
Therapeutic Targets of KRAS in Colorectal Cancer


Abstract
:Simple Summary
Abstract
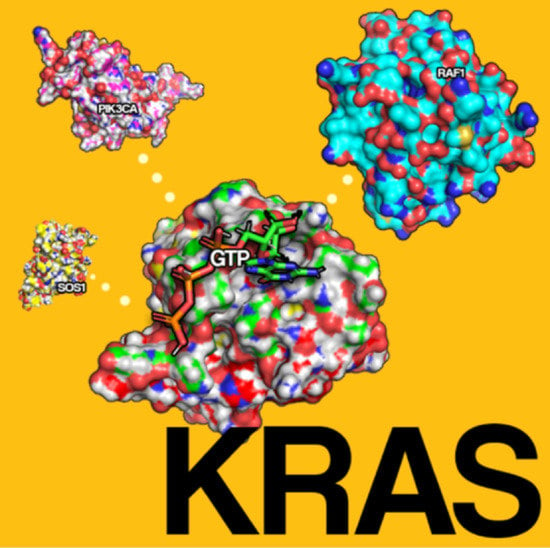
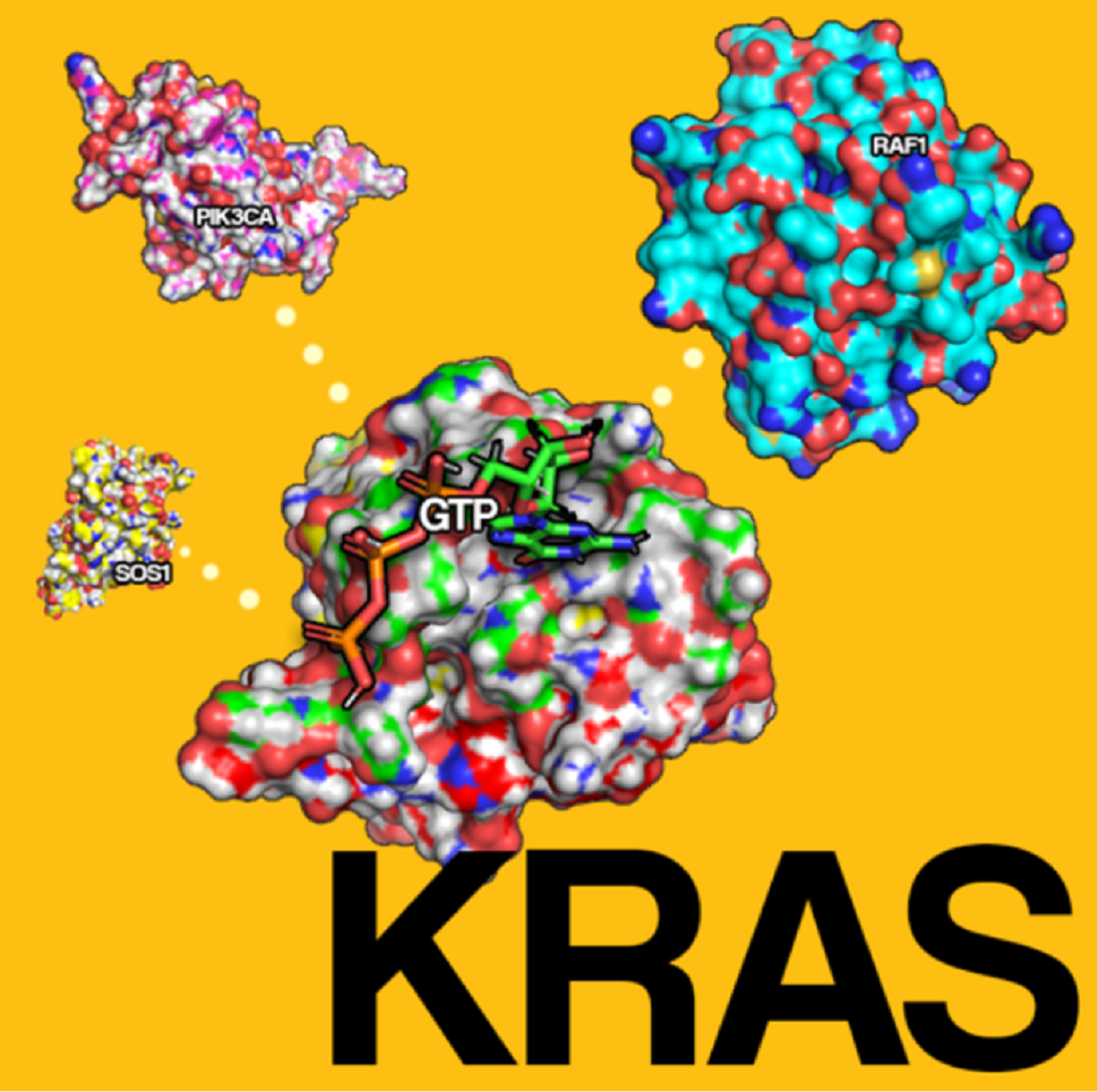
1. Introduction
2. Discovery of RAS
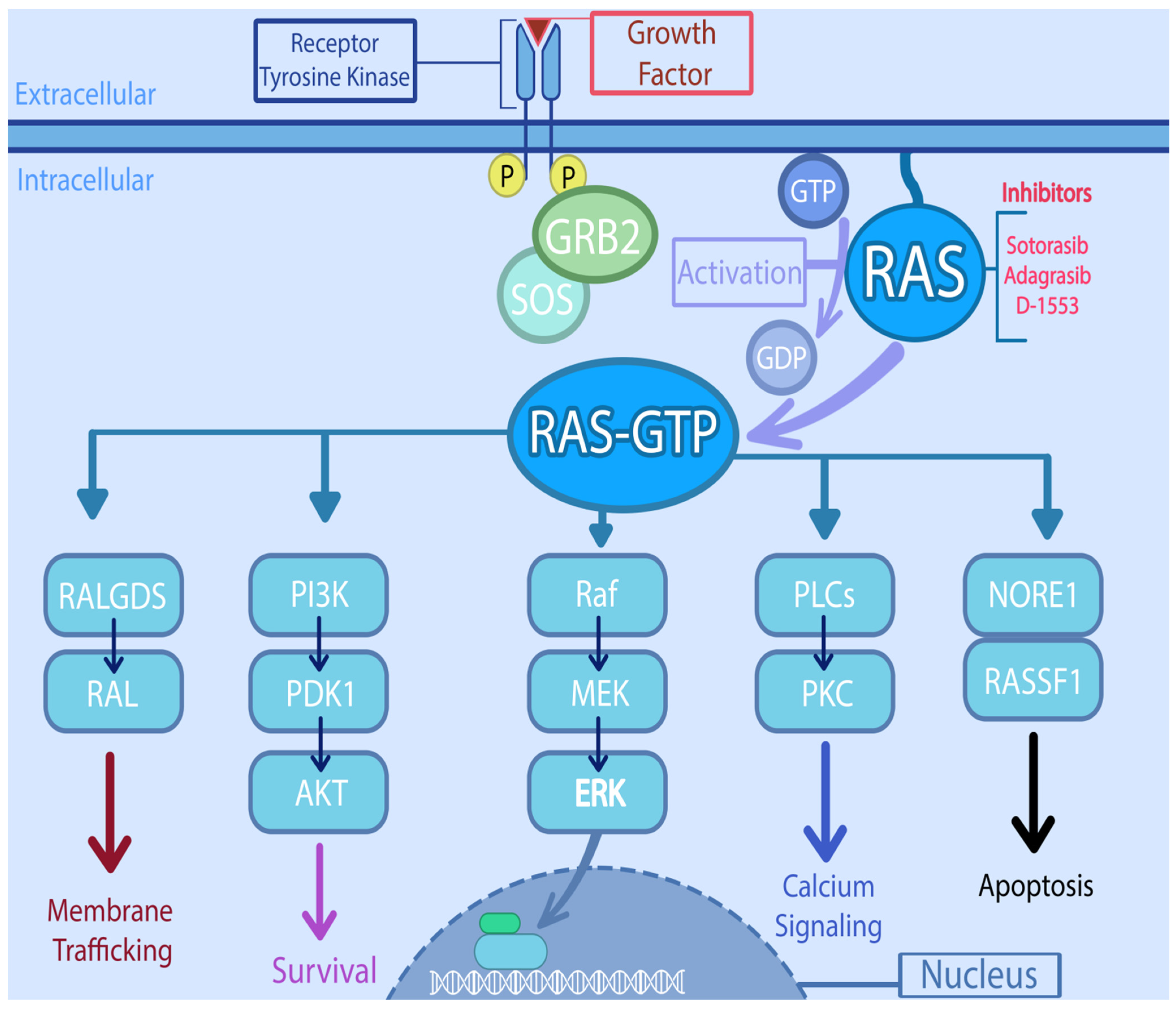
3. Physiology of RAS
4. Mutations Involving RAS in CRC
5. Targeting KRAS
5.1. KRAS Directed
5.2. AMG 510
5.3. MRTX849
5.4. MRTX1133
5.5. PLK-1 Inhibition
5.6. KR12
5.7. SHP2 Inhibition
6. Targeting Membrane Association
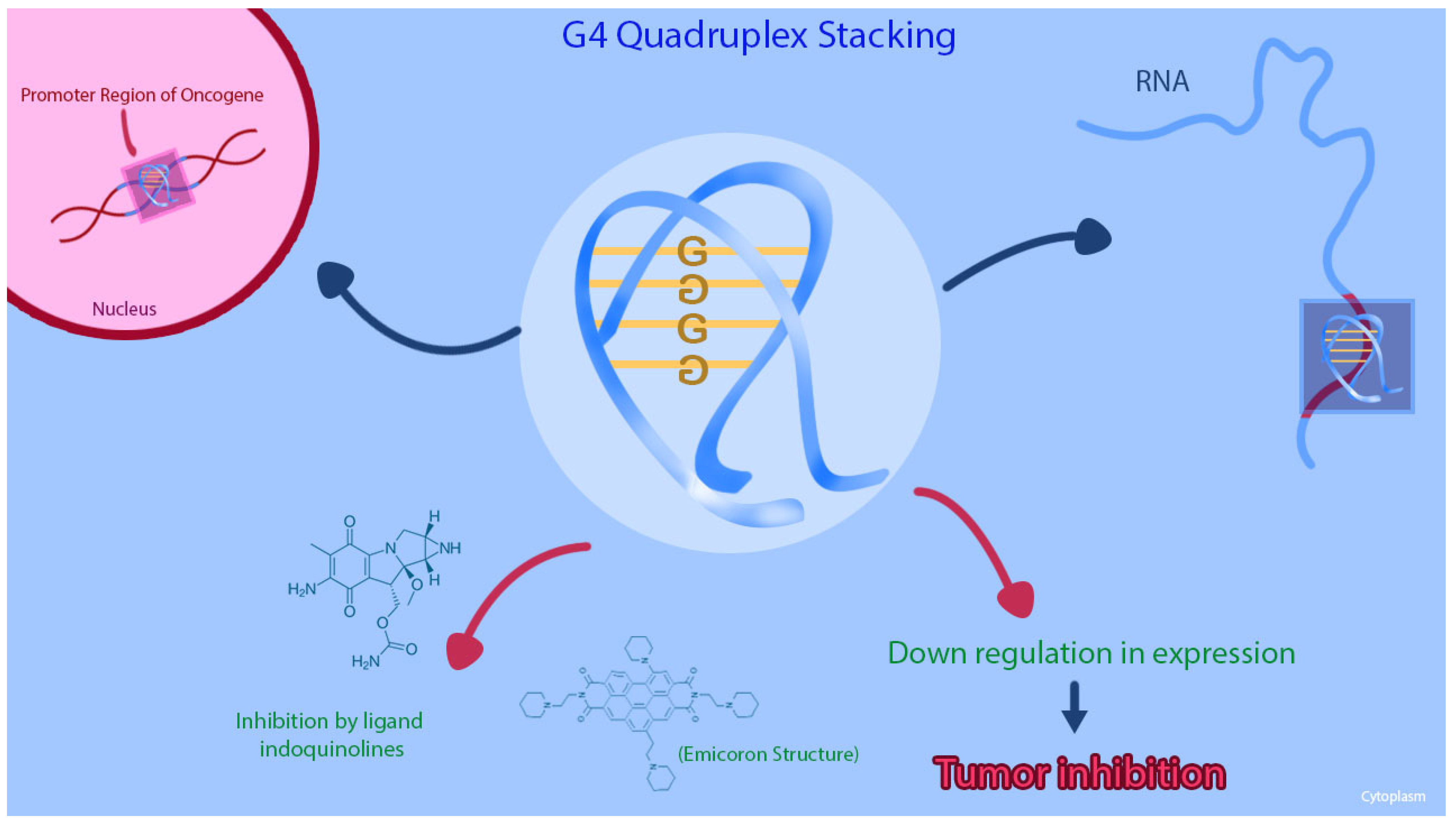
Targeting of G4 Structures
7. Indirect Approaches
7.1. PDEδ Inhibition
7.2. Targeting NRF2/Oxidative and Glutaminolysis
7.3. Oncolytic Virus Induced Autophagy
8. Combined Inhibition of Downstream Pathways
8.1. AZD4785
8.2. AZD6244
8.3. MEK and P13K/mTOR Combination
9. Developing Therapies
9.1. MiRNA as Potential Drug Candidates
9.2. MYC Inhibition
9.3. T Cell-Mediated Therapy
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Islami, F.; Ward, E.M.; Sung, H.; Cronin, K.A.; Tangka, F.K.L.; Sherman, R.L.; Zhao, J.; Anderson, R.N.; Henley, S.J.; Yabroff, K.R.; et al. Annual Report to the Nation on the Status of Cancer, Part 1: National Cancer Statistics. J. Natl. Cancer Inst. 2021, 113, 1648–1669. [Google Scholar] [CrossRef]
- Loeb, L.A.; Springgate, C.F.; Battula, N. Errors in DNA Replication as a Basis of Malignant Changes. Cancer Res. 1974, 34, 2311–2321. [Google Scholar] [PubMed]
- Pino, M.S.; Chung, D.C. The Chromosomal Instability Pathway in Colon Cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer—The stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pancione, M.; Remo, A.; Colantuoni, V. Genetic and Epigenetic Events Generate Multiple Pathways in Colorectal Cancer Progression. Pathol. Res. Int. 2012, 2012, 509348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Vecchia, S.; Sebastián, C. Metabolic pathways regulating colorectal cancer initiation and progression. Semin. Cell Dev. Biol 2020, 98, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, Y.; Vikis, H.G.; Johnson, L.; Liu, G.; Li, J.; Anderson, M.W.; Sills, R.C.; Hong, H.L.; Devereux, T.R.; et al. Wildtype Kras2 can inhibit lung carcinogenesis in mice. Nat. Genet. 