
A Miniaturized Platform for Multiplexed Drug Response Imaging in Live Tumors

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
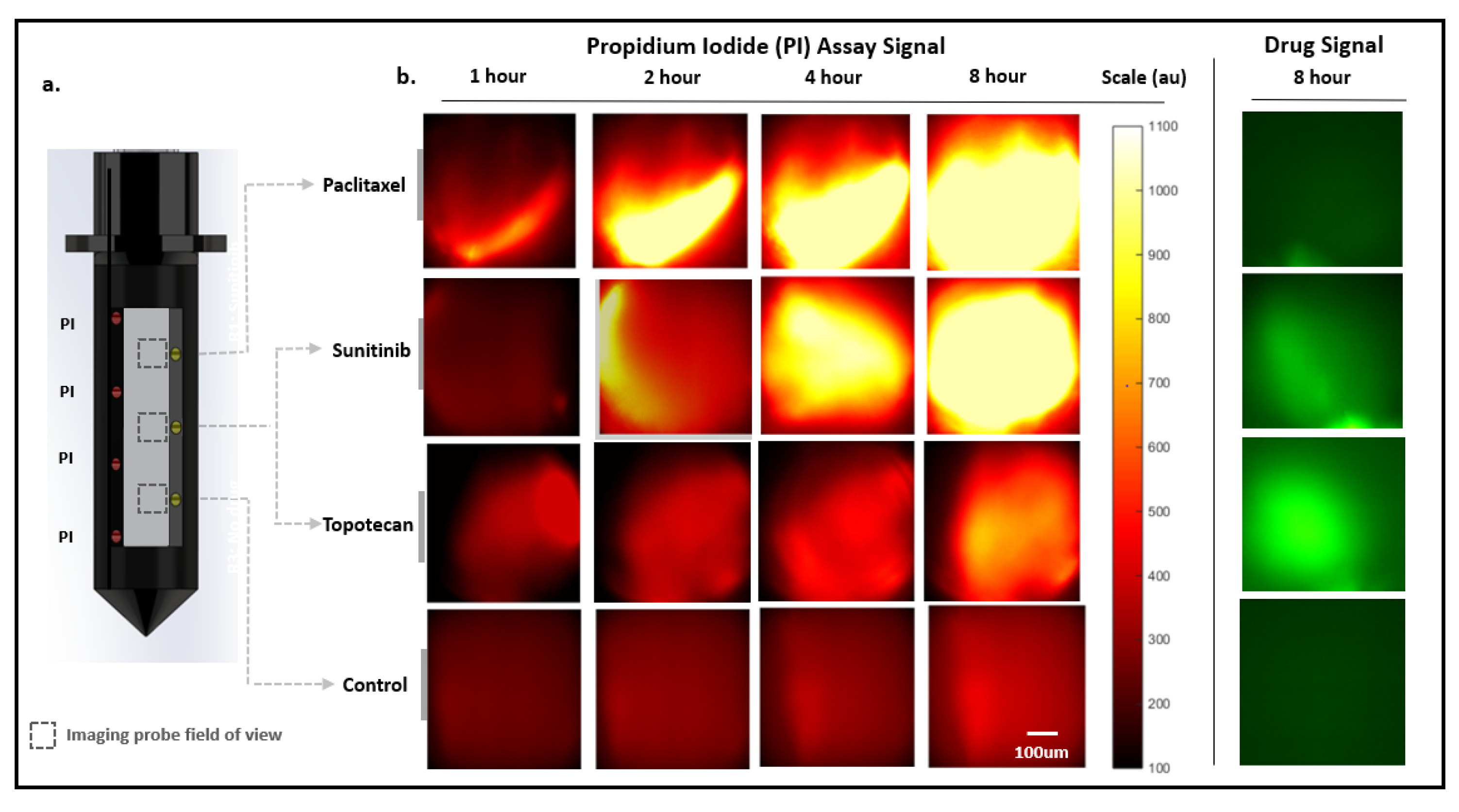
2. Results
2.1. Design and Development of the LIT-IMD Probe
2.2. Miniaturized 2-Color Fluorescence Microscopy (M-2CFM) System Development
2.2.1. Customization and Set-up
2.2.2. Imaging System Characterization
2.3. Drug Response Assessment Proof-of-Concept in Murine Tumor Model
2.3.1. Serial Miniaturized 2-Color Fluorescence Microscopy (M-2CFM) of Drug Diffusion and Response
2.3.2. Correlation with Gold-Standard Cell Death Assessment Methods
3. Materials and Methods
3.1. Design and Development of the LIT-IMD Probe
3.2. Miniaturized 2-Color Fluorescence Microscopy (M-2CFM) System Development
3.2.1. System Customization and Set-up
3.2.2. Imaging System Characterization
3.3. Drug Response Assessment Proof-of-Concept in Murine Tumor Model
3.3.1. Animal Model
3.3.2. Drug and Assay Formulation for Localized Delivery
3.3.3. Tissue Processing for ex vivo Correlation
3.3.4. Conventional Imaging and Image Analysis
3.3.5. Automated Image Classification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Weber, W.A. Assessing tumor response to therapy. J. Nucl. Med. 2009, 50 (Suppl. 1), S1–S10. [Google Scholar] [CrossRef] [Green Version]
- Nishino, M. Tumor Response Assessment for Precision Cancer Therapy: Response Evaluation Criteria in Solid Tumors and Beyond. Am. Soc. Clin. Oncol. Educ. B. 2018, 38, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Jonas, O.; Landry, H.M.; Fuller, J.E.; Santini, J.T.; Baselga, J.; Tepper, R.I.; Cima, M.J.; Langer, R. An implantable microdevice to perform high-throughput in vivo drug sensitivity testing in tumors. Sci. Transl. Med. 2015, 7, 284ra57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonas, O.; Oudin, M.J.; Kosciuk, T.; Whitman, M.; Gertler, F.B.; Cima, M.J.; Flaherty, K.T.; Langer, R. Parallel in-vivo assessment of drug phenotypes at various time points during systemic BRAF inhibition reveals tumor adaptation and altered treatment vulnerabilities. Clin. Cancer Res. 2016, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonas, O.; Calligaris, D.; Methuku, K.R.; Poe, M.M.; Francois, J.P.; Tranghese, F.; Changelian, A.; Sieghart, W.; Ernst, M.; Pomeranz Krummel, D.A.; et al. First in vivo testing of compounds targeting group 3 medulloblastomas using an implantable microdevice as a new paradigm for drug development. J. Biomed. Nanotechnol. 2016, 12, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Implantable Microdevice in Primary Brain Tumors-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04135807?term=oliver+jonas&draw=2&rank=1 (accessed on 24 July 2020).
- Microdevice for Evaluating Drug Response in Site in Lung Lesions-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03972228?term=oliver+jonas&draw=2&rank=2 (accessed on 24 July 2020).
- Kim, J.K.; Lee, W.M.; Kim, P.; Choi, M.; Jung, K.; Kim, S.; Yun, S.H. Fabrication and operation of GRIN probes for in vivo fluorescence cellular imaging of internal organs in small animals. Nat. Protoc. 2012, 7, 1456–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eser, S.; Messer, M.; Eser, P.; Von Werder, A.; Seidler, B.; Bajbouj, M.; Vogelmann, R.; Meining, A.; Von Burstin, J.; Algül, H.; et al. In vivo diagnosis of murine pancreatic intraepithelial neoplasia and early-stage pancreatic cancer by molecular imaging. Proc. Natl. Acad. Sci. USA, 2011; 108, 9945–9950. [Google Scholar] [CrossRef] [Green Version]
- Levene, M.J.; Dombeck, D.A.; Kasischke, K.A.; Molloy, R.P.; Webb, W.W. In Vivo Multiphoton Microscopy of Deep Brain Tissue. J. Neurophysiol. 2004, 91, 1908–1912. [Google Scholar] [CrossRef] [Green Version]
- Kepp, O.; Galluzzi, L.; Lipinski, M.; Yuan, J.; Kroemer, G. Cell death assays for drug discovery. Nat. Rev. Drug Discov. 2011, 10, 221–237. [Google Scholar] [CrossRef]
- Gómez-Cabañas, L.; Delgado-Martín, C.; López-Cotarelo, P.; Escribano-Diaz, C.; Alonso-c, L.M.; Riol-Blanco, L.; Rodríguez-Fernández, J.L. Detecting apoptosis of leukocytes in mouse lymph nodes. Nat. Protoc. 2014, 9, 1102–1112. [Google Scholar] [CrossRef] [PubMed]
- Bao, G.; Rhee, W.J.; Tsourkas, A. Fluorescent Probes for Live-Cell RNA Detection. Annu. Rev. Biomed. Eng. 2009, 11, 25–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Ma, Z.; Wu, X.; Mao, S.; Yang, Y.; Tan, J.; Krueger, C.J.; Chen, A.K. A molecular beacon-based approach for live-cell imaging of RNA transcripts with minimal target engineering at the single-molecule level. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowley, L.C.; Scott, A.P.; Marfell, B.J.; Boughaba, J.A.; Chojnowski, G.; Waterhouse, N.J. Measuring cell death by propidium iodide uptake and flow cytometry. Cold Spring Harb. Protoc. 2016, 2016, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Vitale, M.; Zamai, L.; Mazzotti, G.; Cataldi, A.; Falcieri, E. Differential kinetics of propidium iodide uptake in apoptotic and necrotic thymocytes. Histochemistry 1993, 100, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Bhagavatula, S.K.; Upadhyaya, K.; Miller, B.J.; Bursch, P.; Lammers, A.; Cima, M.J.; Silverman, S.G.; Jonas, O. An interventional image-guided microdevice implantation and retrieval method for in-vivo drug response assessment. Med. Phys. 2019, 46, 5134–5143. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.; Khalsa, B.; Lord, B.; Sandrasegaran, K.; Lall, C. Planting the seeds of success: CT-guided gold seed fiducial marker placement to guide robotic radiosurgery. J. Med. Imaging Radiat. Oncol. 2013, 57, 207–211. [Google Scholar] [CrossRef]
- Folli, S.; Wagnieres, G.; Pelegrin, A.; Calmes, J.M.; Braichotte, D.; Buchegger, F.; Chalandon, Y.; Hardman, N.; Heusser, C.; Givel, J.C. Immunophotodiagnosis of colon carcinomas in patients injected with fluoresceinated chimeric antibodies against carcinoembryonic antigen. Proc. Natl. Acad. Sci. USA 1992, 89, 7973–7977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouma, B.E.; Villiger, M.; Otsuka, K.; Oh, W.-Y. Intravascular optical coherence tomography [Invited]. Biomed. Opt. Express 2017, 8, 2660. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, M.; Yang, L.; Zhang, Y.; Yuan, J.; Liu, Q.; Hou, X.; Fu, L. A Confocal Endoscope for Cellular Imaging. Engineering 2015, 1, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Dilipkumar, A.; Al-Shemmary, A.; Kreiß, L.; Cvecek, K.; Carlé, B.; Knieling, F.; Gonzales Menezes, J.; Thoma, O.; Schmidt, M.; Neurath, M.; et al. Label-Free Multiphoton Endomicroscopy for Minimally Invasive In Vivo Imaging. Adv. Sci. 2019, 6, 1801735. [Google Scholar] [CrossRef] [Green Version]
- Jonas, O.; Kang, J.W.; Singh, S.P.; Lammers, A.; Nguyen, F.T.; Dasari, R.R.; So, P.T.C.; Langer, R.; Cima, M. In vivo detection of drug-induced apoptosis in tumors using Raman spectroscopy. Analyst 2018, 143, 4836–4839. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.; Klein, O.J.; Wang, H.; Evans, C.L. Longitudinal, label-free, quantitative tracking of cell death and viability in a 3D tumor model with OCT. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Farhat, G.; Yang, V.X.D.; Czarnota, G.J.; Kolios, M.C. Detecting cell death with optical coherence tomography and envelope statistics. J. Biomed. Opt. 2011, 16, 026017. [Google Scholar] [CrossRef] [PubMed]
- Briley, W.E.; Bondy, M.H.; Randeria, P.S.; Dupper, T.J.; Mirkin, C.A. Quantification and real-time tracking of RNA in live cells using Sticky-flares. Proc. Natl. Acad. Sci. USA 2015, 112, 9591–9595. [Google Scholar] [CrossRef] [Green Version]
- Toptygina, P.; Zakirov, R.S.; Kapitanova, K.S.; Semikina, E.L. Detection of Small Subsets of CD4+ Lymphocytes with SmartFlare Nanoprobes. Bull. Exp. Biol. Med. 2019, 168, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Schaafsma, B.E.; Mieog, J.S.D.; Hutteman, M.; Van Der Vorst, J.R.; Kuppen, P.J.K.; Löwik, C.W.G.M.; Frangioni, J.V.; Van De Velde, C.J.H.; Vahrmeijer, A.L. The clinical use of indocyanine green as a near-infrared fluorescent contrast agent for image-guided oncologic surgery. J. Surg. Oncol. 2011, 104, 323–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vahrmeijer, A.L.; Hutteman, M.; van der Vorst, J.R.; van de Velde, C.J.H.; Faangioni, J.V. Image-guided cancer surgery using near-infrared fluorescence. Nat. Rev. Clin. Oncol. 2013, 10, 507–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbeek, F.P.R.; Van Der Vorst, J.R.; Schaafsma, B.E.; Swijnenburg, R.J.; Gaarenstroom, K.N.; Elzevier, H.W.; Van De Velde, C.J.H.; Frangioni, J.V.; Vahrmeijer, A.L. Intraoperative near infrared fluorescence guided identification of the ureters using low dose methylene blue: A first in human experience. J. Urol. 2013, 190, 574–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakrishnan, R.; Assudani, D.; Nagaraj, S.; Hunter, T.; Cho, H.; Antonia, S.; Altiok, S.; Celis, E.; Gabrilovich, D. Chemotherapy enhances tumor cell susceptibility to CTL-mediated killing during cancer immunotherapy in mice. J. Clin. Investig. 2010, 120, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- Cubas, R.; Moskalenko, M.; Cheung, J.; Yang, M.; McNamara, E.; Xiong, H.; Hoves, S.; Ries, C.H.; Kim, J.; Gould, S. Chemotherapy Combines Effectively with Anti–PD-L1 Treatment and Can Augment Antitumor Responses. J. Immunol. 2018, 201, 2273–2286. [Google Scholar] [CrossRef] [Green Version]
- Sivakumar, R.; Chan, M.; Shin, J.S.; Nishida-Aoki, N.; Kenerson, H.L.; Elemento, O.; Beltran, H.; Yeung, R.; Gujral, T.S. Organotypic tumor slice cultures provide a versatile platform for immuno-oncology and drug discovery. Oncoimmunology 2019, 8, e1670019. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamal-Hanjani, M.; Quezada, S.A.; Larkin, J.; Swanton, C. Translational implications of tumor heterogeneity. Clin. Cancer Res. 2015, 21, 1258–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | Topotecan (p-Value) | Paclitaxel (p-Value) | Sunitinib (p-Value) | |
|---|---|---|---|---|
| 1 h | 170.9 ± 44 | 261.9 ± 81.3 (0.24) | 293.8 ± 27.0 (0.063) | 217.3 ± 27.9 (0.70) |
| 2 h | 191.7 ± 61.1 | 294.6 ± 37.7 (0.67) | 507.0 ± 111.6 (0.028) | 409.35 ± 149.6 (0.16) |
| 4 h | 191.5 ± 63.9 | 398.1 ± 94.5 (0.13) | 645.4 ± 78.2 (0.0026) | 761.2 ± 121.8 (0.0014) |
| 8 h | 210.1 ± 92.3 | 556.6 ± 154.6 (<0.001) | 802.9 ± 55.1 (<0.001) | 877.9 ± 28.9 (<0.001) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhagavatula, S.; Thompson, D.; Ahn, S.W.; Upadhyaya, K.; Lammers, A.; Deans, K.; Dominas, C.; Ferland, B.; Valvo, V.; Liu, G.; et al. A Miniaturized Platform for Multiplexed Drug Response Imaging in Live Tumors. Cancers 2021, 13, 653. https://doi.org/10.3390/cancers13040653
Bhagavatula S, Thompson D, Ahn SW, Upadhyaya K, Lammers A, Deans K, Dominas C, Ferland B, Valvo V, Liu G, et al. A Miniaturized Platform for Multiplexed Drug Response Imaging in Live Tumors. Cancers. 2021; 13(4):653. https://doi.org/10.3390/cancers13040653
Chicago/Turabian StyleBhagavatula, Sharath, Devon Thompson, Sebastian W. Ahn, Kunj Upadhyaya, Alex Lammers, Kyle Deans, Christine Dominas, Benjamin Ferland, Veronica Valvo, Guigen Liu, and et al. 2021. "A Miniaturized Platform for Multiplexed Drug Response Imaging in Live Tumors" Cancers 13, no. 4: 653. https://doi.org/10.3390/cancers13040653