerkvliet, N.; Morris, V.L.; Chambers, A.F.; Groom, A.C. Multistep nature of metastatic inefficiency: Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol. 1998, 153, 865–873. [Google Scholar] [CrossRef]
- Ottewell, P.D.; Wang, N.; Brown, H.K.; Reeves, K.J.; Fowles, C.A.; Croucher, P.I.; Eaton, C.L.; Holen, I. Zoledronic acid has differential antitumor activity in the pre- and postmenopausal bone microenvironment in vivo. Clin. Cancer Res. 2014, 20, 2922–2932. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Reeves, K.J.; Brown, H.K.; Fowles, A.C.; Docherty, F.E.; Ottewell, P.D.; Croucher, P.I.; Holen, I.; Eaton, C.L. The frequency of osteolytic bone metastasis is determined by conditions of the soil, not the number of seeds; evidence from in vivo models of breast and prostate cancer. J. Exp. Clin. Cancer Res. 2015, 34, 124. [Google Scholar] [CrossRef] [Green Version]
- Allocca, G.; Hughes, R.; Wang, N.; Brown, H.K.; Ottewell, P.D.; Brown, N.J.; Holen, I. The bone metastasis niche in breast cancer-potential overlap with the haematopoietic stem cell niche in vivo. J. Bone Oncol. 2019, 17, 100244. [Google Scholar] [CrossRef]
- Bixel, M.G.; Kusumbe, A.P.; Ramasamy, S.K.; Sivaraj, K.K.; Butz, S.; Vestweber, D.; Adams, R.H. Flow Dynamics and HSPC Homing in Bone Marrow Microvessels. Cell Rep. 2017, 18, 1804–1816. [Google Scholar] [CrossRef] [Green Version]
- Kunisaki, Y.; Bruns, I.; Scheiermann, C.; Ahmed, J.; Pinho, S.; Zhang, D.; Mizoguchi, T.; Wei, Q.; Lucas, D.; Ito, K.; et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 2013, 502, 637–643. [Google Scholar] [CrossRef] [Green Version]
- Kusumbe, A.P.; Ramasamy, S.K.; Itkin, T.; Mae, M.A.; Langen, U.H.; Betsholtz, C.; Lapidot, T.; Adams, R.H. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature 2016, 532, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Sacchetti, B.; Funari, A.; Michienzi, S.; Di Cesare, S.; Piersanti, S.; Saggio, I.; Tagliafico, E.; Ferrari, S.; Robey, P.G.; Riminucci, M.; et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 2007, 131, 324–336. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Niu, C.; Ye, L.; Huang, H.; He, X.; Tong, W.G.; Ross, J.; Haug, J.; Johnson, T.; Feng, J.Q.; et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 2003, 425, 836–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusumbe, A.P.; Ramasamy, S.K.; Starsichova, A.; Adams, R.H. Sample preparation for high-resolution 3D confocal imaging of mouse skeletal tissue. Nat. Protoc. 2015, 10, 1904–1914. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.; Chen, X.; Hunter, K.D.; Hobbs, J.K.; Holen, I.; Brown, N.J. Bone marrow osteoprogenitors are depleted whereas osteoblasts are expanded independent of the osteogenic vasculature in response to zoledronic acid. FASEB J. 2019, 33, 12768–12779. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordon-Cardo, C.; Guise, T.A.; Massague, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Mu, E.; Wei, Y.; Riethdorf, S.; Yang, Q.; Yuan, M.; Yan, J.; Hua, Y.; Tiede, B.J.; Lu, X.; et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging alpha4beta1-positive osteoclast progenitors. Cancer Cell 2011, 20, 701–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caino, M.C.; Chae, Y.C.; Vaira, V.; Ferrero, S.; Nosotti, M.; Martin, N.M.; Weeraratna, A.; O’Connell, M.; Jernigan, D.; Fatatis, A.; et al. Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. J. Clin. Investig. 2013, 123, 2907–2920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humtsoe, J.O.; Kramer, R.H. Differential epidermal growth factor receptor signaling regulates anchorage-independent growth by modulation of the PI3K/AKT pathway. Oncogene 2010, 29, 1214–1226. [Google Scholar] [CrossRef] [Green Version]
