
Derivation and Application of Molecular Signatures to Prostate Cancer: Opportunities and Challenges


Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Prostate Cancer
1.2. Transcription Factors and Activity Signatures
2. Transcription Factors in Prostate Cancer
2.1. Transcription Factor Centred Cignatures: Reverse vs Forward Translation
2.2. Chromatin Landscape
3. Integration of Multiple Datasets: Combining Forward and Reverse Translation
3.1. Integrative Analytics: Models for Signature Generation
3.2. Combining Signatures: Dealing with an Increasing Data Load
3.3. Tools to Normalise Increasing Number of Activity Signatures
4. Multi-Omics and Evolution
4.1. Co-Activation Signatures
4.2. Mutational Burden
5. Conclusions and Future Perspectives
5.1. Beyond TFs
5.2. Thinking about Signature Validity and Dynamic Disease Progression
5.3. Attribution of Activity Weight Within a Signature
5.4. Biobanking and Logistical Considerations
Funding
Conflicts of Interest
References
- Rawla, P. Epidemiology of prostate cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [Green Version]
- Barfeld, S.J.; Itkonen, H.M.; Urbanucci, A.; Mills, I.G. Androgen-regulated metabolism and biosynthesis in prostate cancer. Endocr. Relat. Cancer 2014, 21, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Handle, F.; Prekovic, S.; Helsen, C.; Van den Broeck, T.; Smeets, E.; Moris, L.; Eerlings, R.; Kharraz, S.E.L.; Urbanucci, A.; Mills, I.G.; et al. Drivers of AR indifferent anti-androgen resistance in prostate cancer cells. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, E.V.; Reece, K.M.; Ley, A.M.; Troutman, S.M.; Sissung, T.M.; Price, D.K.; Chau, C.H.; Figg, W.D. Dual targeting of the androgen receptor and hypoxia-inducible factor 1α pathways synergistically inhibits castration-resistant prostate cancer cellss. Mol. Pharmacol. 2015, 87, 1006–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, A.B.; Denko, N.; Barton, M.C. Hypoxia induces a novel signature of chromatin modifications and global repression of transcription. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2008, 640, 174–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushweller, J.H. Targeting transcription factors in cancer—From undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W. Transcription factors and cancer: An overview. Toxicology 2002, 181–182, 131–141. [Google Scholar] [CrossRef]
- Huilgol, D.; Venkataramani, P.; Nandi, S.; Bhattacharjee, S. Transcription factors that govern development and disease: An achilles heel in cancer. Genes 2019, 10, 794. [Google Scholar] [CrossRef] [Green Version]
- Barfeld, S.J.; Fazli, L.; Persson, M.; Marjavaara, L.; Urbanucci, A.; Kaukoniemi, K.M.; Rennie, P.S.; Ceder, Y.; Chabes, A.; Visakorpi, T.; et al. Myc-dependent purine biosynthesis affects nucleolar stress and therapy response in prostate cancer. Oncotarget 2015, 6, 12587–12602. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Bennett, N.C.; Gardiner, R.A.; Hooper, J.D.; Johnson, D.W.; Gobe, G.C. Molecular cell biology of androgen receptor signalling. Int. J. Biochem. Cell Biol. 2010, 42, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer. I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1941, 1, 293–297. [Google Scholar]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.G.; Chang, S.L.; Erho, N.; Yu, M.; Lehrer, J.; Alshalalfa, M.; Speers, C.; Cooperberg, M.R.; Kim, W.; Ryan, C.J.; et al. Associations of luminal and basal subtyping of prostate cancer with prognosis and response to androgen deprivation therapy. JAMA Oncol. 2017, 3, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.A.; Sokolov, A.; Uzunangelov, V.; Baertsch, R.; Newton, Y.; Graim, K.; Mathis, C.; Cheng, D.; Stuart, J.M.; Witte, O.N. A basal stem cell signature identifies aggressive prostate cancer phenotypes. Proc. Natl. Acad. Sci. USA 2015, 112, E6544–E6552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knillova, J.; Kolar, Z. The significance of key regulators of apoptosis in the development and prognosis of prostate carcinoma. I. Proteins of the Bcl-2 family and protein p53. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2003, 147, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, M.G.B.; Bibby, B.A.S.; Yang, L.; Lo, F.; Warren, A.Y.; Shukla, D.; Osborne, M.; Hadfield, J.; Carroll, T.; Stark, R.; et al. Independence of HIF1a and androgen signaling pathways in prostate cancer. BMC Cancer 2020, 20, 469. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Nenseth, H.Z.; Qu, S.; Kuzu, O.F.; Frahnow, T.; Simon, L.; Greene, S.; Zeng, Q.; Fazli, L.; Rennie, P.S.; et al. IRE1α-XBP1s pathway promotes prostate cancer by activating c-MYC signaling. Nat. Commun. 2019, 10, 323. [Google Scholar] [CrossRef] [Green Version]