2001, 29, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.W.; Reynolds, S.H.; You, M.; Maronpot, R.M. Role of proto-oncogene activation in carcinogenesis. Environ. Health Perspect. 1992, 98, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Kranenburg, O. The KRAS oncogene: Past, present, and future. Biochim. Biophys. Acta 2005, 1756, 81–82. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L.; Fearon, E.R.; Hamilton, S.R.; Vries, M.V.d.; van Boom, J.H.; van der Eb, A.J.; Vogelstein, B. Prevalence of ras gene mutations in human colorectal cancers. Nature 1987, 327, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Bazan, V.; Agnese, V.; Corsale, S.; Calò, V.; Valerio, M.R.; Latteri, M.A.; Vieni, S.; Grassi, N.; Cicero, G.; Dardanoni, G.; et al. Specific TP53 and/or Ki-ras mutations as independent predictors of clinical outcome in sporadic colorectal adenocarcinomas: Results of a 5-year Gruppo Oncologico dell’Italia Meridionale (GOIM) prospective study. Ann. Oncol. 2005, 16, iv50–iv55. [Google Scholar] [CrossRef] [PubMed]
- Überall, I.; Kolář, Z.; Trojanec, R.; Berkovcová, J.; Hajdúch, M. The status and role of ErbB receptors in human cancer. Exp. Mol. Pathol. 2008, 84, 79–89. [Google Scholar] [CrossRef]
- Scolnick, E.M.; Rands, E.; Williams, D.; Parks, W.P. Studies on the nucleic acid sequences of Kirsten sarcoma virus: A model for formation of a mammalian RNA-containing sarcoma virus. J. Virol. 1973, 12, 458–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, J.J. An unidentified virus which causes the rapid production of tumours in mice. Nature 1964, 204, 1104–1105. [Google Scholar] [CrossRef] [PubMed]
- Stehelin, D.; Varmus, H.E.; Bishop, J.M.; Vogt, P.K. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature 1976, 260, 170–173. [Google Scholar] [CrossRef]