- Jo, H.; Jia, Y.; Subramanian, K.K.; Hattori, H.; Luo, H.R. Cancer cell-derived clusterin modulates the phosphatidylinositol 3’-kinase-Akt pathway through attenuation of insulin-like growth factor 1 during serum deprivation. Mol. Cell Biol. 2008, 28, 4285–4299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simoni, R.D.; Hill, R.L.; Vaughan, M. On glutathione. II. A thermostable oxidation-reduction system (Hopkins, F. G., and Dixon, M. (1922) J. Biol. Chem. 54, 527-563). J. Biol. Chem. 2002, 277, e13. [Google Scholar] [PubMed]
- Griffith, O.W. Mechanism of action, metabolism, and toxicity of buthionine sulfoximine and its higher homologs, potent inhibitors of glutathione synthesis. J. Biol. Chem. 1982, 257, 13704–13712. [Google Scholar] [CrossRef]
- Korah, R.; Boots, M.; Wieder, R. Integrin alpha5beta1 promotes survival of growth-arrested breast cancer cells: An in vitro paradigm for breast cancer dormancy in bone marrow. Cancer Res. 2004, 64, 4514–4522. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, N.; Puzon-McLaughlin, W.; Irie, A.; Morikawa, Y.; Kakudo, K.; Takada, Y. Induction of experimental bone metastasis in mice by transfection of integrin alpha 4 beta 1 into tumor cells. Am. J. Pathol. 1996, 148, 55–61. [Google Scholar]
- McCabe, N.P.; De, S.; Vasanji, A.; Brainard, J.; Byzova, T.V. Prostate cancer specific integrin alphavbeta3 modulates bone metastatic growth and tissue remodeling. Oncogene 2007, 26, 6238–6243. [Google Scholar] [CrossRef] [Green Version]
- Boyerinas, B.; Zafrir, M.; Yesilkanal, A.E.; Price, T.T.; Hyjek, E.M.; Sipkins, D.A. Adhesion to osteopontin in the bone marrow niche regulates lymphoblastic leukemia cell dormancy. Blood 2013, 121, 4821–4831. [Google Scholar] [CrossRef] [Green Version]
- Ottewell, P.D.; Wang, N.; Brown, H.K.; Fowles, C.A.; Croucher, P.I.; Eaton, C.L.; Holen, I. OPG-Fc inhibits ovariectomy-induced growth of disseminated breast cancer cells in bone. Int. J. Cancer 2015, 137, 968–977. [Google Scholar] [CrossRef]
- Becerikli, M.; Jaurich, H.; Schira, J.; Schulte, M.; Dobele, C.; Wallner, C.; Abraham, S.; Wagner, J.M.; Dadras, M.; Kneser, U.; et al. Age-dependent alterations in osteoblast and osteoclast activity in human cancellous bone. J. Cell Mol. Med. 2017, 21, 2773–2781. [Google Scholar] [CrossRef] [Green Version]
- Lips, P.; Courpron, P.; Meunier, P.J. Mean wall thickness of trabecular bone packets in the human iliac crest: Changes with age. Calcif. Tissue Res. 1978, 26, 13–17. [Google Scholar] [CrossRef]
- Donahue, H.J.; Saunders, M.M.; Li, Z.; Mastro, A.M.; Gay, C.V.; Welch, D.R. A potential role for gap junctions in breast cancer metastasis to bone. J. Musculoskelet. Neuronal Interact. 2003, 3, 156–161. [Google Scholar] [PubMed]
- Kolb, A.D.; Shupp, A.B.; Mukhopadhyay, D.; Marini, F.C.; Bussard, K.M. Osteoblasts are “educated” by crosstalk with metastatic breast cancer cells in the bone tumor microenvironment. Breast Cancer Res. 2019, 21, 31. [Google Scholar] [CrossRef] [Green Version]
- Yu-Lee, L.Y.; Lee, Y.C.; Pan, J.; Lin, S.C.; Pan, T.; Yu, G.; Hawke, D.H.; Pan, B.F.; Lin, S.H. Bone secreted factors induce cellular quiescence in prostate cancer cells. Sci. Rep. 2019, 9, 18635. [Google Scholar] [CrossRef] [PubMed]
- Yu-Lee, L.Y.; Yu, G.; Lee, Y.C.; Lin, S.C.; Pan, J.; Pan, T.; Yu, K.J.; Liu, B.; Creighton, C.J.; Rodriguez-Canales, J.; et al. Osteoblast-Secreted Factors Mediate Dormancy of Metastatic Prostate Cancer in the Bone via Activation of the TGFbetaRIII-p38MAPK-pS249/T252RB Pathway. Cancer Res. 2018, 78, 2911–2924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, P.; Saunders, M.M.; Li, Z.; Zhou, Z.; Sheaffer, N.; Kunze, E.L.; Samant, R.S.; Welch, D.R.; Donahue, H.J. Breast cancer metastatic potential: Correlation with increased heterotypic gap junctional intercellular communication between breast cancer cells and osteoblastic cells. Int. J. Cancer 2004, 111, 693–697. [Google Scholar] [CrossRef]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef] [Green Version]
- Isnaini, I.; Permatasari, N.; Mintaroem, K.; Prihardina, B.; Widodo, M.A. Oxidants-Antioxidants Profile in the Breast Cancer Cell Line MCF-7. Asian Pac. J. Cancer Prev. 2018, 19, 3175–3178. [Google Scholar] [CrossRef] [Green Version]
- Mobley, J.A.; Brueggemeier, R.W. Estrogen receptor-mediated regulation of oxidative stress and DNA damage in breast cancer. Carcinogenesis 2004, 25, 3–9. [Google Scholar] [CrossRef]
- Berger, C.E.; Qian, Y.; Liu, G.; Chen, H.; Chen, X. p53, a target of estrogen receptor (ER) alpha, modulates DNA damage-induced growth suppression in ER-positive breast cancer cells. J. Biol. Chem. 2012, 287, 30117–30127. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.Z.; Cherian, M.G. Potential role of p53 on metallothionein induction in human epithelial breast cancer cells. Br. J. Cancer 2002, 87, 1019–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiaraviglio, L.; Kirby, J.E. Evaluation of impermeant, DNA-binding dye fluorescence as a real-time readout of eukaryotic cell toxicity in a high throughput screening format. Assay Drug Dev. Technol. 2014, 12, 219–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirons, G.T.; Fawcett, J.J.; Crissman, H.A. TOTO and YOYO: New very bright fluorochromes for DNA content analyses by flow cytometry. Cytometry 1994, 15, 129–140. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hughes, R.; Chen, X.; Cowley, N.; Ottewell, P.D.; Hawkins, R.J.; Hunter, K.D.; Hobbs, J.K.; Brown, N.J.; Holen, I. Osteoblast-Derived Paracrine and Juxtacrine Signals Protect Disseminated Breast Cancer Cells from Stress. Cancers 2021, 13, 1366. https://doi.org/10.3390/cancers13061366
Hughes R, Chen X, Cowley N, Ottewell PD, Hawkins RJ, Hunter KD, Hobbs JK, Brown NJ, Holen I. Osteoblast-Derived Paracrine and Juxtacrine Signals Protect Disseminated Breast Cancer Cells from Stress. Cancers. 2021; 13(6):1366. https://doi.org/10.3390/cancers13061366
Chicago/Turabian StyleHughes, Russell, Xinyue Chen, Natasha Cowley, Penelope D. Ottewell, Rhoda J. Hawkins, Keith D. Hunter, Jamie K. Hobbs, Nicola J. Brown, and Ingunn Holen. 2021. "Osteoblast-Derived Paracrine and Juxtacrine Signals Protect Disseminated Breast Cancer Cells from Stress" Cancers 13, no. 6: 1366. https://doi.org/10.3390/cancers13061366