- Doultsinos, D.; Mills, I. The role of the androgen receptor as a driver and mitigator of cellular stress. J. Mol. Endocrinol. 2020, 1. [Google Scholar] [CrossRef]
- Wu, X.; Scott, H.; Carlsson, S.V.; Sjoberg, D.D.; Cerundolo, L.; Lilja, H.; Prevo, R.; Rieunier, G.; Macaulay, V.; Higgins, G.S.; et al. Increased EZH2 expression in prostate cancer is associated with metastatic recurrence following external beam radiotherapy. Prostate 2019, 79, 1079–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melling, N.; Thomsen, E.; Tsourlakis, M.C.; Kluth, M.; Hube-Magg, C.; Minner, S.; Koop, C.; Graefen, M.; Heinzer, H.; Wittmer, C.; et al. Overexpression of enhancer of zeste homolog 2 (EZH2) characterizes an aggressive subset of prostate cancers and predicts patient prognosis independently from pre- and postoperatively assessed clinicopathological parameters. Carcinogenesis 2015, 36, 1333–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorodetska, I.; Lukiyanchuk, V.; Peitzsch, C.; Kozeretska, I.; Dubrovska, A. BRCA1 and EZH2 cooperate in regulation of prostate cancer stem cell phenotype. Int. J. Cancer 2019, 145, 2974–2985. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbé, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 2013, 155, 1309–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hankey, W.; Chen, Z.; Wang, Q. Shaping chromatin states in prostate cancer by pioneer transcription factors. Cancer Res. 2020, 80, 2427–2436. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.W.; Pan, Y.; Tsai, B.L.; Xie, Z.; Demirdjian, L.; Xiao, W.; Yang, H.T.; Zhang, Y.; Lin, C.H.; Cheng, D.; et al. Pathway-guided analysis identifies Myc-dependent alternative pre-mRNA splicing in aggressive prostate cancers. Proc. Natl. Acad. Sci. USA 2020, 117, 5269–5279. [Google Scholar] [CrossRef] [Green Version]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.C.; Wongvipat, J.; Ku, S.Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53-and RB1-deficient prostate cancer. Science 2017, 355. [Google Scholar] [CrossRef] [Green Version]
- Zou, M.; Toivanen, R.; Mitrofanova, A.; Floch, N.; Hayati, S.; Sun, Y.; Le Magnen, C.; Chester, D.; Mostaghel, E.A.; Califano, A.; et al. Transdifferentiation as a mechanism of treatment resistance in a mouse model of castration-resistant prostate cancer. Cancer Discov. 2017, 7, 736–749. [Google Scholar] [CrossRef] [Green Version]
- Bishop, J.L.; Thaper, D.; Vahid, S.; Davies, A.; Ketola, K.; Kuruma, H.; Jama, R.; Nip, K.M.; Angeles, A.; Johnson, F.; et al. The master neural transcription factor BRN2 is an androgen receptor–suppressed driver of neuroendocrine differentiation in prostate cancer. Cancer Discov. 2017, 7, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Risbridger, G.P.; Toivanen, R.; Taylor, R.A. Preclinical models of prostate cancer: Patient-derived xenografts, organoids, and other explant models. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbadawy, M.; Abugomaa, A.; Yamawaki, H.; Usui, T.; Sasaki, K. Development of prostate cancer organoid culture models in basic medicine and translational research. Cancers 2020, 12, 777. [Google Scholar] [CrossRef] [Green Version]
- Kretschmer, A.; Tilki, D. Biomarkers in prostate cancer—Current clinical utility and future perspectives. Crit. Rev. Oncol. Hematol. 2017, 120, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.A.; Cooperberg, M.R.; Magi-Galluzzi, C.; Simko, J.P.; Falzarano, S.M.; Maddala, T.; Chan, J.M.; Li, J.; Cowan, J.E.; Tsiatis, A.C.; et al. A 17-gene assay to predict prostate cancer aggressiveness in the context of gleason grade heterogeneity, tumor multifocality, and biopsy undersampling. Eur. Urol. 2014, 66, 550–560. [Google Scholar] [CrossRef] [Green Version]
- Knezevic, D.; Goddard, A.D.; Natraj, N.; Cherbavaz, D.B.; Clark-Langone, K.M.; Snable, J.; Watson, D.; Falzarano, S.M.; Magi-Galluzzi, C.; Klein, E.A.; et al. Analytical validation of the Oncotype DX prostate cancer assay—A clinical RT-PCR assay optimized for prostate needle biopsies. BMC Genom. 2013, 14, 690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuzick, J.; Swanson, G.P.; Fisher, G.; Brothman, A.R.; Berney, D.M.; Reid, J.E.; Mesher, D.; Speights, V.O.; Stankiewicz, E.; Foster, C.S.; et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: A retrospective study. Lancet Oncol. 2011, 12, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Kollmeyer, T.M.; Morlan, B.W.; Anderson, S.K.; Bergstralh, E.J.; Davis, B.J.; Asmann, Y.W.; Klee, G.G.; Ballman, K.V.; Jenkins, R.B. A tissue biomarker panel predicting systemic progression after psa recurrence post-definitive prostate cancer therapy. PLoS ONE 2008, 3, e2318. [Google Scholar] [CrossRef] [Green Version]
- Donovan, M.J.; Noerholm, M.; Bentink, S.; Belzer, S.; Skog, J.; O’Neill, V.; Cochran, J.S.; Brown, G.A. A molecular signature of PCA3 and ERG exosomal RNA from non-DRE urine is predictive of initial prostate biopsy result. Prostate Cancer Prostatic Dis. 2015, 18, 370–375. [Google Scholar] [CrossRef]
- Leyten, G.H.J.M.; Hessels, D.; Smit, F.P.; Jannink, S.A.; De Jong, H.; Melchers, W.J.G.; Cornel, E.B.; De Reijke, T.M.; Vergunst, H.; Kil, P.; et al. Identification of a candidate gene panel for the early diagnosis of prostate cancer. Clin. Cancer Res. 2015, 21, 3061–3070. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Roberts, D.; Takhar, M.; Erho, N.; Bibby, B.A.S.; Thiruthaneeswaran, N.; Bhandari, V.; Cheng, W.C.; Haider, S.; McCorry, A.M.B.; et al. Development and validation of a 28-gene hypoxia-related prognostic signature for localized prostate cancer. EBioMedicine 2018, 31, 182–189. [Google Scholar] [CrossRef] [Green Version]
- Urbanucci, A.; Barfeld, S.J.; Kytölä, V.; Itkonen, H.M.; Coleman, I.M.; Vodák, D.; Sjöblom, L.; Sheng, X.; Tolonen, T.; Minner, S.; et al. Androgen receptor deregulation drives bromodomain-mediated chromatin alterations in prostate cancer. Cell Rep. 2017, 19, 2045–2059. [Google Scholar] [CrossRef] [Green Version]
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Rodrigues, D.N.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G.; et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Investig. 2019, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luca, B.A.; Moulton, V.; Ellis, C.; Connell, S.P.; Rewer, D.S.; Cooper, C.S. Convergence of prognostic gene signatures suggests underlying mechanisms of human prostate cancer progression. Genes 2020, 11, 802. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.M.; Knight, L.A.; McCavigan, A.M.; Logan, G.E.; Berge, V.; Sherif, A.; Pandha, H.; Warren, A.Y.; Davidson, C.; Uprichard, A.; et al. Molecular subgroup of primary prostate cancer presenting with metastatic biology. Eur. Urol. 2017, 72, 509–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valcarcel-Jimenez, L.; Macchia, A.; Martín-Martín, N.; Cortazar, A.R.; Schaub-Clerigué, A.; Pujana-Vaquerizo, M.; Fernández-Ruiz, S.; Lacasa-Viscasillas, I.; Santos-Martin, A.; Loizaga-Iriarte, A.; et al. Integrative analysis of transcriptomics and clinical data uncovers the tumor-suppressive activity of MITF in prostate cancer. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ou, Z.; Sun, Y.; Yeh, S.; Wang, X.; Long, J.; Chang, C. Androgen receptor promotes melanoma metastasis via altering the miRNA-539-3p/USP13/MITF/AXL signals. Oncogene 2017, 36, 1644–1654. [Google Scholar] [CrossRef]
- Berger, A.; Brady, N.J.; Bareja, R.; Robinson, B.; Conteduca, V.; Augello, M.A.; Puca, L.; Ahmed, A.; Dardenne, E.; Lu, X.; et al. N-Myc-mediated epigenetic reprogramming drives lineage plasticity in advanced prostate cancer. J. Clin. Investig. 2019, 129, 3924–3940. [Google Scholar] [CrossRef]
- Labrecque, M.P.; Coleman, I.M.; Brown, L.G.; True, L.D.; Kollath, L.; Lakely, B.; Nguyen, H.M.; Yang, Y.C.; Da Costa, R.M.G.; Kaipainen, A.; et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J. Clin. Investig. 2019, 129, 4492–4505. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Hu, Z.; Phatak, M.; Reichard, J.; Freudenberg, J.M.; Sivaganesan, S.; Medvedovic, M. Genome-wide signatures of transcription factor activity: Connecting transcription factors, disease, and small molecules. PLoS Comput. Biol. 2013, 9, e1003198. [Google Scholar] [CrossRef]
- Mapelli, S.N.; Albino, D.; Mello-Grand, M.; Shinde, D.; Scimeca, M.; Bonfiglio, R.; Bonanno, E.; Chiorino, G.; Garcia-Escudero, R.; Catapano, C.V.; et al. A novel prostate cell type-specific gene signature to interrogate prostate tumor differentiation status and monitor therapeutic response (Running title: Phenotypic classification of prostate tumors). Cancers 2020, 12, 176. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.L.; Greer, C.B.; Chen, J.F.; Arnal-Estapé, A.; Cao, J.; Yan, Q.; Nguyen, D.X. Specific chromatin landscapes and transcription factors couple breast cancer subtype with metastatic relapse to lung or brain. BMC Med. Genom. 2020, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Zhou, C.; Li, X.; Barnes, S.D.; Deng, S.; Hoover, E.; Chen, C.C.; Lee, Y.S.; Zhang, Y.; Wang, C.; et al. Loss of CHD1 promotes heterogeneous mechanisms of resistance to AR-targeted therapy via chromatin dysregulation. Cancer Cell 2020, 37, 584–598. [Google Scholar] [CrossRef] [PubMed]
- Augello, M.A.; Liu, D.; Deonarine, L.D.; Robinson, B.D.; Huang, D.; Stelloo, S.; Blattner, M.; Doane, A.S.; Wong, E.W.P.; Chen, Y.; et al. CHD1 loss alters AR binding at lineage-specific enhancers and modulates distinct transcriptional programs to drive prostate tumorigenesis. Cancer Cell 2019, 35, 603–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomerantz, M.M.; Qiu, X.; Zhu, Y.; Takeda, D.Y.; Pan, W.; Baca, S.C.; Gusev, A.; Korthauer, K.D.; Severson, T.M.; Ha, G.; et al. Prostate cancer reactivates developmental epigenomic programs during metastatic progression. Nat. Genet. 2020, 52, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; MacDonald, T.; et al. N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Xu, L.; Chang, Y.; Zeng, T.; Chen, X.; Wang, A.; Groth, J.; Foo, W.C.; Liang, C.; Hu, H.; et al. N-Myc promotes therapeutic resistance development of neuroendocrine prostate cancer by differentially regulating miR-421/ATM pathway. Mol. Cancer 2019, 18, 11. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Qin, C.; Zhao, Y.; Zou, L.; Zhao, H.; Cheng, C. Gene signatures associated with genomic aberrations predict prognosis in neuroblastoma. Cancer Commun. 2020, 40, 105–118. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.; Lyons, C.A.; Walker, S.M.; McQuaid, S.; Hynes, S.O.; Mitchell, D.M.; Pang, B.; Logan, G.E.; McCavigan, A.M.; O’Rourke, D.; et al. Validation of a Metastatic Assay using biopsies to improve risk stratification in patients with prostate cancer treated with radical radiation therapy. Ann. Oncol. 2018, 29, 215–222. [Google Scholar] [CrossRef]
- Yu, J.; Yu, J.; Rhodes, D.R.; Tomlins, S.A.; Cao, X.; Chen, G.; Mehra, R.; Wang, X.; Ghosh, D.; Shah, R.B.; et al. A polycomb repression signature in metastatic prostate cancer predicts cancer outcome. Cancer Res. 2007, 67, 10657–10663. [Google Scholar] [CrossRef] [Green Version]
- Lupien, M.; Eeckhoute, J.; Meyer, C.A.; Wang, Q.; Zhang, Y.; Li, W.; Carroll, J.S.; Liu, X.S.; Brown, M. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell 2008, 132, 958–970. [Google Scholar] [CrossRef] [Green Version]
- Parolia, A.; Cieslik, M.; Chu, S.C.; Xiao, L.; Ouchi, T.; Zhang, Y.; Wang, X.; Vats, P.; Cao, X.; Pitchiaya, S.; et al. Distinct structural classes of activating FOXA1 alterations in advanced prostate cancer. Nature 2019, 571, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Adams, E.J.; Karthaus, W.R.; Hoover, E.; Liu, D.; Gruet, A.; Zhang, Z.; Cho, H.; DiLoreto, R.; Chhangawala, S.; Liu, Y.; et al. FOXA1 mutations alter pioneering activity, differentiation and prostate cancer phenotypes. Nature 2019, 571, 408–412. [Google Scholar] [CrossRef] [PubMed]
- Ware, K.E.; Garcia-Blanco, M.A.; Armstrong, A.J.; Dehm, S.M. Biologic and clinical significance of androgen receptor variants in castration resistant prostate cancer. Endocr. Relat. Cancer 2014, 21, 87–103. [Google Scholar] [CrossRef]
- Boukovala, M.; Spetsieris, N.; Weldon, J.A.; Tsikkinis, A.; Hoang, A.; Aparicio, A.; Tu, S.M.; Araujo, J.C.; Zurita, A.J.; Corn, P.G.; et al. A candidate androgen signalling signature predictive of response to abiraterone acetate in men with metastatic castration-resistant prostate cancer. Eur. J. Cancer 2020, 127, 67–75. [Google Scholar] [CrossRef]
- Schacht, T.; Oswald, M.; Eils, R.; Eichmüller, S.B.; König, R. Estimating the activity of transcription factors by the effect on their target genes. Bioinformatics 2014, 30, 401–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, I.S.; Margolin, A.; Califano, A. hARACNe: Improving the accuracy of regulatory model reverse engineering via higher-order data processing inequality tests. Interface Focus 2013, 3, 20130011. [Google Scholar] [CrossRef] [PubMed]
- Faith, J.J.; Hayete, B.; Thaden, J.T.; Mogno, I.; Wierzbowski, J.; Cottarel, G.; Kasif, S.; Collins, J.J.; Gardner, T.S. Large-scale mapping and validation of escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007, 5, e8. [Google Scholar] [CrossRef]
- Stephenson, A.J.; Smith, A.; Kattan, M.W.; Satagopan, J.; Reuter, V.E.; Scardino, P.T.; Gerald, W.L. Integration of gene expression profiling and clinical variables to predict prostate carcinoma recurrence after radical prostatectomy. Cancer 2005, 104, 290–298. [Google Scholar] [CrossRef]
- Kourou, K.; Exarchos, T.P.; Exarchos, K.P.; Karamouzis, M.V.; Fotiadis, D.I. Machine learning applications in cancer prognosis and prediction. Comput. Struct. Biotechnol. J. 2015, 13, 8–17. [Google Scholar] [CrossRef] [Green Version]
- Fleck, J.L.; Pavel, A.B.; Cassandras, C.G. Integrating mutation and gene expression cross-sectional data to infer cancer progression. BMC Syst. Biol. 2016, 10, 12. [Google Scholar] [CrossRef] [Green Version]
- Shafi, A.; Nguyen, T.; Peyvandipour, A.; Draghici, S. GSMA: An approach to identify robust global and test gene signatures using meta-analysis. Bioinformatics 2020, 36, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Rydenfelt, M.; Klinger, B.; Klünemann, M.; Blüthgen, N. SPEED2: Inferring upstream pathway activity from differential gene expression. Nucleic Acids Res. 2020, 48, W307–W312. [Google Scholar] [CrossRef]
- Dhawan, A.; Barberis, A.; Cheng, W.C.; Domingo, E.; West, C.; Maughan, T.; Scott, J.G.; Harris, A.L.; Buffa, F.M. Guidelines for using sigQC for systematic evaluation of gene signatures. Nat. Protoc. 2019, 14, 1377–1400. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The molecular signatures database hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantini, L.; Calzone, L.; Martignetti, L.; Rydenfelt, M.; Blüthgen, N.; Barillot, E.; Zinovyev, A. Classification of gene signatures for their information value and functional redundancy. npj Syst. Biol. Appl. 2018, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauman, G.; Breau, R.H.; Kamel-Reid, S.; Louie, A.V.; Pautlere, S. Ontario health technology assessment series: Prolaris cell cycle progression test for localized prostate cancer: A health technology assessment. Ont. Health Technol. Assess. Ser. 2017, 17, 1–75. [Google Scholar]
- Tretiakova, M.S.; Wei, W.; Boyer, H.D.; Newcomb, L.F.; Hawley, S.; Auman, H.; Vakar-Lopez, F.; McKenney, J.K.; Fazli, L.; Simko, J.; et al. Prognostic value of Ki67 in localized prostate carcinoma: A multi-institutional study of >1000 prostatectomies. Prostate Cancer Prostatic Dis. 2016, 19, 264–270. [Google Scholar] [CrossRef] [Green Version]
- Heemers, H.V.; Schmidt, L.J.; Sun, Z.; Regan, K.M.; Anderson, S.K.; Duncan, K.; Wang, D.; Liu, S.; Ballman, K.V.; Tindall, D.J. Identification of a clinically relevant androgen-dependent gene signature in prostate cancer. Cancer Res. 2011, 71, 1978–1988. [Google Scholar] [CrossRef] [Green Version]
- Akamatsu, S.; Wyatt, A.W.; Lin, D.; Lysakowski, S.; Zhang, F.; Kim, S.; Tse, C.; Wang, K.; Mo, F.; Haegert, A.; et al. The placental gene PEG10 promotes progression of neuroendocrine prostate cancer. Cell Rep. 2015, 12, 922–936. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Montoya, A.; Lamb, A.D.; Russell, R.; Carroll, T.; Jurmeister, S.; Galeano-Dalmau, N.; Massie, C.E.; Boren, J.; Bon, H.; Theodorou, V.; et al. HES6 drives a critical AR transcriptional programme to induce castration-resistant prostate cancer through activation of an E2F1-mediated cell cycle network. EMBO Mol. Med. 2014, 6, 651–661. [Google Scholar] [CrossRef] [Green Version]
- Luca, B.A.; Brewer, D.S.; Edwards, D.R.; Edwards, S.; Whitaker, H.C.; Merson, S.; Dennis, N.; Cooper, R.A.; Hazell, S.; Warren, A.Y.; et al. DESNT: A Poor Prognosis Category of Human Prostate Cancer. Eur. Urol. Focus 2018, 4, 842–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luca, B.A.; Moulton, V.; Ellis, C.; Edwards, D.R.; Campbell, C.; Cooper, R.A.; Clark, J.; Brewer, D.S.; Cooper, C.S. A novel stratification framework for predicting outcome in patients with prostate cancer. Br. J. Cancer 2020, 122, 1467–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, S.; Knudsen, B.S.; Erho, N.; Alshalalfa, M.; Takhar, M.; Ashab, H.A.D.; Davicioni, E.; Karnes, R.J.; Klein, E.A.; Den, R.B.; et al. Integrated classification of prostate cancer reveals a novel luminal subtype with poor outcome. Cancer Res. 2016, 76, 4948–4958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Alonso, L.; Iorio, F.; Matchan, A.; Fonseca, N.; Jaaks, P.; Peat, G.; Pignatelli, M.; Falcone, F.; Benes, C.H.; Dunham, I.; et al. Transcription factor activities enhance markers of drug sensitivity in cancer. Cancer Res. 2018, 78, 769–780. [Google Scholar] [CrossRef] [Green Version]
- Berglund, A.E.; Welsh, E.A.; Eschrich, S.A. Characteristics and validation techniques for PCA-based gene-expression signatures. Int. J. Genom. 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Fox, N.S.; Starmans, M.H.W.; Haider, S.; Lambin, P.; Boutros, P.C. Ensemble analyses improve signatures of tumour hypoxia and reveal inter-platform differences. BMC Bioinform. 2014, 15, 170. [Google Scholar] [CrossRef] [Green Version]
- Gene Expression in Patients with Metastatic Prostate Cancer Receiving CYP-17 Inhibition Therapy—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01953640 (accessed on 20 December 2020).
- Arriaga, J.M.; Panja, S.; Alshalalfa, M.; Zhao, J.; Zou, M.; Giacobbe, A.; Madubata, C.J.; Kim, J.Y.; Rodriguez, A.; Coleman, I.; et al. A MYC and RAS co-activation signature in localized prostate cancer drives bone metastasis and castration resistance. Nat. Cancer 2020, 1, 1082–1096. [Google Scholar] [CrossRef]
- Bhandari, V.; Hoey, C.; Liu, L.Y.; Lalonde, E.; Ray, J.; Livingstone, J.; Lesurf, R.; Shiah, Y.J.; Vujcic, T.; Huang, X.; et al. Molecular landmarks of tumor hypoxia across cancer types. Nat. Genet. 2019, 51, 308–318. [Google Scholar] [CrossRef]
- Lalonde, E.; Ishkanian, A.S.; Sykes, J.; Fraser, M.; Ross-Adams, H.; Erho, N.; Dunning, M.J.; Halim, S.; Lamb, A.D.; Moon, N.C.; et al. Tumour genomic and microenvironmental heterogeneity for integrated prediction of 5-year biochemical recurrence of prostate cancer: A retrospective cohort study. Lancet Oncol. 2014, 15, 1521–1532. [Google Scholar] [CrossRef]
- Luoto, K.R.; Kumareswaran, R.; Bristow, R.G. Tumor hypoxia as a driving force in genetic instability. Genome Integr. 2013, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Betts, G.N.J.; Eustace, A.; Patiar, S.; Valentine, H.R.; Irlam, J.; Ramachandran, A.; Merve, A.; Homer, J.J.; Möller-Levet, C.; Buffa, F.M.; et al. Prospective technical validation and assessment of intra-tumour heterogeneity of a low density array hypoxia gene profile in head and neck squamous cell carcinoma. Eur. J. Cancer 2013, 49, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Buffa, F.M.; Harris, A.L.; West, C.M.; Miller, C.J. Large meta-analysis of multiple cancers reveals a common, compact and highly prognostic hypoxia metagene. Br. J. Cancer 2010, 102, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Lendahl, U.; Lee, K.L.; Yang, H.; Poellinger, L. Generating specificity and diversity in the transcriptional response to hypoxia. Nat. Rev. Genet. 2009, 10, 821–832. [Google Scholar] [CrossRef]
- Ragnum, H.B.; Vlatkovic, L.; Lie, A.K.; Axcrona, K.; Julin, C.H.; Frikstad, K.M.; Hole, K.H.; Seierstad, T.; Lyng, H. The tumour hypoxia marker pimonidazole reflects a transcriptional programme associated with aggressive prostate cancer. Br. J. Cancer 2015, 112, 382–390. [Google Scholar] [CrossRef] [Green Version]
- Toustrup, K.; Sørensen, B.S.; Nordsmark, M.; Busk, M.; Wiuf, C.; Alsner, J.; Overgaard, J. Development of a hypoxia gene expression classifier with predictive impact for hypoxic modification of radiotherapy in head and neck cancer. Cancer Res. 2011, 71, 5923–5931. [Google Scholar] [CrossRef] [Green Version]
- Winter, S.C.; Buffa, F.M.; Silva, P.; Miller, C.; Valentine, H.R.; Turley, H.; Shah, K.A.; Cox, G.J.; Corbridge, R.J.; Homer, J.J.; et al. Relation of a hypoxia metagene derived from head and neck cancer to prognosis of multiple cancers. Cancer Res. 2007, 67, 3441–3449. [Google Scholar] [CrossRef] [Green Version]
- Topatana, W.; Juengpanich, S.; Li, S.; Cao, J.; Hu, J.; Lee, J.; Suliyanto, K.; Ma, D.; Zhang, B.; Chen, M.; et al. Advances in synthetic lethality for cancer therapy: Cellular mechanism and clinical translation. J. Hematol. Oncol. 2020, 13, 118. [Google Scholar] [CrossRef]
- Turkington, R.C.; Knight, L.A.; Blayney, J.K.; Secrier, M.; Douglas, R.; Parkes, E.E.; Sutton, E.K.; Stevenson, L.; McManus, D.; Halliday, S.; et al. Immune activation by DNA damage predicts response to chemotherapy and survival in oesophageal adenocarcinoma. Gut 2019, 68, 1918–1927. [Google Scholar] [CrossRef]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-dependent innate immune signaling by s-phase-specific DNA damage in breast cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, C.J.; Walker, S.; McCabe, N.; Hill, L.; Parkes, E.; Jain, S.; O’Rourke, D.; Kennedy, R. An innate immune response to intrinsic DNA damage predicts resistance to docetaxel in prostate cancer. Ann. Oncol. 2016, 27, 256. [Google Scholar] [CrossRef]
- Ma, M.; Ghosh, S.; Tavernari, D.; Katarkar, A.; Clocchiatti, A.; Mazzeo, L.; Samarkina, A.; Epiney, J.; Yu, Y.R.; Ho, P.C.; et al. Sustained androgen receptor signaling is a determinant of melanoma cell growth potential and tumorigenesis. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef]
- Wengner, A.M.; Scholz, A.; Haendler, B. Targeting DNA damage response in prostate and breast cancer. Int. J. Mol. Sci. 2020, 21, 8273. [Google Scholar] [CrossRef]
- A Clinical Study Evaluating The Benefit of Adding Rucaparib to Enzalutamide for Men With Metastatic Prostate Cancer That Has Become Resistant to Testosterone-Deprivation Therapy—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04455750 (accessed on 17 December 2020).
- Hussain, M.; Daignault, S.; Twardowski, P.; Albany, C.; Stein, M.N.; Kunju, L.P.; Robinson, D.R.; Cooney, K.A.; Montgomery, R.B.; Antonarakis, E.S.; et al. Abiraterone + prednisone (Abi) +/- veliparib (Vel) for patients (pts) with metastatic castration-resistant prostate cancer (CRPC): NCI 9012 updated clinical and genomics data. J. Clin. Oncol. 2017, 35, 5001. [Google Scholar] [CrossRef]
- Choudhury, A.D.; Xie, W.; Parikh, M.; Lee, D.; Kessler, E.R.; Einstein, D.J.; Kochupurakkal, B.; Mouw, K.W.; Van Allen, E.M.; Doyle, L.A.; et al. A phase II study of M6620 in combination with carboplatin compared with docetaxel in combination with carboplatin in metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2020, 38, 5597. [Google Scholar] [CrossRef]
- Rathkopf, D.E.; Autio, K.A.; Antonarakis, E.S.; Cheng, H.H.; Arauz, G.; Slack, A.; Hullings, M.; Scher, H.I.; Feng, F.Y.; Knudsen, K.E. c15-160: Enzalutamide (ENZA) plus CC-115 in men with metastatic castration-resistant prostate cancer (mCRPC): A phase 1b Prostate Cancer Clinical Trials Consortium study. J. Clin. Oncol. 2018, 36, 5045. [Google Scholar] [CrossRef]
- Riches, L.C.; Trinidad, A.G.; Hughes, G.; Jones, G.N.; Hughes, A.M.; Thomason, A.G.; Gavine, P.; Cui, A.; Ling, S.; Stott, J.; et al. Pharmacology of the ATM inhibitor AZD0156: Potentiation of irradiation and olaparib responses preclinically. Mol. Cancer Ther. 2020, 19, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez, Y.; Segura, V.; Marín-Béjar, O.; Athie, A.; Marchese, F.P.; González, J.; Bujanda, L.; Guo, S.; Matheu, A.; Huarte, M. Genome-wide analysis of the human p53 transcriptional network unveils a lncRNA tumour suppressor signature. Nat. Commun. 2014, 5, 5812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, Q.; Johnson, B.A.; Osunkoya, A.O.; Lai, Y.H.; Zhou, W.; Abramovitz, M.; Xia, M.; Bouzyk, M.B.; Nam, R.K.; Sugar, L.; et al. Protein-coding and microRNA biomarkers of recurrence of prostate cancer following radical prostatectomy. Am. J. Pathol. 2011, 179, 46–54. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Ma, X.; Yang, M.; Wang, M.; Li, L.; Huang, T. Transcription factor profiling to predict recurrence-free survival in breast cancer: Development and validation of a nomogram to optimize clinical management. Front. Genet. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Lhomond, S.; Avril, T.; Dejeans, N.; Voutetakis, K.; Doultsinos, D.; McMahon, M.; Pineau, R.; Obacz, J.; Papadodima, O.; Jouan, F.; et al. Dual IRE1 RNase functions dictate glioblastoma development. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Lindner, P.; Jirmo, A.C.; Maus, U.; Illig, T.; Deluca, D.S. A comparison of curated gene sets versus transcriptomics-derived gene signatures for detecting pathway activation in immune cells. BMC Bioinform. 2020, 21, 28. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Bao, X.; Weischenfeldt, J.; Schaefer, C.; Rogowski, P.; Schmidt-Hegemann, N.-S.; Unger, K.; Lauber, K.; Wang, X.; Buchner, A.; et al. A novel gene signature-based model predicts biochemical recurrence-free survival in prostate cancer patients after radical prostatectomy. Cancers 2020, 12, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyshkovskiy, A.; Bozaykut, P.; Borodinova, A.A.; Gerashchenko, M.V.; Ables, G.P.; Garratt, M.; Khaitovich, P.; Clish, C.B.; Miller, R.A.; Gladyshev, V.N. Identification and application of gene expression signatures associated with lifespan extension. Cell Metab. 2019, 30, 573–593. [Google Scholar] [CrossRef] [PubMed]
- Burgess, D.J. Spatial transcriptomics coming of age. Nat. Rev. Genet. 2019, 20, 317. [Google Scholar] [CrossRef]
- Isaacs, J.T.; Coffey, D.S. Adaptation versus selection as the mechanism responsible for the relapse of prostatic cancer to androgen ablation therapy as studied in the dunning R-3327-H adenocarcinoma. Cancer Res. 1981, 41, 5070–5074. [Google Scholar]
- Clocchiatti, A.; Ghosh, S.; Procopio, M.G.; Mazzeo, L.; Bordignon, P.; Ostano, P.; Goruppi, S.; Bottoni, G.; Katarkar, A.; Levesque, M.; et al. Androgen receptor functions as transcriptional repressor of cancer-associated fibroblast activation. J. Clin. Investig. 2018, 128, 5465–5478. [Google Scholar] [CrossRef] [Green Version]
- IMPRESS Leads the Way for Cancer Precision Medicine—Oslo Cancer Cluster. Available online: https://oslocancercluster.no/2020/10/13/impress-leads-the-way-for-cancer-precision-medicine/ (accessed on 20 December 2020).
- Tomlins, S.A.; Alshalalfa, M.; Davicioni, E.; Erho, N.; Yousefi, K.; Zhao, S.; Haddad, Z.; Den, R.B.; Dicker, A.P.; Trock, B.J.; et al. Characterization of 1577 primary prostate cancers reveals novel biological and clinicopathologic insights into molecular subtypes. Eur. Urol. 2015, 68, 555–567. [Google Scholar] [CrossRef]
- Mills, I.G. Molecular subtyping of prostate cancer: A partnership model. Eur. Urol. 2015, 68, 568–569. [Google Scholar] [CrossRef] [PubMed]
- Home | BioS Project. Available online: https://www.bios-project.eu/site/ (accessed on 20 December 2020).





{kind=link}
{kind=link}
{kind=link}
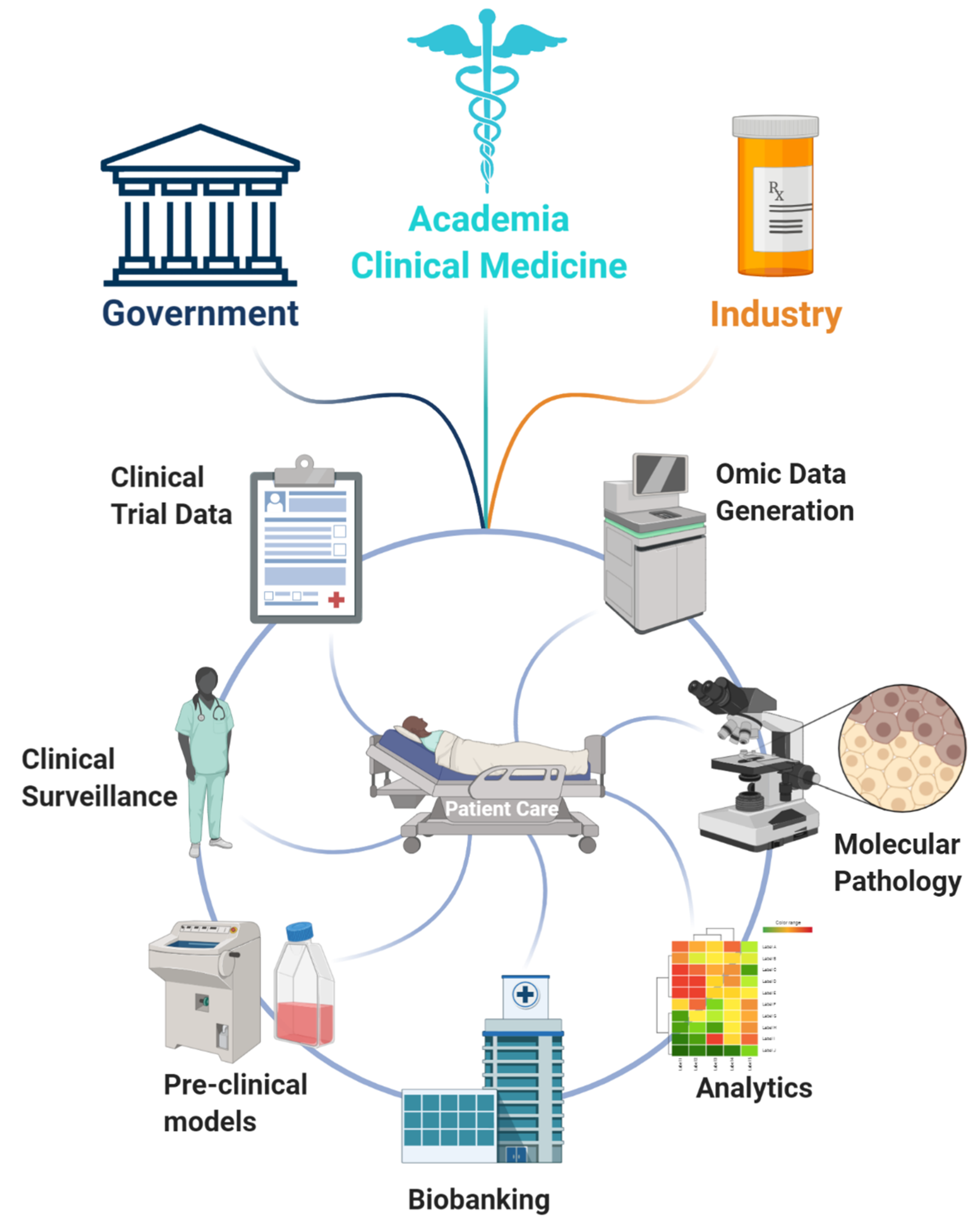{kind=link}
| Scheme | Signature | Mechanism | Translation | Ref. |
|---|---|---|---|---|
| Commercial | Oncotype DX | 12 genes related to androgen metabolism, cellular organization, proliferation, and stromal response plus 5 reference genes | Reverse | [34,35] |
| Prolaris | 31 cell cycle genes | Reverse | [36] | |
| Decipher | 22 genes involved in proliferation, structure, immune modulation, cell cycle and androgen signaling | Reverse | [37] | |
| ExoDX | Urinary-derived exosomal gene signature based on PCA3 and ERG RNA levels | Reverse | [38] | |
| SelectMDx | 2 gene signature (DLX1 and HOXC6) | Reverse | [39] | |
| Academic | Hypoxia-28 | 6 hypoxia and 11 PCa signature overlap | Combinatorial | [40] |
| BROMO-10 | 10 gene signature from bromodomain inhibitor treated cells and PCa cohorts | Forward | [41] | |
| AR-v7 | Variant correlation with 59 gene CRPC signature | Reverse | [42] | |
| SIG-DENT SIG-HES6 | Overlapping PCa-related signatures | Combinatorial | [43] | |
| Primary PCa metastatic assay | 70 gene signature to predict risk of biochemical recurrence | Reverse | [44] | |
| AR–MITF–Myc | Combination of signatures to prognosticate PCa survival | Forward | [45,46] | |
| MycN-EZH2 | Transcriptome/cistrome/interactome N-Myc-derived signature in advanced PCa | Forward | [47] | |
| mCRPC-26 | Treatment-resistant CRPC classification | Combinatorial | [48] |
| Table | Therapeutic Combination | Clinical Trial Identifier | Ref |
|---|---|---|---|
| PARP-1 | Rucaparib/enzalutamide | NCT04455750 | [106] |
| PARP-1 | Veliparib/abiraterone | NCT01576172 | [107] |
| ATR | Berzosertib/carboplatin/docetaxel | NCT03517969 | [108] |
| DNA-PK | CC-115/enzalutamide | NCT02833883 | [109] |
| ATM | AZD0156/FOLFIRI | NCT02588105 | [110] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doultsinos, D.; Mills, I.G. Derivation and Application of Molecular Signatures to Prostate Cancer: Opportunities and Challenges. Cancers 2021, 13, 495. https://doi.org/10.3390/cancers13030495
Doultsinos D, Mills IG. Derivation and Application of Molecular Signatures to Prostate Cancer: Opportunities and Challenges. Cancers. 2021; 13(3):495. https://doi.org/10.3390/cancers13030495
Chicago/Turabian StyleDoultsinos, Dimitrios, and Ian G. Mills. 2021. "Derivation and Application of Molecular Signatures to Prostate Cancer: Opportunities and Challenges" Cancers 13, no. 3: 495. https://doi.org/10.3390/cancers13030495