- Román, M.; Baraibar, I.; López, I.; Nadal, E.; Rolfo, C.; Vicent, S.; Gil-Bazo, I. KRAS oncogene in non-small cell lung cancer: Clinical perspectives on the treatment of an old target. Mol. Cancer 2018, 17, 33. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.; Marshall, C.J.; Spurr, N.K.; Weiss, R.A. Identification of transforming gene in two human sarcoma cell lines as a new member of the ras gene family located on chromosome 1. Nature 1983, 303, 396–400. [Google Scholar] [CrossRef]
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The Ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jančík, S.; Drábek, J.; Radzioch, D.; Hajdúch, M. Clinical Relevance of KRAS in Human Cancers. J. Biomed. Biotechnol. 2010, 2010, 150960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancock, J.F.; Magee, A.I.; Childs, J.E.; Marshall, C.J. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 1989, 57, 1167–1177. [Google Scholar] [CrossRef]
- Seger, R.; Krebs, E.G. The MAPK signaling cascade. FASEB J. 1995, 9, 726–735. [Google Scholar] [CrossRef]
- Lambert, J.M.; Lambert, Q.T.; Reuther, G.W.; Malliri, A.; Siderovski, D.P.; Sondek, J.; Collard, J.G.; Der, C.J. Tiam1 mediates Ras activation of Rac by a PI(3)K-independent mechanism. Nat. Cell Biol. 2002, 4, 621–625. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Vander Heiden, M.G.; McCormick, F. The Metabolic Landscape of RAS-Driven Cancers from biology to therapy. Nat. Cancer 2021, 2, 271–283. [Google Scholar] [CrossRef]
- Vaughn, C.P.; Zobell, S.D.; Furtado, L.V.; Baker, C.L.; Samowitz, W.S. Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes Chromosomes Cancer 2011, 50, 307–312. [Google Scholar] [CrossRef]
- Li, Z.-N.; Zhao, L.; Yu, L.-F.; Wei, M.-J. BRAF and KRAS mutations in metastatic colorectal cancer: Future perspectives for personalized therapy. Gastroenterol. Rep. 2020, 8, 192–205. [Google Scholar] [CrossRef]
- Zocche, D.M.; Ramirez, C.; Fontao, F.M.; Costa, L.D.; Redal, M.A. Global impact of KRAS mutation patterns in FOLFOX treated metastatic colorectal cancer. Front. Genet. 2015, 6, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M.; González, S.; Risques, R.A.; Marcuello, E.; Mangues, R.; Germà, J.R.; Herman, J.G.; Capellà, G.; Peinado, M.A. K-ras and p16 aberrations confer poor prognosis in human colorectal cancer. J. Clin. Oncol. 2001, 19, 299–304. [Google Scholar] [CrossRef]
- Lito, P.; Solomon, M.; Li, L.-S.; Hansen, R.; Rosen, N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 2016, 351, 604–608. [Google Scholar] [CrossRef] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Fakih, M.; Desai, J.; Kuboki, Y.; Strickler, J.H.; Price, T.J.; Durm, G.A.; Falchook, G.S.; Denlinger, C.S.; Krauss, J.C.; Shapiro, G.; et al. CodeBreak 100: Activity of AMG 510, a novel small molecule inhibitor of KRASG12C, in patients with advanced colorectal cancer. J. Clin. Oncol. 2020, 38, 4018. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabari, J.K.; Park, H.; Tolcher, A.W.; Ou, S.-H.I.; Garon, E.B.; George, B.; Janne, P.A.; Moody, S.E.; Tan, E.Y.; Sen, S.K.; et al. KRYSTAL-2: A phase I/II trial of adagrasib (MRTX849) in combination with TNO155 in patients with advanced solid tumors with KRAS G12C mutation. J. Clin. Oncol. 2021, 39, TPS146. [Google Scholar] [CrossRef]
- Xie, M.; Xu, X.; Fan, Y. KRAS-Mutant Non-Small Cell Lung Cancer: An Emerging Promisingly Treatable Subgroup. Front. Oncol. 2021, 11, 672612. [Google Scholar] [CrossRef]
- Therapeutics, M. Mirati Therapeutics Reports Investigational Adagrasib (MRTX849) Preliminary Data Demonstrating Tolerability and Durable Anti-Tumor Activity as Well as Initial MRTX1133 Preclinical Data. 2020. Available online: https://ir.mirati.com/press-releases/press-release-details/2020/Mirati-Therapeutics-Reports-Investigational-Adagrasib-MRTX849-Preliminary-Data-Demonstrating-Tolerability-and-Durable-Anti-Tumor-Activity-as-well-as-Initial-MRTX1133-Preclinical-Data/default.aspx (accessed on 28 October 2021).
- Addeo, A.; Banna, G.L.; Friedlaender, A. KRAS G12C Mutations in NSCLC: From Target to Resistance. Cancers 2021, 13, 2541. [Google Scholar] [CrossRef]
- de Cárcer, G.; Manning, G.; Malumbres, M. From Plk1 to Plk5: Functional evolution of polo-like kinases. Cell Cycle 2011, 10, 2255–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, N.; Hamanaka, R.; Yoshimatsu, J.; Miyakawa, I. Polo-like kinases (Plks) and cancer. Oncogene 2005, 24, 287–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimino, S.K.; Eng, C. Up-and-Coming Experimental Drug Options for Metastatic Colorectal Cancer. J. Exp. Pharm. 2020, 12, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Jayachandran, P.; Elliott, A.; Battaglin, F.; Lou, E.; Zhang, W.; Soni, S.; Arai, H.; Wang, J.; Millstein, J.; Lockhart, A.C.; et al. 473P PLK1 expression and KRAS mutations in colorectal cancer. Ann. Oncol. 2020, 31, S442. [Google Scholar] [CrossRef]
- Valsasina, B.; Beria, I.; Alli, C.; Alzani, R.; Avanzi, N.; Ballinari, D.; Cappella, P.; Caruso, M.; Casolaro, A.; Ciavolella, A.; et al. NMS-P937, an orally available, specific small-molecule polo-like kinase 1 inhibitor with antitumor activity in solid and hematologic malignancies. Mol. Cancer 2012, 11, 1006–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Einstein, D.J.; Choudhury, A.D.; Saylor, P.J.; Patterson, J.C.; Croucher, P.; Ridinger, M.; Erlander, M.G.; Yaffe, M.B.; Bubley, G. A phase II study of onvansertib in combination with abiraterone and prednisone in patients with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2021, 39, TPS186. [Google Scholar] [CrossRef]
- Hiraoka, K.; Inoue, T.; Taylor, R.D.; Watanabe, T.; Koshikawa, N.; Yoda, H.; Shinohara, K.; Takatori, A.; Sugimoto, H.; Maru, Y.; et al. Inhibition of KRAS codon 12 mutants using a novel DNA-alkylating pyrrole-imidazole polyamide conjugate. Nat. Commun. 2015, 6, 6706. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.Q.; Lin, Q.; Zhuang, X.; Cai, L.L.; Ruan, R.S.; Lu, Z.X.; Tzeng, C.M. Structure, function, and pathogenesis of SHP2 in developmental disorders and tumorigenesis. Curr. Cancer Drug Targets 2014, 14, 567–588. [Google Scholar] [CrossRef]
- Cunnick, J.M.; Meng, S.; Ren, Y.; Desponts, C.; Wang, H.G.; Djeu, J.Y.; Wu, J. Regulation of the mitogen-activated protein kinase signaling pathway by SHP2. J. Biol. Chem. 2002, 277, 9498–9504. [Google Scholar] [CrossRef] [Green Version]
- Maroun, C.R.; Naujokas, M.A.; Holgado-Madruga, M.; Wong, A.J.; Park, M. The tyrosine phosphatase SHP-2 is required for sustained activation of extracellular signal-regulated kinase and epithelial morphogenesis downstream from the met receptor tyrosine kinase. Mol. Cell. Biol. 2000, 20, 8513–8525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.J.; O’Rourke, D.M.; Feng, G.S.; Johnson, G.R.; Wang, Q.; Greene, M.I. The tyrosine phosphatase SHP-2 is required for mediating phosphatidylinositol 3-kinase/Akt activation by growth factors. Oncogene 2001, 20, 6018–6025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, M.; Yu, D.H.; Feng, G.S. Shp-2 tyrosine phosphatase functions as a negative regulator of the interferon-stimulated Jak/STAT pathway. Mol. Cell Biol 1999, 19, 2416–2424. [Google Scholar] [CrossRef] [Green Version]
- Patsoukis, N.; Duke-Cohan, J.S.; Chaudhri, A.; Aksoylar, H.-I.; Wang, Q.; Council, A.; Berg, A.; Freeman, G.J.; Boussiotis, V.A. Interaction of SHP-2 SH2 domains with PD-1 ITSM induces PD-1 dimerization and SHP-2 activation. Commun. Biol. 2020, 3, 128. [Google Scholar] [CrossRef]
- Zito, C.I.; Qin, H.; Blenis, J.; Bennett, A.M. SHP-2 Regulates Cell Growth by Controlling the mTOR/S6 Kinase 1 Pathway. J. Biol. Chem. 2007, 282, 6946–6953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, C.K. The SHP-2 tyrosine phosphatase: Signaling mechanisms and biological functions. Cell Res. 2000, 10, 279–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Bu, H.; Zhou, J.; Yang, C.-Y.; Zhang, H. Recent Advances of SHP2 Inhibitors in Cancer Therapy: Current Development and Clinical Application. J. Med. Chem. 2020, 63, 11368–11396. [Google Scholar] [CrossRef]
- Fedele, C.; Li, S.; Teng, K.W.; Foster, C.; Peng, D.; Ran, H.; Mita, P.; Geer, M.; Hattori, T.; Koide, A.; et al. SHP2 Inhibition Abrogates Adaptive Resistance to KRASG12C-Inhibition and Remodels the Tumor Microenvironment of KRAS-Mutant Tumors. bioRxiv 2020. bioRxiv:2020.2005.2030.125138. [Google Scholar] [CrossRef]
- Wu, F.; Niu, K.; Cui, Y.; Li, C.; Lyu, M.; Ren, Y.; Chen, Y.; Deng, H.; Huang, L.; Zheng, S.; et al. Genome-wide analysis of DNA G-quadruplex motifs across 37 species provides insights into G4 evolution. Commun. Biol. 2021, 4, 98. [Google Scholar] [CrossRef]
- Brooks, T.A.; Kendrick, S.; Hurley, L. Making sense of G-quadruplex and i-motif functions in oncogene promoters. FEBS J. 2010, 277, 3459–3469. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Tang, Q.; Li, Y.; Zhang, Y.; Zhao, C.; Yan, J.; You, H. Folding/unfolding kinetics of G-quadruplexes upstream of the P1 promoter of the human BCL-2 oncogene. J. Biol. Chem. 2019, 294, 5890–5895. [Google Scholar] [CrossRef]
- Lavrado, J.; Borralho, P.M.; Ohnmacht, S.A.; Castro, R.E.; Rodrigues, C.M.; Moreira, R.; dos Santos, D.J.; Neidle, S.; Paulo, A. Synthesis, G-quadruplex stabilisation, docking studies, and effect on cancer cells of indolo[3,2-b]quinolines with one, two, or three basic side chains. ChemMedChem 2013, 8, 1648–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porru, M.; Artuso, S.; Salvati, E.; Bianco, A.; Franceschin, M.; Diodoro, M.G.; Passeri, D.; Orlandi, A.; Savorani, F.; D’Incalci, M.; et al. Targeting G-Quadruplex DNA Structures by EMICORON Has a Strong Antitumor Efficacy against Advanced Models of Human Colon Cancer. Mol. Cancer 2015, 14, 2541–2551. [Google Scholar] [CrossRef] [Green Version]
- Porru, M.; Zizza, P.; Franceschin, M.; Leonetti, C.; Biroccio, A. EMICORON: A multi-targeting G4 ligand with a promising preclinical profile. Biochim. Biophys. Acta 2017, 1861, 1362–1370. [Google Scholar] [CrossRef]
- Chandra, A.; Grecco, H.E.; Pisupati, V.; Perera, D.; Cassidy, L.; Skoulidis, F.; Ismail, S.A.; Hedberg, C.; Hanzal-Bayer, M.; Venkitaraman, A.R.; et al. Erratum: The GDI-like solubilizing factor PDEδ sustains the spatial organization and signalling of Ras family proteins. Nat. Cell Biol. 2012, 14, 329. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Liu, X.-h.; Zhang, K.; Chen, C.-K.; Frederick, J.M.; Prestwich, G.D.; Baehr, W. Photoreceptor cGMP Phosphodiesterase δ Subunit (PDEδ) Functions as a Prenyl-binding Protein. J. Biol. Chem. 2004, 279, 407–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papke, B.; Murarka, S.; Vogel, H.A.; Martín-Gago, P.; Kovacevic, M.; Truxius, D.C.; Fansa, E.K.; Ismail, S.; Zimmermann, G.; Heinelt, K.; et al. Identification of pyrazolopyridazinones as PDEδ inhibitors. Nat. Commun. 2016, 7, 11360. [Google Scholar] [CrossRef]
- Martín-Gago, P.; Fansa, E.K.; Klein, C.H.; Murarka, S.; Janning, P.; Schürmann, M.; Metz, M.; Ismail, S.; Schultz-Fademrecht, C.; Baumann, M.; et al. A PDE6δ-KRas Inhibitor Chemotype with up to Seven H-Bonds and Picomolar Affinity that Prevents Efficient Inhibitor Release by Arl2. Angew. Chem. Int. Ed. Engl. 2017, 56, 2423–2428. [Google Scholar] [CrossRef]
- Klein, C.H.; Truxius, D.C.; Vogel, H.A.; Harizanova, J.; Murarka, S.; Martín-Gago, P.; Bastiaens, P.I.H. PDEδ inhibition impedes the proliferation and survival of human colorectal cancer cell lines harboring oncogenic KRas. Int. J. Cancer 2019, 144, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Maitra, R.; Ghalib, M.H.; Goel, S. Reovirus: A targeted therapeutic--progress and potential. Mol. Cancer Res. 2012, 10, 1514–1525. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, K.J.; Ademi, C.; Bertram, S.; Schmid, K.W.; Baba, H.A. Prognostic relevance of autophagy-related markers LC3, p62/sequestosome 1, Beclin-1 and ULK1 in colorectal cancer patients with respect to KRAS mutational status. World J. Surg. Oncol 2016, 14, 189. [Google Scholar] [CrossRef] [Green Version]
- Jiffry, J.; Thavornwatanayong, T.; Rao, D.; Fogel, E.J.; Saytoo, D.; Nahata, R.; Guzik, H.; Chaudhary, I.; Augustine, T.; Goel, S.; et al. Oncolytic Reovirus (pelareorep) Induces Autophagy in KRAS-mutated Colorectal Cancer. Clin. Cancer Res. 2021, 27, 865–876. [Google Scholar] [CrossRef]
- Goel, S.; Ocean, A.J.; Parakrama, R.Y.; Ghalib, M.H.; Chaudhary, I.; Shah, U.; Viswanathan, S.; Kharkwal, H.; Coffey, M.; Maitra, R. Elucidation of Pelareorep Pharmacodynamics in A Phase I Trial in Patients with KRAS-Mutated Colorectal Cancer. Mol. Cancer Ther. 2020, 19, 1148–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, L.F.; Sebolt-Leopold, J.; Meyer, M.B. CI-1040 (PD184352), a targeted signal transduction inhibitor of MEK (MAPKK). Semin. Oncol. 2003, 30, 105–116. [Google Scholar] [CrossRef]
- Carracedo, A.; Pandolfi, P.P. The PTEN–PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, S.J.; Revenko, A.S.; Hanson, L.L.; Ellston, R.; Staniszewska, A.; Whalley, N.; Pandey, S.K.; Revill, M.; Rooney, C.; Buckett, L.K.; et al. Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Jones, D. The long march of antisense. Nat. Rev. Drug Discov. 2011, 10, 401–402. [Google Scholar] [CrossRef]
- Yeh, T.C.; Marsh, V.; Bernat, B.A.; Ballard, J.; Colwell, H.; Evans, R.J.; Parry, J.; Smith, D.; Brandhuber, B.J.; Gross, S.; et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin. Cancer Res. 2007, 13, 1576–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, B.R.; Logie, A.; McKay, J.S.; Martin, P.; Steele, S.; Jenkins, R.; Cockerill, M.; Cartlidge, S.; Smith, P.D. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: Mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol. Cancer 2007, 6, 2209–2219. [Google Scholar] [CrossRef] [Green Version]
- Bennouna, J.; Lang, I.; Valladares-Ayerbes, M.; Boer, K.; Adenis, A.; Escudero, P.; Kim, T.-Y.; Pover, G.M.; Morris, C.D.; Douillard, J.-Y. A Phase II, open-label, randomised study to assess the efficacy and safety of the MEK1/2 inhibitor AZD6244 (ARRY-142886) versus capecitabine monotherapy in patients with colorectal cancer who have failed one or two prior chemotherapeutic regimens. Investig. New Drugs 2011, 29, 1021–1028. [Google Scholar] [CrossRef]
- Ciombor, K.K.; Bekaii-Saab, T. Selumetinib for the treatment of cancer. Expert Opin. Investig. Drugs 2015, 24, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Tolcher, A.W.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Smith, L.S.; Gunn, S.; Smetzer, L.; Mays, T.A.; Kaiser, B.; et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin. Cancer Res. 2012, 18, 2316–2325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitts, T.M.; Newton, T.P.; Bradshaw-Pierce, E.L.; Addison, R.; Arcaroli, J.J.; Klauck, P.J.; Bagby, S.M.; Hyatt, S.L.; Purkey, A.; Tentler, J.J.; et al. Dual pharmacological targeting of the MAP kinase and PI3K/mTOR pathway in preclinical models of colorectal cancer. PLoS ONE 2014, 9, e113037. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, E.; Troiani, T.; D’Aiuto, E.; Morgillo, F.; Vitagliano, D.; Capasso, A.; Costantino, S.; Ciuffreda, L.P.; Merolla, F.; Vecchione, L.; et al. Antitumor activity of pimasertib, a selective MEK 1/2 inhibitor, in combination with PI3K/mTOR inhibitors or with multi-targeted kinase inhibitors in pimasertib-resistant human lung and colorectal cancer cells. Int. J. Cancer 2013, 133, 2089–2101. [Google Scholar] [CrossRef]
- E, J.; Xing, J.; Gong, H.; He, J.; Zhang, W. Combine MEK inhibition with PI3K/mTOR inhibition exert inhibitory tumor growth effect on KRAS and PIK3CA mutation CRC xenografts due to reduced expression of VEGF and matrix metallopeptidase-9. Tumor Biol. 2015, 36, 1091–1097. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal. Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.; Liang, H.; Uzair Ur, R.; Wang, Y.; Zhang, W.; Zhou, Y.; Chen, S.; Yu, M.; Cui, S.; Liu, M.; et al. miR-96 promotes cell proliferation, migration and invasion by targeting PTPN9 in breast cancer. Sci. Rep. 2016, 6, 37421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Guo, X.; Zhang, H.; Xiang, Y.; Chen, J.; Yin, Y.; Cai, X.; Wang, K.; Wang, G.; Ba, Y.; et al. Role of miR-143 targeting KRAS in colorectal tumorigenesis. Oncogene 2009, 28, 1385–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Dang, J.; Chang, K.Y.; Yau, E.; Aza-Blanc, P.; Moscat, J.; Rana, T.M. miR-1298 Inhibits Mutant KRAS-Driven Tumor Growth by Repressing FAK and LAMB3. Cancer Res. 2016, 76, 5777–5787. [Google Scholar] [CrossRef] [Green Version]
- Soucek, L.; Whitfield, J.R.; Sodir, N.M.; Massó-Vallés, D.; Serrano, E.; Karnezis, A.N.; Swigart, L.B.; Evan, G.I. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013, 27, 504–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soucek, L.; Whitfield, J.; Martins, C.P.; Finch, A.J.; Murphy, D.J.; Sodir, N.M.; Karnezis, A.N.; Swigart, L.B.; Nasi, S.; Evan, G.I. Modelling Myc inhibition as a cancer therapy. Nature 2008, 455, 679–683. [Google Scholar] [CrossRef] [Green Version]
- Blake, D.R.; Vaseva, A.V.; Hodge, R.G.; Kline, M.P.; Gilbert, T.S.K.; Tyagi, V.; Huang, D.; Whiten, G.C.; Larson, J.E.; Wang, X.; et al. Application of a MYC degradation screen identifies sensitivity to CDK9 inhibitors in KRAS-mutant pancreatic cancer. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Ischenko, I.; Zhi, J.; Hayman, M.J.; Petrenko, O. KRAS-dependent suppression of MYC enhances the sensitivity of cancer cells to cytotoxic agents. Oncotarget 2017, 8, 17995–18009. [Google Scholar] [CrossRef]
- Gupta, P.; Zhang, Y.K.; Zhang, X.Y.; Wang, Y.J.; Lu, K.W.; Hall, T.; Peng, R.; Yang, D.H.; Xie, N.; Chen, Z.S. Voruciclib, a Potent CDK4/6 Inhibitor, Antagonizes ABCB1 and ABCG2-Mediated Multi-Drug Resistance in Cancer Cells. Cell. Physiol. Biochem. 2018, 45, 1515–1528. [Google Scholar] [CrossRef] [PubMed]
- Wiley, S.E.; Su, Y.; Ge, Y. Abstract 1962: Voruciclib, a CDK9 inhibitor, downregulates MYC and inhibits proliferation of KRAS mutant cancers in preclinical models. Cancer Res. 2021, 81, 1962. [Google Scholar] [CrossRef]
- Lu, Y.-C.; Yao, X.; Crystal, J.S.; Li, Y.F.; El-Gamil, M.; Gross, C.; Davis, L.; Dudley, M.E.; Yang, J.C.; Samuels, Y.; et al. Efficient Identification of Mutated Cancer Antigens Recognized by T Cells Associated with Durable Tumor Regressions. Clin. Cancer Res. 2014, 20, 3401–3410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- T-Cell Transfer Therapy Targeting Mutant KRAS. N. Engl. J. Med. 2017, 376, e11. [CrossRef] [PubMed]
- Tran, E.; Robbins, P.F.; Lu, Y.-C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Clinical Trial | Drug | Target | Cancer Type | Estimated Enrollment (N) | NCT ID |
|---|---|---|---|---|---|
| Phase I | MRTX849 | KRAS G12C inhibitor | KRAS G12C mutant cancers | 565 | NCT03785249 |
| Phase 1 | KRAS TCR | Anti-KRAS G12D engineered T-cells | KRAS G12D Mutated cancer | 70 | NCT03745326 |
| Phase 1 | KRAS TCR | Anti-KRAS G12 V engineered T-cells | KRAS G12V Mutated cancer | 110 | NCT03190941 |
| Phase 1 | GDC-6036+/− Atezolizumab, Cetuximab, Bevacizumab, Erlotinib | KRAS G12C Mutation | Advanced or Metastatic Solid Tumors With a KRAS G12C Mutation | 342 | NCT0444987 |
| Phase 1 | BBP-398 | SHP2 inhibitor | MAPK pathway or RTK driven advanced solid tumors | 60 | NCT04528836 |
| Phase 1b/2 | Onvansertib (PCM-075) + FOLFIRI + bevacizumab | PLK-1 inhibitor | Metastatic CRC with KRAS mutation | 44 | NCT03829410 |
| Phase 1b/2 | SX-682 +/−nivolumab | CXCR1/2 inhibitor | Metastatic CRC, RAS mutated | 53 | NCT04599140 |
| Phase 1 | JNJ-74699157 | KRAS G12 C | KRAS mutated advanced solid tumor | 10 | NCT0400630 |
| Phase 1 | mRNA-5671/V941 +/−pembrolizumab | KRAS vaccine | KRAS mutant CRC, NSCLC and PDAC | 100 | NCT03948763 |
| Phase 1 | D-1553 | KRAS G12C inhibitor | KRAS mutated CRC and NSCLC | 200 | NCT04585035 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, S.; Garrel, S.; Gerber, M.; Maitra, R.; Goel, S. Therapeutic Targets of KRAS in Colorectal Cancer. Cancers 2021, 13, 6233. https://doi.org/10.3390/cancers13246233
Rahman S, Garrel S, Gerber M, Maitra R, Goel S. Therapeutic Targets of KRAS in Colorectal Cancer. Cancers. 2021; 13(24):6233. https://doi.org/10.3390/cancers13246233
Chicago/Turabian StyleRahman, Shafia, Shimon Garrel, Michael Gerber, Radhashree Maitra, and Sanjay Goel. 2021. "Therapeutic Targets of KRAS in Colorectal Cancer" Cancers 13, no. 24: 6233. https://doi.org/10.3390/cancers13246233