
Targeting Oncogenic Gαq/11 in Uveal Melanoma

Department of Biochemistry and Molecular Biology, Sidney Kimmel Medical College, Thomas Jefferson University, Philadelphia, PA 19107, USA
*
Author to whom correspondence should be addressed.
Cancers 2021, 13(24), 6195; https://doi.org/10.3390/cancers13246195
Submission received: 7 November 2021
/
Revised: 2 December 2021
/
Accepted: 6 December 2021
/
Published: 9 December 2021
(This article belongs to the Special Issue Metastatic Uveal Melanoma)
{kind=link}
{kind=link}
Abstract
:Simple Summary
Uveal melanoma is a deadly form of eye cancer with a high rate of metastasis. Once metastasis occurs, patients are often left with a short survival time, since there is no FDA standard of care for the metastatic disease. Uveal melanoma develops from mutations mainly in proteins involved in the Gq/11 signaling pathway, which drives pathogenesis. This review article aims to summarize pre-clinical and clinical studies that have attempted to understand and treat the disease by inhibiting the Gq/11 signaling pathway. We discuss the limited success of treatments focused on downstream targets of the Gq/11 pathway and evaluate the effectiveness and feasibility of treating the disease by directly inhibiting Gq/11.
Abstract
Uveal melanoma is the most common intraocular cancer in adults and arises from the transformation of melanocytes in the uveal tract. While treatment of the primary tumor is often effective, 36–50% of patients develop metastatic disease primarily to the liver. While various strategies have been used to treat the metastatic disease, there remain no effective treatments that improve survival. Significant insight has been gained into the pathways that are altered in uveal melanoma, with mutually exclusive activating mutations in the GNAQ and GNA11 genes being found in over 90% of patients. These genes encode the alpha subunits of the hetetrotrimeric G proteins, Gq and G11, and mutations result in activation of several important signaling pathways, including phospholipase C and activation of the transcription factor YAP. In this review, we discuss current efforts to target various signaling pathways in the treatment of uveal melanoma including recent efforts to target Gq and G11 in mouse models. While selective targeting of Gq and G11 provides a potential therapeutic strategy to treat uveal melanoma, it is evident that improved inhibitors and methods of delivery are needed.
1. Introduction
Uveal melanoma (UM) is the most common intraocular tumor in adults, and, as the second most common form of melanoma, accounts for approximately 5% of all melanomas. The median age at diagnosis is about 62 years, and risk factors include fair skin, light eye color (green or blue), ocular melanocytosis, dysplastic nevus syndrome, germline BRCA1-associated protein 1 (BAP1) mutations, and welding as an occupation [1,2,3,4,5]. UM arises from melanocytes contained in the choroid, ciliary body, and iris (together known as the uvea) of the ocular cavity, and has a high propensity to metastasize [6,7]. As a result, 36–50% of patients with UM develop metastasis, predominantly to the liver, even when the primary tumor has successfully been eliminated [3,6,8,9,10,11]. While the primary tumor can be successfully treated via enucleation, laser therapy, radiotherapy, or surgical resection, many experts believe that the metastatic spread of UM is unpreventable, as it has likely already taken place by the time the primary tumor is detected [8]. For instance, only 4% of patients show detectable metastasis at the time of diagnosis of the primary tumor, but up to half of all UM patients will develop metastatic disease. This suggests UM develops small, undetectable metastases at an early stage of tumorigenesis, as early as 5 years prior to primary tumor detection, that remain dormant or quiescent for years, classifying UM as a systemic disease requiring some form of systemic therapy [12,13,14]. Once macro-metastasis develops, patients have a poor prognosis with a median overall survival time of less than 1 year as there are currently no effective therapies with a significant impact on survival [6,15,16,17,18,19].
To date, there is no FDA standard of care for metastatic UM. Systemic treatments that have been successful in treating cutaneous melanoma (CM), such as chemotherapy and immunotherapy, have largely failed to produce similar positive results in UM patients. UM response rates to chemotherapy drugs such as dacarbazine, temozolomide, cisplatin, treosulfan, and fotemustine range from 0% to 15%, and no agent has successfully prolonged survival [20,21,22,23,24]. Moreover, immune checkpoint inhibitors of cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and programmed cell death-1 (PD-1), which significantly improve survival outcomes for patients with advanced CM, have shown no clinical benefit in patients with metastatic UM [25,26,27,28]. Additionally, while BRAF inhibitors show efficacy in treating CM patients, UM remains unresponsive to similar treatments, despite both malignancies originating from melanocytes derived from the neural-crest [7,29]. These differences may be explained by the observation that UM has a distinctly different etiology from CM. The main oncogenic BRAF and NRAS driver mutations in CM are not typically found in UM [30,31,32,33,34], and while 80% of CM show a UV radiation mutational signature, there is no observed evidence of the same in UM [26,35]. Furthermore, UM has an incredibly low mutational burden compared to CM and most other cancer types [36,37]. With an improved understanding of the genetic differences between cutaneous melanoma and uveal melanoma, there is a clear need for more specific or targeted treatment strategies for UM, as advances in CM treatment will be unlikely to confer the same results in UM.
2. Driver Mutations Involved in UM
UM predominantly involves mutually exclusive activating mutations in the GNAQ or GNA11 genes that encode the highly conserved Gαq and Gα11 subunits of the heterotrimeric G-proteins [31,38,39]. The introduction of the oncogenic Gαq/11 mutants into human or mouse melanocytes results in anchorage-independent growth and gives rise to heavily pigmented tumors in mice [39,40,41]. Interestingly, while about 93% of cases contain oncogenic Gαq or Gα11, cases without such mutations often harbor activating mutations in other genes linked to the Gq pathway. Activating mutations in PLCB4, which encodes for the canonical Gq/11 effector phospholipase C β (PLCβ), or Cysteinyl Leukotriene Receptor 2 (CYSLTR2), a G protein-coupled receptor (GPCR) activator of Gq, are observed in 2.5% and 4% of cases, respectively [26,36,42,43]. Taken together, it has become clear that aberrant signaling via the Gq pathway is the main driver of uveal melanoma.
3. Gαq/11 Proteins and Their Mutations in UM
Gαq and Gα11 proteins are members of the Gq subfamily of Gα proteins, are 90% homologous at the amino acid level, and are ubiquitously expressed. To maintain homeostasis in mammalian cells, the activation and deactivation of G-proteins is regulated by tight control over GDP/GTP exchange and GTP hydrolysis rates. Ligand-activated GPCRs, such as CysLTR2, act as guanine nucleotide exchange factors (GEFs) by stimulating the exchange of GDP for GTP on the Gα subunit of the heterotrimeric G-protein. Upon binding to GTP, the Gα subunit changes conformation, dissociates from the Gβγ dimer, and interacts with downstream effectors, such as PLCβ and ARF6 [44,45,46,47,48,49]. Gα proteins contain a Ras-like GTPase domain, wherein important amino acid residues provide the subunit with intrinsic GTP hydrolytic activity, which is accelerated by the interaction of GTPase-activating proteins (GAPs), such as regulators of G protein signaling (RGS). GTP hydrolysis to GDP then terminates the Gα signaling, allowing the subunit to reassociate with Gβγ and return to an inactive state [44,50,51].
The Gαq/11 mutations observed in uveal melanoma primarily occur at the Q209 and R183 residues [31,38,39]. Both mutational hotspots are in the GTPase domain and are critical for stabilizing the transition state for GTP hydrolysis. When mutated, the protein loses its GTPase activity and is rendered constitutively active in the GTP-bound state, leading to aberrant proliferative signaling [38,39,47,52,53,54]. Q209 mutations (Gαq/11Q209L/P) are 13 times more frequent than R183 mutations (Gαq/11R183C) in UM. While Q209 mutations effectively impede GTPase activity, R183 mutants still retain the ability to hydrolyze GTP, although at a reduced catalytic rate, through interactions with RGS proteins, and still respond to receptor stimulation [39,50,55]. Since Gαq/11Q209 mutants appear incapable of hydrolyzing GTP even in the presence of RGS proteins, aberrant signaling is increased, leading to a more severe phenotype [52].
4. Inhibition of Signaling Pathways Downstream of Activated Gαq/11
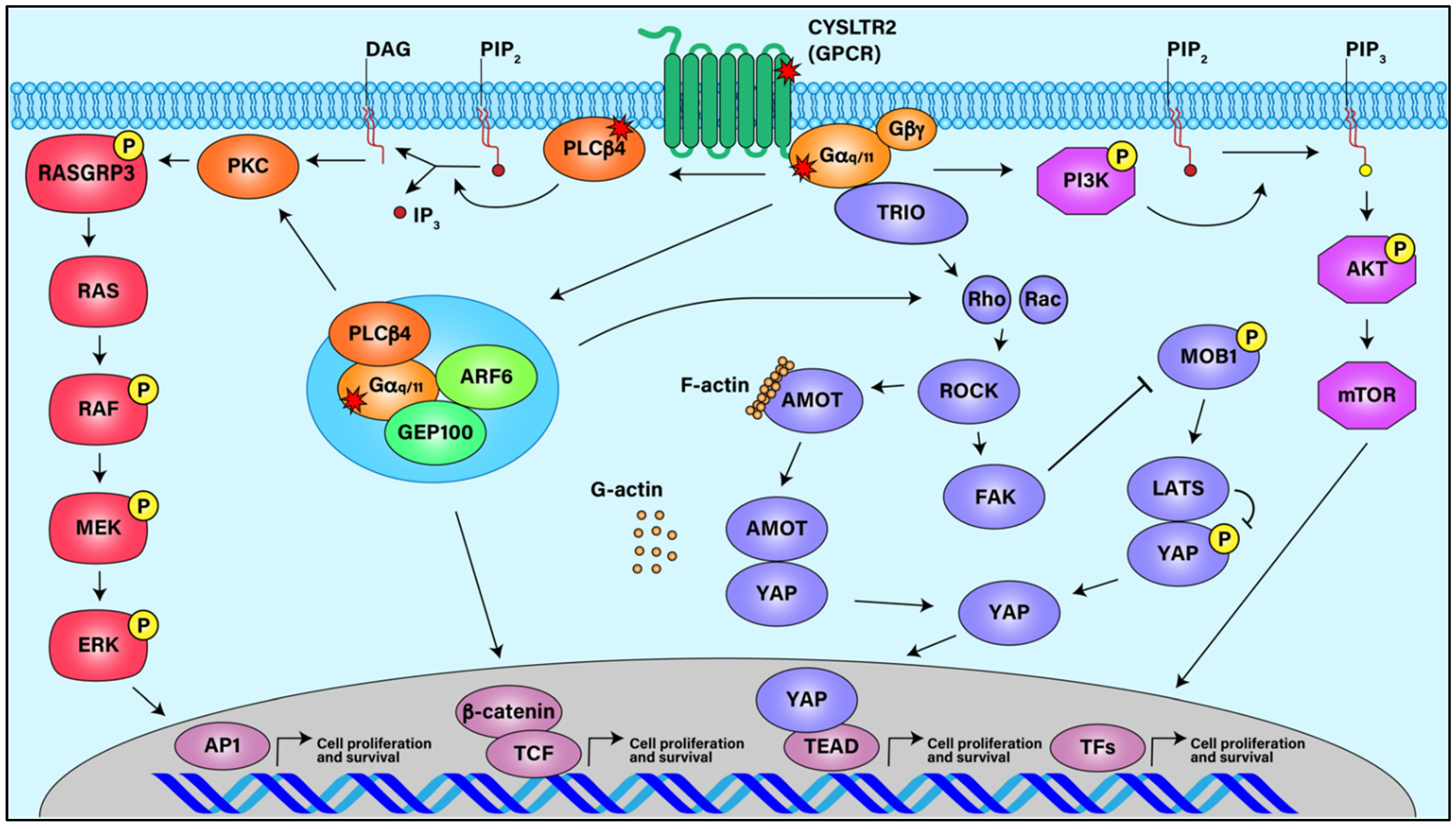
Due to the redundancy of their signaling pathways, oncogenic mutations in either Gαq or Gα11 appear to cause similar cellular oncogenic properties leading to the pathogenesis of UM (Figure 1). For example, GNAQ and GNA11 mutations are responsible for the upregulation of mitogen-activated protein kinase (MAPK) pathway, RAF/MEK/ERK, in the absence of BRAF mutations in UM [30,56,57,58]. Knockdown of Gαq in cell lines derived from primary or metastatic UM results in decreased MAPK signaling [38]. Additional Gq/11 stimulated signaling pathways that promote tumorigenesis have been implicated in UM. Oncogenic Gαq/11 leads to aberrant Akt signaling through PI3K/AKT/MTOR and increased activation of small GTPases RhoA and Rac1, which promote cell growth through JNK, p38, and yes-associated protein (YAP)-directed transcription of growth promoting genes [40,41,59,60,61]. To date, most of the efforts to treat UM have mainly been focused on inhibiting the downstream signaling pathways of oncogenic Gαq/11 and have provided limited success in improving progression-free survival of UM patients.
4.1. ARF6 Inhibition
ADP-ribosylation factor 6 (ARF6) is a small G-protein that has been shown to be an immediate downstream effector of an activated GNAQ/GEP100 complex. ARF6 acts as a proximal node of Gαq signaling to induce all the downstream pathways as well as β-catenin signaling. ARF6 mediates the trafficking of Gαq from the cell membrane to cytoplasmic vesicles and β-catenin to the nucleus. Inhibition of ARF6, by the ARF6 specific small-molecule inhibitor, NAV-2729, reduces uveal melanoma cell proliferation and tumorigenesis in a mouse model [48]. Therefore, ARF6 is considered a potential therapeutic target downstream of oncogenic Gαq.
4.2. PKC Inhibition
Activated Gαq canonically triggers the protein kinase C (PKC) pathway through diacylglycerol produced by PLCβ activation [44]. Attempts to inhibit PKC in UM have shown promise. The pan-PKC inhibitor, AEB071 (sotrastaurin), at low micromolar concentrations, significantly inhibits the growth of UM cells harboring GNAQ mutations, through G1 arrest and apoptosis, and had little effect on UM cells carrying wild-type GNAQ [62]. A phase one study of AEB071 resulted in one achieved partial response, while 47% of 118 participants achieved disease stabilization and a median progression-free survival of 15.4 weeks although with some adverse effects [63]. This suggests there is some clinical benefit of PKC inhibition and a new phase one study using the PKC inhibitor LXS196 is currently underway (NCT02601378).
While PKC inhibitors inhibit MAPK signaling and induce G1 arrest in UM cells, they fail to induce tumor regression in xenograft models. As a result, combination treatments of PKC and MEK inhibitors have been attempted and have shown synergistic effects on tumor regression in a UM in vivo model [64]. However, a phase one-b/two study of a PKC and MEK combination treatment of patients with metastatic UM was halted before the beginning of phase two (NCT01801358).
4.3. MAPK Pathway Inhibition
The drugs that have been used most often in an attempt to inhibit downstream Gq/11 signaling in UM are MEK inhibitors, because, similar to CM, MAPK signaling levels are elevated in most UM [57]. Early studies showed MEK inhibitors, such as selumetinib or TAK-733, successfully inhibit UM cell proliferation and viability in vitro [65]. These studies resulted in several clinical trials with various MEK inhibitors, the first of which was a comparison of selumetinib versus chemotherapy treatment, which showed improved progression-free survival (15.9 vs. 7 weeks) but did not significantly improve overall survival (9.1 vs. 11.8 months), while treatment-related adverse events were observed in 97% of patients treated with selumetinib [66]. There is currently recruitment for a new phase one trial to test the effect of intermittent selumetinib administration. The purpose of this study is to test higher drug doses, to more completely block the MAPK pathway and prevent the development of drug resistance mechanisms within the tumor. Intermittent administration of the drug may also reduce side effects (NCT02768766). A phase one study using TAK-733 also showed limited antitumor activity in patients with advanced solid tumors [67]. A recent review analyzed 590 case records from six eligible clinical trials, including the study mentioned above [66]; a phase three study of selumetinib in combination with dacarbazine [68]; phase two studies of selumetinib monotherapy versus temozolomide [69], as well as trametinib, with or without GSK2141795 [70]; a phase one study of trametinib [71]; and a terminated phase one study of AEB071 and MEK162 (NCT01801358). The conclusion is that UM is poorly responsive to MEK inhibitors, regardless of the inhibiting agent and combination partner [72]. The failure of MEK inhibitors to provide meaningful effects on overall survival and tumor growth may be explained by resistance mechanisms that are enabled by the growth factors present in the liver tumor microenvironment [73,74]. However, these results have not deterred investigators from continuing to search for effective combinations with selumetinib. A preclinical study of selumetinib in combination with the ERK inhibitor AZ6197, or the mTORC1/2 inhibitor AZD2014, displayed significant efficacy in two PDX mouse models of UM [75]. Another recent study suggests chloroquine or hydroxychloroquine sensitizes GNAQ/11-mutant UM to MEK inhibition by trametinib, through the inhibition of autophagy or lysosome function. This combination treatment synergistically inhibited tumor growth in metastatic mouse models [76].
While MAPK signaling is significantly upregulated in UM, and is often the target of potential therapeutic interventions, the mechanism of how oncogenic Gαq/11 activates the MAPK pathway is incompletely understood. The Ras guanyl releasing protein (RASGRP3) is highly expressed in Gαq/11-driven tumors, acts as a critical node for ERK activation, and is activated via the PKC δ- and ε-isomers downstream of activated Gαq/11 in UM [77,78]. RASGRP3 is required for growth and Ras-MAPK activation in UM cells, as knockdown of the gene transcript leads to decreased MAPK signaling and reduced growth in vitro and in vivo [77,78]. Therefore, RASGRP3 is considered a potential therapeutic target downstream of Gαq/11 and represents an additional avenue to MAPK inhibition specific to UM.
4.4. PI3K/AKT/MTOR Inhibition
Another activated pathway downstream of oncogenic Gαq/11 that has been therapeutically targeted is the PI3K/AKT/MTOR pathway. This pathway is often targeted in combination with MEK or PKC inhibition. One study showed that neither MEK nor PI3K inhibition by trametinib or GSK2126458 alone was sufficient to induce apoptosis in the majority of UM cell lines, but the combination of MEK and PI3K inhibitor treatment resulted in a significant induction of apoptosis in a GNAQ/11 mutant-dependent manner [79]. Another study co-targeted PI3K and PKC inhibition with BYL719 and AEB071, which showed synergistic inhibition of cell proliferation and apoptotic cell death in UM cells, as well as a significant reduction in tumor growth, in a xenograft model [80]. As a result, a phase one clinical trial of the efficacy and safety of BYL719 and AEB071 drug combination in metastatic UM has begun, but its status is unknown (NCT02273219). Others have targeted AKT in combination with MEK inhibition, which showed induced activation of AMP-activated protein kinase, resulted in the synergistic induction of autophagic cell death in UM cells, and inhibited UM tumor growth in xenograft mouse models [81]. This evidence led to a phase two clinical trial, mentioned previously [70], of trametinib with or without the AKT inhibitor GSK795, which failed to improve progression-free survival compared to selumetinib treatment alone. Additionally, MTOR has also been targeted in UM studies. A phase two clinical trial tested the combination of the MTOR inhibitor everolimus, and the somatostatin receptor agonist pasireotide, in metastatic UM patients. The combination showed little benefit and significant side-effects [82]. A preclinical study found some success in the combination treatment of the MTOR inhibitor everolimus and the PI3K inhibitor GDC0941, which provided increased apoptosis in UM tumors in two PDX models [83]. Another study showed synergistic effects on tumor regression in UM PDX models with a combination of the mTORC1 inhibitor RAD001 and the PKC inhibitor AEB071 [84].
4.5. YAP Inhibition
The upregulation of yes-associated protein (YAP) driven by oncogenic Gαq/11 in UM is a recent observation [40,41,85]. YAP is a co-transcriptional regulator involved in the cell-growth-regulating Hippo pathway, which, when dephosphorylated, translocates from the cytoplasm into the nucleus, where it associates with TEA domain transcription factor (TEAD) to promote the transcription of growth promoting genes [86,87,88,89]. Studies have shown that knockdown of Gαq/11 inactivates YAP, while YAP is required for mutant Gαq/11 driven tumorigenesis [40,41]. Further studies indicated that YAP dephosphorylation in UM occurs through the Gαq/11 activation of Trio, a guanine nucleotide exchange factor for the small G-proteins, RhoA and Rac1, independent of the Hippo pathway [41]. Additionally, the YAP inhibitor, verteporfin, can inhibit oncogenic Gαq/11 UM cell growth in mice [40,41,90]. Although these original studies suggest YAP as a potential therapeutic target of UM, there has since been conflicting studies that suggest YAP may not be as crucial to UM tumorigenesis as originally thought. One study investigating the association between YAP activity and clinical outcome concluded that the effect of YAP on the development, growth, and invasion of UM in patients is less than previously found in experimental studies [91]. Another study investigated the susceptibility of melanoma cell lines to YAP inhibition by verteporfin and found that while most UM cell lines responded in vitro to verteporfin, high risk metastatic UM lines (BAP1-negative) did not. Therefore, the mutational background is an important predictor of response to YAP inhibition by verteporfin, suggesting that not all UM cell lines are susceptible to YAP inhibition [92].
4.6. FAK Inhibition
Related to the Hippo pathway and YAP signaling, a recent study showed that Gαq activates focal adhesion kinase (FAK), and that FAK activity is essential for YAP activation and UM cell growth. Using an integrated bioinformatics pipeline, FAK was identified as a candidate synthetic lethal gene with GNAQ activation. FAK inhibition, by small molecules VS-4718 or PF562771, suppresses YAP activation in vivo and prevents UM cell growth [93]. Unfortunately, MAPK signaling via oncogenic Gαq provides resistance to FAK inhibition in UM cells. Therefore, a study wherein UM cells were treated with a combination of the MEK inhibitor trametinib and the FAK inhibitor VS-4718 showed a synergistic effect on metastatic UM tumor growth in a mouse model [94]. This study led to a phase two clinical trial, which is currently recruiting metastatic UM patients, testing the efficacy of the combination treatment of the RAF/MEK inhibitor VS-6766 and the FAK inhibitor defactinib (VS-6063) (NCT04720417). Another recent study suggests that PLCβ/PKC activity, but not FAK/YAP, is elevated in UM cell lines as a consequence of Gαq pathway mutations, and that FAK may not be activated independent of PLCβ activation, as previously suggested [95]. This study further illustrates that PKC/MAPK signaling is essential for UM cell proliferation, and that only combined inhibition of PKC and MEK, not FAK and MEK, or FAK and PKC, synergistically reduces cell viability in UM cells [95]. However, this was only shown in a few UM cell lines, and may not be true in every instance of UM, as there is evidence to the contrary, as previously discussed. However, this study, along with the observation that genetic analyses of UM have failed to identify mutations in the FAK or YAP pathways, suggest that FAK or YAP may not be optimal targets for therapeutic intervention in UM [26,96].
5. Direct Targeting of Gαq/11
Taken altogether, a potential explanation for these unsuccessful therapies is that oncogenic Gαq/11 activates multiple, individually dispensable downstream signaling networks. Inhibition of one, or sometimes two, pathways does not achieve the desired treatment outcome. This raises the possibility that direct inhibition of oncogenic Gαq/11 may be an advantageous and promising therapeutic strategy for UM treatment. While there are no current FDA-approved drugs that directly target Gαq/11, a few compounds, namely YM-254890 (YM) and FR900359 (FR) (Figure 2), that effectively inhibit Gαq/11, have been identified and have shown some promising results in pre-clinical UM studies [97,98].
5.1. Short History of Gq Inhibitors
YM is a cyclic depsipeptide first isolated from Chromobacterium sp. QS3666 in 2003 during a screen for platelet aggregation inhibitors of human plasma [99]. After its isolation, YM was found to reduce the mobilization of intracellular Ca2+ by selective inhibition of the Gq, G11, and G14 proteins, without affecting the signaling of other G-proteins [55]. Gq inhibition by YM was then used as a pharmacological tool to probe Gq signaling in cells and mice. Originally it was determined that YM inhibits ADP-induced platelet aggregation (a Gq-mediated biological response) in vitro, followed by studies that showed that it prevents the formation of thrombosis and neointima in rats and monkeys, and demonstrates antithrombotic and thrombolytic activity [99,100,101,102].
Unfortunately, shortly after its discovery, YM became unavailable to researchers, and in 2012 a worldwide competition for the total synthesis of 1 mg of YM was announced. Due to its complex macrocyclic structure, the total synthesis of YM is not a simple task, but in 2016 the first total synthesis of YM and FR was achieved, albeit with a very low yield [103]. Shortly thereafter, YM became commercially available.
FR, also known as UBO-QIC, is structurally very similar to YM and only differs by the addition of three methyl groups. It was first isolated from the plant Aridisia crenata sims in 1988 during an investigation into possible platelet aggregation inhibitors [104]. It has since been shown that FR is synthesized by Candidatus Burkholderia crenata, a bacterial symbiont of Ardisia crenata Sims [105]. FR is a potent and selective inhibitor of Gαq/11/14, has a vasorelaxant effect in rats, and effectively inhibits agonist-promoted airway contraction in human precision-cut lung slices and rats [106,107,108,109]. FR has also been suggested to inhibit Gβγ signaling following the activation of Gi-coupled receptors [110], although this effect may be due to the involvement of Gαq in Gβγ-mediated activation of PLCβ [111].
FR was commercially available for a short period of time from the König group at the University of Bonn, who isolated it from the dried leaves of A. crenata Sims, but the compound is no longer sold. The König group has managed to biosynthesize FR in cultivable E. coli by expression of the FR nonribosomal peptide synthetase (frs) genes, which are found in Candidatus Burkholderia crenata and are responsible for the production of FR in the endosymbiont [112]. The Wollbrett group sought to improve upon the low yield of FR by this method via genetic engineering of Chromobacterium vaccinii DSM 25,150, with some success [113]. However, chemical synthesis and biosynthesis methods result in very low yields, leaving purification of the compound from natural sources as the main method of production, as is the case for YM.
5.2. Mechanism of Action and Physiochemical Properties of YM and FR
In 2010, the structural basis for inhibition was revealed via an X-ray co-crystal structure of YM bound to a chimeric Gαi/qβγ protein complex, showing that YM acts as a guanosine nucleotide dissociation inhibitor (GDI) [114]. The structure revealed that YM binds to a hydrophobic pocket between linker one and switch I of the Gαq subunit, with minimal contact with Gβγ. The region in which linker one and switch I reside is between the helical and GTPase domains and undergoes large conformational changes between the inactive GDP-bound and active GTP-bound conformations. Upon binding of YM, the GDP-bound state of Gαq is stabilized by suppression of the hinge motion of linker one and switch I, resulting in the inhibition of the GDP/GTP exchange, leaving the Gαq protein “locked” in the GDP-bound, inactive conformation [114].
In an effort to characterize FR, a comprehensive 2015 study used molecular dynamics simulations, and a combination of purified proteins and cell-based assays, to show that FR functions similarly to YM as a GDI of Gαq/11/14 proteins [107]. This same study determined that FR inhibits Gαq activation in a pseudo-irreversible manner, and that its effects are exceptionally resistant to washout in vasorelaxation and cell-based experiments. Mutagenesis studies established that FR and YM bind to the same hydrophobic pocket of Gαq, and that genetically engineered FR-binding sites can be used to create additional Gα proteins, susceptible to FR inhibition, as an investigatory tool [115].
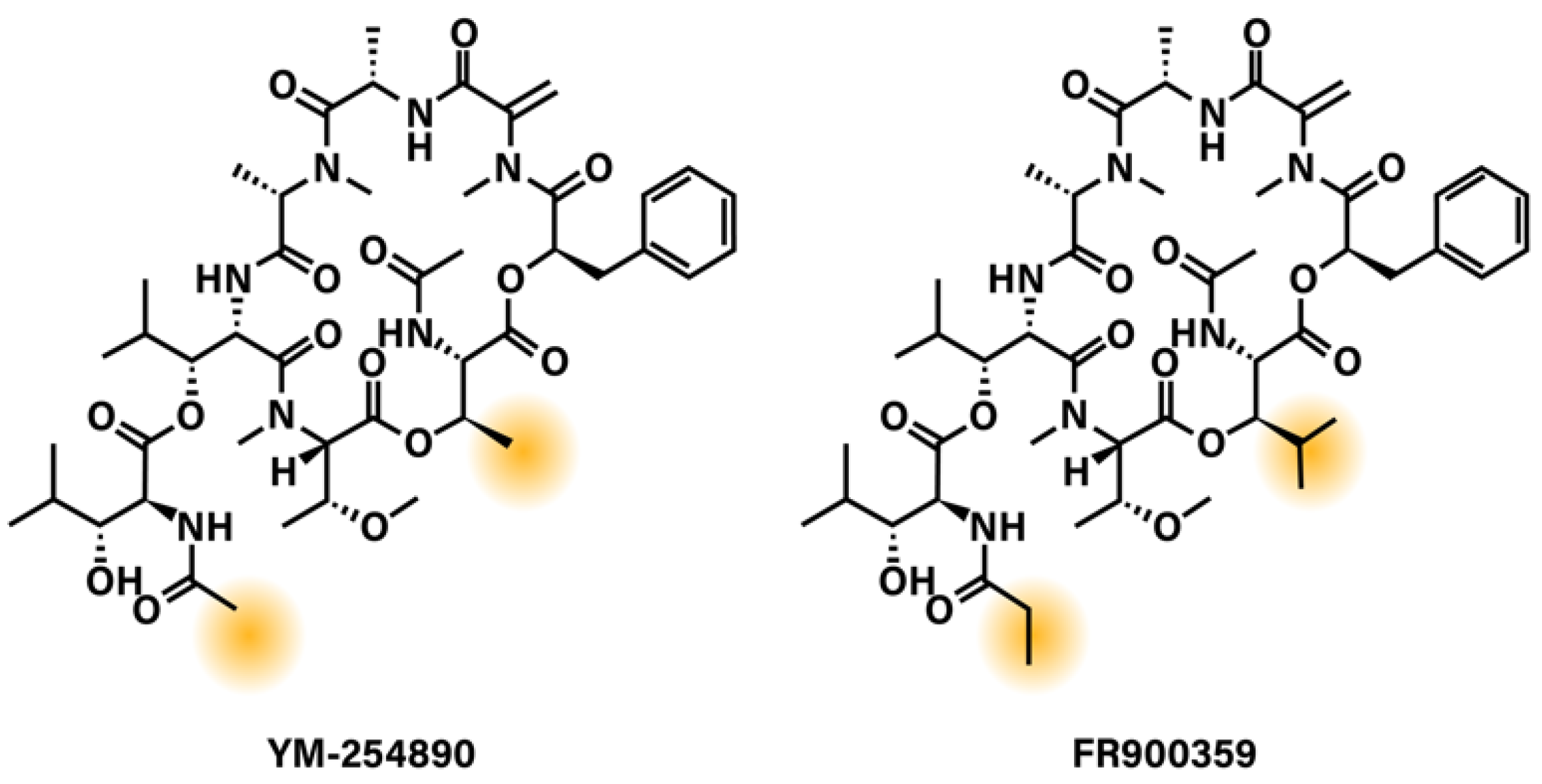
While YM and FR are very similar in their structure and mechanism of action, they do show some biologically distinct activities. FR inhibits Gαq/11 with a 3-fold higher potency than YM, and disruption of FR binding to Gαq is more difficult to achieve than the binding of YM [103,115]. This difference in potency may be explained by physiochemical, kinetic, and molecular characteristics that the two very similar compounds do not share. As explained previously, FR differs from YM by the addition of three methyl groups. These differences make FR slightly larger and more lipophilic than YM. A conformational study showed that while FR exists as a single conformer in aqueous solution, YM exists as two different conformers in a major–minor (3:1) ratio. The major form is the biologically active conformer, while the minor represents a conformer that is unable to inhibit Gαq [116]. This suggests that conformational stability is important to the inhibitory potency of these compounds. Additionally, while both compounds display similar association rates (t1/2(FR) = 3.6 min versus t1/2(YM) = 8.8 min), FR binds pseudo-irreversibly and displays a residence time of 92.1 min, compared to 3.8 min for YM [117]. Molecular docking studies seeking to explain this difference in dissociation rates suggest the additional lipophilic moieties of FR anchor the compound in the binding pocket, similar to “a dowel forming a latch”, while YM lacks those anchor points and may be more readily released from the Gq protein [117]. This suggests that even small changes, which seem insignificant within such a large and complex molecule, can lead to differences in potency and potential pharmacological effects.
5.3. YM and FR Activity in Uveal Melanoma
Since the discovery of YM and FR, as potent Gq/11 inhibitors, it was believed that the two compounds could inhibit only wild-type and R183 mutant proteins, but not Q209 mutants, which are almost always the driver mutations in UM. For example, the inhibitory effect of YM on Gαq Q209L and R183C mutants was first tested in HEK293 cells, co-transfected with a serum response element (SRE)-luciferase reporter gene, and either Gαq Q209L or R183C mutant cDNA. This study showed the constitutive activity of the R183C mutant to be completely suppressed by YM, while that of the Q209L mutant was only modestly affected [55]. A comprehensive characterization of FR as a Gαq/11/14 selective inhibitor found that it was capable of inhibiting the Q209 mutant Gαq/11 [107]. In this study, FR inhibited the growth of a mouse melanoma cell line, Hcmel12, which carries a Gαq-Q209L mutation, and is wild type for B-Raf and N-Ras. FR was also found to effectively inhibit nucleotide binding to purified Gαq-Q209L protein in vitro, suggesting FR directly binds to the mutant Gαq/11 [118]. FR also inhibited the growth and proliferative signaling of UM cells containing oncogenic Gαq/11 proteins [118]. Additional studies investigating similar effects of FR emerged around the same time [93,119,120,121] while research involving YM treatment of UM have followed [95,122].
YM and FR show significant inhibitory activity against every mutant Gαq/11 variant found in UM in transfected HEK293 cell-based experiments [121,122]. Both compounds selectively inhibit cell growth and promote growth arrest of UM cells harboring oncogenic Gαq/11, as well as oncogenic cysteinyl leukotriene receptor 2 (CysLT2RL129Q) [93,95,118,119,120,121,122]. FR, and likely YM, target both primary and metastatic cells from UM tumors [121]. YM and FR selectively inhibit the essential oncogenic signaling of Gαq/11 mutant UM cells. Treatment of UM cells with these Gq/11 inhibitors abolishes MAPK signaling, suppresses Akt activation, prevents YAP localization to the nucleus, and represses FAK activation [93,95,118,119,120,121,122]. Class one UM cells (wild-type for BAP1) begin to express melan-A at the plasma membrane and gain pigmentation when treated with low nanomolar concentrations of FR [118,119,121]. This suggests FR can induce a redifferentiation of these cancer cells, possibly by decreasing Gq/11 activity to a level closer to that of a normal melanocyte. It is believed that this FR-mediated redifferentiation is partly the result of restored function to the polycomb repressive complex 2, which plays a role in the differentiation of embryonic stem cells [119,121]. Concentrations of YM and FR at 100 nM and above selectively induce apoptosis and cell death of UM cells, as evidenced by increased caspase-3 and PARP cleavage, as well as an increased population of cells in the sub-G1 phase [95,118,119]. These Gq/11 inhibitors have shown great promise as potential therapeutics in cell-based assays, but are these results recapitulated in vivo?
To address this, there are some recent studies wherein UM mouse models were treated with YM and FR. The first of which showed that FR (0.5 mg/kg) treatment of severe combined immunodeficient (SCID) mice with subcutaneously grafted GαqQ209P UM cells resulted in 72% tumor growth inhibition after 14 days, whereas the growth of xenografts established from B-Raf mutant melanoma cells was unaffected [120]. A second recent study using subcutaneous UM tumor xenografts of Gα11Q209L and GαqQ209L UM cells, established in NOD-scid-gamma (NSG) mice, showed that FR inhibited the growth of UM tumors at 0.1 and 0.3 mg/kg, but had no effect on the growth of B-Raf mutant melanoma tumors [121]. However, UM tumors resumed growth 16 days after FR treatment was stopped, suggesting that FR only causes reversible, rather than durable, arrest or regression of UM tumors. ERK phosphorylation in xenograft tumors was only reduced by 30% compared to at least 85% observed in vitro, suggesting FR treatment does not completely suppress aberrant MAPK signaling in vivo. However, FR treatment does provide a therapeutic window in which tumors cells are targeted, while healthy cells maintain the correct physiological Gq/11 signaling. It is possible the observed incomplete inhibition of MAPK signaling is the result of a resistance or compensatory mechanism at work in these cells, as reverse-phase protein analysis showed an increase of HER3 and activated STAT3 upon FR treatment of UM cells in vitro [118]. Additionally, a recent study showed that YM, similar to FR, inhibited tumor formation in mice with xenografts of GαqQ209P UM cells [122]. Notably, it was observed during this study that YM does not durably suppress MAPK signaling of UM cells in vitro, and that MAPK signature genes begin to rebound after 24 h. This was similarly observed in tumors treated with YM ex vivo. However, a combination strategy of YM plus an MEK inhibitor was able to prevent reactivation and durably suppress MAPK in UM xenograft mice, while the combination also worked synergistically to inhibit tumor growth and promote tumor regression. Taken together, the current in vivo studies suggest Gq/11 inhibition as a viable therapeutic strategy to treat UM; however, to achieve durable arrest and regression of UM tumors, a combinatorial approach may be warranted.
5.4. Feasibility of Gq/11 Inhibitors as Potential Therapeutics
A recent study that sought to determine if YM and FR are fit for translation, and provides a solid basis for considering future in vivo investigations utilizing YM and FR [123]. It was determined that both YM and FR have high chemical stability under physiological conditions, and both are high clearance compounds in human liver microsomes. However, YM is more stable than FR in human liver microsomes, with short half-lives of 27.3 min and 8.1 min, respectively. Due to the high clearance rate of these compounds, they may not be suitable for systemic application. Importantly, in vivo studies showed that both compounds are only marginally able to cross the blood-brain barrier. However, local administration of these compounds will be preferred, since systemic application can be expected to block Gq/11 signaling throughout the body. In this regard, the Blumer group determined a LD50 of approximately 0.6 mg/kg FR in NSG mice and showed that FR delivered at tolerable doses did not significantly affect heart rate, liver function, or hematopoiesis in xenograft UM models [121].
6. Discussion
Uveal melanomas are divided into class one (low metastatic risk) and class two (high metastatic risk) based on gene expression profiling [124]. While almost all UM patients have aberrations in the Gq/11 pathway, particularly in the Gαq/11 proteins, these mutations occur early in tumorigenesis and are not correlated with molecular class or metastasis [31,125]. Class two tumors are strongly associated with an additional BAP1 mutation or deletion that occurs after Gαq/11 mutation [5]. This additional mutation increases the metastatic potential of UM, but aberrant Gq/11 signaling remains as the main driver of tumorigenesis. Recently, the Blumer group has shown that both class one and class two tumor growth is inhibited by Gq/11 inhibition by FR in UM xenografts [121]. This further strengthens the idea that targeting oncogenic Gαq/11 can be a viable therapeutic strategy in treating all subtypes of UM. Targeting oncogenic Gαq/11 has the additional benefit, over current monotherapy strategies, of inhibiting the multiple signaling networks downstream of Gq/11.
While Gq/11 inhibitors show promise as future therapeutic options, their potential use presents a risk of adverse effects, as they act on both mutated and wild-type Gαq/11 proteins, which are ubiquitously expressed and have important physiological functions [126,127,128,129]. Clearly, compounds that can selectively target mutationally activated Gαq/11 over wild-type Gαq/11 would be ideal as therapeutic agents, but these do not currently exist. However, a growing body of work investigating the important moieties of YM and FR at the molecular level and how these compounds interact with Gαq/11, provides valuable insight into the possible synthesis of such a compound. In an effort to synthesize YM, a few simplified YM analogs were synthesized based on the motifs of YM that are thought to be important in maintaining the compound’s stability and making contact with the Gαq protein [130,131]. Its successful synthesis, in 2016, led to the production of many additional analogs used to probe the structure–activity relationship of YM, by modification of key structural elements of the compound [103,116,132,133]. However, all the analogs are less potent than YM, and it has become clear that even small structural changes result in reduced affinity for Gαq. Many simplified analogs of FR have also been reported, and while a few selectively inhibit Gq/11 at a comparable potency to FR, most have failed to inhibit Gq/11, and none have surpassed the biological activity of FR [116,134,135].
At present, the chemical and synthetic approaches to synthesizing YM and FR have low yields and very complex procedures. Purification of the compounds is the only way to obtain substantial quantities of YM and FR but is time consuming. However, having a method of chemical synthesis provides an avenue to rationally modify these compounds, determine crucial moieties for Gq/11 inhibition, and possibly design compounds that may be able to more specifically target the oncogenic mutant Gαq/11. Some have taken to using biosynthesis methods of creating new YM/FR analogs [136], while others are utilizing feature-based molecular networking techniques to identify new FR analogs produced by other bacterial strains [135,137]. A recent study using tritium-labelled YM and FR compounds in a high throughput competition binding assay discovered novel Gq inhibitors, which inhibited Gq signaling in recombinant cells and primary murine brown adipocytes, resulting in enhanced differentiation, albeit with significantly less potency than FR [117]. Moreover, the group that first synthesized YM and FR has recently reported a new Gq/11 inhibitor, GQ127, to be a potent, stable, and safe small molecule. It exhibits good Gq/11 protein inhibition and antitumor potency in vitro, and significantly inhibits in vivo tumor growth of UM cells [138]. While GQ127 requires significantly higher concentrations compared to YM and FR to observe similar effects on UM cell signaling and tumor growth, it is easily synthesized, displays high oral bioavailability, and shows no obvious side effects in mice. Therefore, it may be an additional structure to build upon for future UM drugs that target Gq/11.
7. Conclusions
Uveal melanoma is a rare melanoma that is biologically distinct from cutaneous melanoma and cannot be treated in a similar manner in the clinic. Despite a high level of success in treating primary UM locally, 36–50% of patients will eventually develop metastasis. To this point, the metastatic disease remains uncurable, and outcomes for patients with UM metastasis are poor [17]. Single agents that target signaling pathways downstream of mutated Gαq/11 are not effective, and there is a desperate need for better treatment options. Combinational therapies that co-target multiple pathways are being investigated. The emergence of the Gq/11 inhibitors YM-254890 and FR900359, and their recent success in treating UM in preclinical studies, suggests that direct inhibition of oncogenic Gαq/11 may be a viable approach, or at least provide a therapeutic window to treat the disease more effectively with an additional drug, such as an MEK inhibitor or an inhibitor of survival signals from the liver microenvironment. However, these Gq/11 inhibitors act on both mutated and wild-type Gαq/11 proteins, which raises safety concerns, as Gq/11 is ubiquitously expressed and has a number of important physiological roles [126,127,128,129]. A local delivery system would be most advantageous, especially one that is able to target the liver, as it is likely that micrometastasis has already formed by the time of initial diagnosis. As we improve our understanding of how these compounds function and interact with Gq/11, the development of a compound that specifically targets the activated mutant Gαq/11 may one day be possible.
Author Contributions
D.L., writing—original draft and editing; J.L.B., writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Dr. Ralph and Marian Falk Medical Research Trust Bank of America, N.A., Trustee and National Institutes of Health awards P01 HL114471 (to J. Benovic) and F31 CA225064 (to D. Lapadula).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Weis, E.; Shah, C.P.; