
Oncolytic Virus Therapy Alters the Secretome of Targeted Glioblastoma Cells

 ,
,
Abstract
:Simple Summary
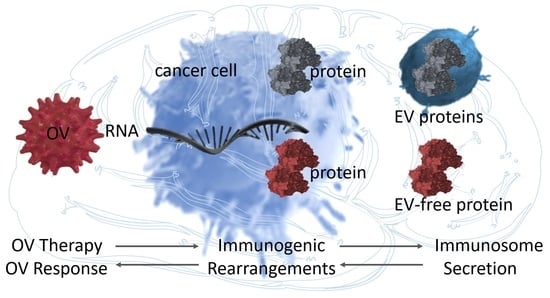
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Viral Particle Preparation
2.3. Viral Infection Assay
2.4. Quantitative PCR
2.5. Immunofluorescence
2.6. Electron Microscopy
2.7. Gene Microarray
2.8. Arrays Data Analysis
2.9. Mass Spectrometry-Based Proteomics
2.10. Proteomic Data Analysis
2.11. Performing Gene Survival Analysis
2.12. Quantification and Statistical Analysis
2.13. Key Resources Table
3. Results
3.1. OV Infection Alters the Transcriptome of Patient-Derived GSCs toward the Extracellular Mode
3.2. OV Infection Leads to Quantitative and Qualitative Changes in Both the EV-Depleted and EV Secretome of GSCs
3.3. Proteins Secreted by Mock- and OV-Infected GSCs End Up in Non-Overlapping EV-Depleted and EV Fractions
3.4. The Re-Configuration of GSCs’ Transcriptome upon OV Infection Predicts the Composition of Their “Immune Mode” Secretome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Drake, C.G.; Lipson, E.J.; Brahmer, J.R. Breathing new life into immunotherapy: Review of melanoma, lung and kidney cancer. Nat. Rev. Clin. Oncol. 2014, 11, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Bovenberg, M.S.S.; Degeling, M.H.; Tannous, B.A. Cell-based Immunotherapy Against Gliomas: From Bench to Bedside. Mol. Ther. 2013, 21, 1297–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Wucherpfennig, K.; Chiocca, E.A. Immunotherapy for glioblastoma: On the sidelines or in the game? Discov. Med. 2017, 24, 201–208. [Google Scholar] [PubMed]
- Iorgulescu, J.B.; Reardon, D.A.; Chiocca, E.A.; Wu, C.J. Immunotherapy for glioblastoma: Going viral. Nat. Med. 2018, 24, 1094–1096. [Google Scholar] [CrossRef]
- Russell, S.J.; Barber, G.N. Oncolytic Viruses as Antigen-Agnostic Cancer Vaccines. Cancer Cell 2018, 33, 599–605. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Swami, U.; Swick, B.; Zakharia, Y.; Milhem, M. Clinical Challenges with Talimogene Laherparepvec: Cured Lymph Nodes Masquerading as Active Melanoma. Case Rep. Oncol. Med. 2019, 2019, 4683531–4683535. [Google Scholar] [CrossRef] [PubMed]
- Lawler, S.E.; Speranza, M.C.; Cho, C.F.; Chiocca, E.A. Oncolytic Viruses in Cancer Treatment: A Review. JAMA Oncol. 2016, 3, 841–849. [Google Scholar] [CrossRef] [Green Version]
- Kambara, H.; Okano, H.; Chiocca, E.A.; Saeki, Y. An Oncolytic HSV-1 Mutant Expressing ICP34.5 under Control of a Nestin Promoter Increases Survival of Animals even when Symptomatic from a Brain Tumor. Cancer Res. 2005, 65, 2832–2839. [Google Scholar] [CrossRef] [Green Version]
- Mao, P.; Joshi, K.; Li, J.; Kim, S.-H.; Li, P.; Santana-Santos, L.; Luthra, S.; Chandran, U.R.; Benos, P.V.; Smith, L.; et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc. Natl. Acad. Sci. USA 2013, 110, 8644–8649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronisz, A.; Rooj, A.K.; Krawczyński, K.; Peruzzi, P.; Salińska, E.; Nakano, I.; Purow, B.; Chiocca, E.A.; Godlewski, J. The nuclear DICER–circular RNA complex drives the deregulation of the glioblastoma cell microRNAome. Sci. Adv. 2020, 6, eabc0221. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, F. Advances and potential pitfalls of oncolytic viruses expressing immunomodulatory transgene therapy for malignant gliomas. Cell Death Dis. 2020, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Alexander, B.M.; Cloughesy, T.F. Adult Glioblastoma. J. Clin. Oncol. 2017, 35, 2402–2409. [Google Scholar] [CrossRef] [PubMed]
- Nakano, I.; Garnier, D.; Minata, M.; Rak, J. Extracellular vesicles in the biology of brain tumour stem cells—Implications for inter-cellular communication, therapy and biomarker development. Semin. Cell Dev. Biol. 2015, 40, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, W.; Freeman, M.L.; Lederman, M.M.; Vasilieva, E.; Romero, R.; Margolis, L. A System of Cytokines Encapsulated in ExtraCellular Vesicles. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Van Niel, G.; d’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Exosomes and tumor-mediated immune suppression. J. Clin. Investig. 2016, 126, 1216–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, F.; Zhou, X.; Fang, M.; Li, H.; Su, P.; Tu, Y.; Zhang, L.; Zhou, F. Extracellular Vesicles in Cancer Immune Microenvironment and Cancer Immunotherapy. Adv. Sci. 2019, 6, 1901779. [Google Scholar] [CrossRef]
- Garofalo, M.; Saari, H.; Somersalo, P.; Crescenti, D.; Kuryk, L.; Aksela, L.; Capasso, C.; Madetoja, M.; Koskinen, K.; Oksanen, T.; et al. Antitumor effect of oncolytic virus and paclitaxel encapsulated in extracellular vesicles for lung cancer treatment. J. Control. Release 2018, 283, 223–234. [Google Scholar] [CrossRef]
- Garofalo, M.; Villa, A.; Rizzi, N.; Kuryk, L.; Rinner, B.; Cerullo, V.; Yliperttula, M.; Mazzaferro, V.; Ciana, P. Extracellular vesicles enhance the targeted delivery of immunogenic oncolytic adenovirus and paclitaxel in immunocompetent mice. J. Control. Release 2019, 294, 165–175. [Google Scholar] [CrossRef]
- Ferguson, M.S.; Lemoine, N.R.; Wang, Y. Systemic Delivery of Oncolytic Viruses: Hopes and Hurdles. Adv. Virol. 2012, 2012, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Melcher, A.; Parato, K.; Rooney, C.M.; Bell, J.C. Thunder and Lightning: Immunotherapy and Oncolytic Viruses Collide. Mol. Ther. 2011, 19, 1008–1016. [Google Scholar] [CrossRef]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef]
- Ening, J.; Ewakimoto, H. Oncolytic herpes simplex virus-based strategies: Toward a breakthrough in glioblastoma therapy. Front. Microbiol. 2014, 5, 303. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Xu, R.; Greening, D.W.; Zhu, H.-J.; Takahashi, N.; Simpson, R.J. Extracellular vesicle isolation and characterization: Toward clinical application. J. Clin. Investig. 2016, 126, 1152–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, P.; Mani, S. Methodologies to decipher the cell secretome. Biochim. Biophys. Acta Bioenerg. 2013, 1834, 2226–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Butler, J.M. Short Tandem Repeat Analysis for Human Identity Testing. Curr. Protoc. Hum. Genet. 2004, 41, 14.8.1–14.8.22. [Google Scholar] [CrossRef]
- Desai, P.; Person, S. Incorporation of the Green Fluorescent Protein into the Herpes Simplex Virus Type 1 Capsid. J. Virol. 1998, 72, 7563–7568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krisky, D.M.; Wolfe, D.; Goins, W.F.; Marconi, P.C.; Ramakrishnan, R.; Mata, M.; Rouse, R.J.D.; Fink, D.J.; Glorioso, J.C. Deletion of multiple immediate–early genes from herpes simplex virus reduces cytotoxicity and permits long-term gene expression in neurons. Gene Ther. 1998, 5, 1593–1603. [Google Scholar] [CrossRef] [Green Version]
- Goins, W.F.; Huang, S.; Cohen, J.B.; Glorioso, J.C. Engineering HSV-1 Vectors for Gene Therapy. Adv. Struct. Saf. Stud. 2014, 1144, 63–79. [Google Scholar] [CrossRef]
- Bronisz, A.; Wang, Y.; Nowicki, M.O.; Peruzzi, P.; Ansari, K.I.; Ogawa, D.; Balaj, L.; de Rienzo, G.; Mineo, M.; Nakano, I.; et al. Extracellular Vesicles Modulate the Glioblastoma Microenvironment via a Tumor Suppression Signaling Network Directed by miR-1. Cancer Res. 2014, 74, 738–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronisz, A.; Godlewski, J.; Wallace, J.A.; Merchant, A.; Nowicki, M.; Mathsyaraja, H.; Srinivasan, R.; Trimboli, A.J.; Martin, C.K.; Li, F.; et al. Reprogramming of the tumour microenvironment by stromal PTEN-regulated miR-320. Nat. Cell Biol. 2011, 14, 159–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Wong, W.H. Model-based analysis of oligonucleotide arrays: Expression index computation and outlier detection. Proc. Natl. Acad. Sci. USA 2000, 98, 31–36. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Chambers, M.C.; MacLean, B.; Burke, R.D.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.D.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Bjornson, R.D.; Carriero, N.J.; Colangelo, C.; Shifman, M.; Cheung, K.-H.; Miller, P.L.; Williams, K. X!! Tandem, an Improved Method for Running X!Tandem in Parallel on Collections of Commodity Computers. J. Proteome Res. 2008, 7, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Craig, R.; Beavis, R.C. TANDEM: Matching proteins with tandem mass spectra. Bioinformatics 2004, 20, 1466–1467. [Google Scholar] [CrossRef]
- MacLean, B.; Eng, J.K.; Beavis, R.C.; McIntosh, M. General framework for developing and evaluating database scoring algorithms using the TANDEM search engine. Bioinformatics 2006, 22, 2830–2832. [Google Scholar] [CrossRef] [Green Version]
- Geer, L.Y.; Markey, S.P.; Kowalak, J.A.; Wagner, L.; Xu, M.; Maynard, D.M.; Yang, X.; Shi, W.; Bryant, S.H. Open Mass Spectrometry Search Algorithm. J. Proteome Res. 2004, 3, 958–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slotta, D.J.; McFarland, M.A.; Markey, S.P. MassSieve: Panning MS/MS peptide data for proteins. Proteomics 2010, 10, 3035–3039. [Google Scholar] [CrossRef] [Green Version]
- Sturm, M.; Bertsch, A.; Gröpl, C.; Hildebrandt, A.; Hussong, R.; Lange, E.; Pfeifer, N.; Schulz-Trieglaff, O.; Zerck, A.; Reinert, K.; et al. OpenMS—An open-source software framework for mass spectrometry. BMC Bioinform. 2008, 9, 1–11. [Google Scholar] [CrossRef]
- Vapnik, V.N. An overview of statistical learning theory. IEEE Trans. Neural Netw. 1999, 10, 988–999. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-Y.; Sun, B.-Y.; Zhu, Z.-H.; Chang, E.T.; To, K.-F.; Jiang, H.; Kam, M.K.-M.; Chen, G.; Cheah, S.-L.; Lee, M.; et al. Eight-Signature Classifier for Prediction of Nasopharyngeal Carcinoma Survival. J. Clin. Oncol. 2011, 29, 4516–4525. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xie, J.; Han, Z.; Liu, W.; Xi, S.; Huang, L.; Huang, W.; Lin, T.; Zhao, L.; Hu, Y.; et al. Immunomarker Support Vector Machine Classifier for Prediction of Gastric Cancer Survival and Adjuvant Chemotherapeutic Benefit. Clin. Cancer Res. 2018, 24, 5574–5584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; Decarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [Green Version]
- Ricklefs, F.; Mineo, M.; Rooj, A.K.; Nakano, I.; Charest, A.; Weissleder, R.; Breakefield, X.O.; Chiocca, E.A.; Godlewski, J.; Bronisz, A. Extracellular Vesicles from High-Grade Glioma Exchange Diverse Pro-oncogenic Signals That Maintain Intratumoral Heterogeneity. Cancer Res. 2016, 76, 2876–2881. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.P.; Kim, E.Y.; Badr, C.E.; Weissleder, R.; Mempel, T.R.; Tannous, B.A.; Breakefield, X.O. Visualization and tracking of tumour extracellular vesicle delivery and RNA translation using multiplexed reporters. Nat. Commun. 2015, 6, 7029. [Google Scholar] [CrossRef] [PubMed]
- Li, C. Automating dChip: Toward reproducible sharing of microarray data analysis. BMC Bioinform. 2008, 9, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Celiku, O.; Johnson, S.; Zhao, S.; Camphausen, K.; Shankavaram, U. Visualizing Molecular Profiles of Glioblastoma with GBM-BioDP. PLoS ONE 2014, 9, e101239. [Google Scholar] [CrossRef]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro. Oncol. 2017, 19, 139–141. [Google Scholar] [CrossRef] [Green Version]
- Orre, L.M.; Vesterlund, M.; Pan, Y.; Arslan, T.; Zhu, Y.; Woodbridge, A.F.; Frings, O.; Fredlund, E.; Lehtiö, J. SubCellBarCode: Proteome-wide Mapping of Protein Localization and Relocalization. Mol. Cell 2019, 73, 166–182.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rooj, A.K.; Ricklefs, F.; Mineo, M.; Nakano, I.; Chiocca, E.A.; Bronisz, A.; Godlewski, J. MicroRNA-Mediated Dynamic Bidirectional Shift between the Subclasses of Glioblastoma Stem-like Cells. Cell Rep. 2017, 19, 2026–2032. [Google Scholar] [CrossRef] [Green Version]
- Godlewski, J.; Ferrer-Luna, R.; Rooj, A.K.; Mineo, M.; Ricklefs, F.; Takeda, Y.S.; Nowicki, M.O.; Salińska, E.; Nakano, I.; Lee, H.; et al. MicroRNA Signatures and Molecular Subtypes of Glioblastoma: The Role of Extracellular Transfer. Stem Cell Rep. 2017, 8, 1497–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mineo, M.; Ricklefs, F.; Rooj, A.K.; Lyons, S.M.; Ivanov, P.; Ansari, K.I.; Nakano, I.; Chiocca, E.A.; Godlewski, J.; Bronisz, A. The Long Non-coding RNA HIF1A-AS2 Facilitates the Maintenance of Mesenchymal Glioblastoma Stem-like Cells in Hypoxic Niches. Cell Rep. 2016, 15, 2500–2509. [Google Scholar] [CrossRef] [Green Version]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Ma, R.; Russell, L.; Yoo, J.Y.; Han, J.; Cui, H.; Yi, P.; Zhang, J.; Nakashima, H.; Dai, H.; et al. An oncolytic herpesvirus expressing E-cadherin improves survival in mouse models of glioblastoma. Nat. Biotechnol. 2018, 37, 45–54. [Google Scholar] [CrossRef]
- Belcaid, Z.; Berrevoets, C.; Choi, J.; van Beelen, E.; Stavrakaki, E.; Pierson, T.; Kloezeman, J.; Routkevitch, D.; van der Kaaij, M.; van der Ploeg, A.; et al. Low-dose oncolytic adenovirus therapy overcomes tumor-induced immune suppression and sensitizes intracranial gliomas to anti-PD-1 therapy. Neuro-Oncol. Adv. 2020, 2. [Google Scholar] [CrossRef]
- Koch, M.S.; Lawler, S.E.; Chiocca, E.A. HSV-1 Oncolytic Viruses from Bench to Bedside: An Overview of Current Clinical Trials. Cancers 2020, 12, 3514. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Yu, J.S.; Lukas, R.V.; Solomon, I.H.; Ligon, K.L.; Nakashima, H.; Triggs, D.A.; Reardon, D.A.; Wen, P.; Stopa, B.M.; et al. Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: Results of a phase 1 trial. Sci. Transl. Med. 2019, 11, eaaw5680. [Google Scholar] [CrossRef]
- Passaro, C.; Alayo, Q.; de Laura, I.; McNulty, J.; Grauwet, K.; Ito, H.; Bhaskaran, V.; Mineo, M.; Lawler, S.E.; Shah, K.; et al. Arming an Oncolytic Herpes Simplex Virus Type 1 with a Single-chain Fragment Variable Antibody against PD-1 for Ex-perimental Glioblastoma Therapy. Clin. Cancer Res. 2018, 25, 290–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, M.; Fan, J.; Lyon, C.; Wan, M.; Hu, Y. Role of Extracellular Vesicles in Viral and Bacterial Infections: Pathogenesis, Diagnostics, and Therapeutics. Theranostics 2018, 8, 2709–2721. [Google Scholar] [CrossRef]
- Zhang, H.; Freitas, D.; Kim, H.S.; Fabijanic, K.; Li, Z.; Chen, H.; Mark, M.T.; Molina, H.; Benito-Martin, A.; Bojmar, L.; et al. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat. Cell Biol. 2018, 20, 332–343. [Google Scholar] [CrossRef]
- Wieckowski, E.U.; Visus, C.; Szajnik, M.; Szczepanski, M.J.; Storkus, W.J.; Whiteside, T.L. Tumor-Derived Microvesicles Promote Regulatory T Cell Expansion and Induce Apoptosis in Tumor-Reactive Activated CD8+ T Lymphocytes. J. Immunol. 2009, 183, 3720–3730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pucci, F.; Garris, C.; Lai, C.P.; Newton, A.; Pfirschke, C.; Engblom, C.; Alvarez, D.; Sprachman, M.; Evavold, C.; Magnuson, A.; et al. SCS macrophages suppress melanoma by restricting tumor-derived vesicle-B cell interactions. Science 2016, 352, 242–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katzmann, D.J.; Babst, M.; Emr, S.D. Ubiquitin-Dependent Sorting into the Multivesicular Body Pathway Requires the Function of a Conserved Endosomal Protein Sorting Complex, ESCRT-I. Cell 2001, 106, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Bianco, F.; Perrotta, C.; Novellino, L.; Francolini, M.; Riganti, L.; Menna, E.; Saglietti, L.; Schuchman, E.H.; Furlan, R.; Clementi, E.; et al. Acid sphingomyelinase activity triggers microparticle release from glial cells. EMBO J. 2009, 28, 1043–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; de Geest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan–syntenin–ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Mitchell, J.P.; Court, J.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes selectively impair lymphocyte responses to interleukin-2. Cancer Res. 2007, 67, 7458–7466. [Google Scholar] [CrossRef] [Green Version]
- Ricklefs, F.L.; Alayo, Q.; Krenzlin, H.; Mahmoud, A.B.; Speranza, M.C.; Nakashima, H.; Hayes, J.L.; Lee, K.; Balaj, L.; Passaro, C.; et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci. Adv. 2018, 4, eaar2766. [Google Scholar] [CrossRef] [Green Version]
- Raab-Traub, N.; Dittmer, N.R.-T.D.P. Viral effects on the content and function of extracellular vesicles. Nat. Rev. Genet. 2017, 15, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Kucharzewska, P.; Christianson, H.C.; Welch, J.E.; Svensson, K.J.; Fredlund, E.; Ringnér, M.; Mörgelin, M.; Bourseau-Guilmain, E.; Bengzon, J.; Belting, M. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc. Natl. Acad. Sci. USA 2013, 110, 7312–7317. [Google Scholar] [CrossRef] [Green Version]
- Downes, N.L.; Laham-Karam, N.; Kaikkonen, M.U.; Ylä-Herttuala, S. Differential but Complementary HIF1α and HIF2α Transcriptional Regulation. Mol. Ther. 2018, 26, 1735–1745. [Google Scholar] [CrossRef] [Green Version]
- Burkholder, B.; Huang, R.-Y.; Burgess, R.; Luo, S.; Jones, V.S.; Zhang, W.; Lv, Z.-Q.; Gao, C.-Y.; Wang, B.-L.; Zhang, Y.-M.; et al. Tumor-induced perturbations of cytokines and immune cell networks. Biochim. Biophys. Acta Bioenerg. 2014, 1845, 182–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noman, M.Z.; Messai, Y.; Carre, T.; Akalay, I.; Meron, M.; Janji, B.; Hasmim, M.; Chouaib, S. Microenvironmental Hypoxia Orchestrating the Cell Stroma Cross Talk, Tumor Progression and Antitumor Response. Crit. Rev. Immunol. 2011, 31, 357–377. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Wu, A.; Kong, L.-Y.; Wang, Y.; Fuller, G.; Fokt, I.; Melillo, G.; Priebe, W.; Heimberger, A.B. Hypoxia Potentiates Glioma-Mediated Immunosuppression. PLoS ONE 2011, 6, e16195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, H.; Kaufmann, J.K.; Wang, P.-Y.; Nguyen, T.; Speranza, M.-C.; Kasai, K.; Okemoto, K.; Otsuki, A.; Nakano, I.; Fernandez, S.; et al. Histone deacetylase 6 inhibition enhances oncolytic viral replication in glioma. J. Clin. Investig. 2015, 125, 4269–4280. [Google Scholar] [CrossRef] [Green Version]
- Lawler, S.E. Cytomegalovirus and glioblastoma; controversies and opportunities. J. Neuro-Oncol. 2015, 123, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Wollmann, G.; Ozduman, K.; Pol, A.N.V.D. Oncolytic Virus Therapy of Glioblastoma Multiforme—Concepts and Candidates. Cancer J. 2012, 18, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Martikainen, M.; Essand, M. Virus-Based Immunotherapy of Glioblastoma. Cancers 2019, 11, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Godlewski, J.; Farhath, M.; Ricklefs, F.L.; Passaro, C.; Kiel, K.; Nakashima, H.; Chiocca, E.A.; Bronisz, A. Oncolytic Virus Therapy Alters the Secretome of Targeted Glioblastoma Cells. Cancers 2021, 13, 1287. https://doi.org/10.3390/cancers13061287
Godlewski J, Farhath M, Ricklefs FL, Passaro C, Kiel K, Nakashima H, Chiocca EA, Bronisz A. Oncolytic Virus Therapy Alters the Secretome of Targeted Glioblastoma Cells. Cancers. 2021; 13(6):1287. https://doi.org/10.3390/cancers13061287
Chicago/Turabian StyleGodlewski, Jakub, Mohamed Farhath, Franz L. Ricklefs, Carmela Passaro, Klaudia Kiel, Hiroshi Nakashima, E. Antonio Chiocca, and Agnieszka Bronisz. 2021. "Oncolytic Virus Therapy Alters the Secretome of Targeted Glioblastoma Cells" Cancers 13, no. 6: 1287. https://doi.org/10.3390/cancers13061287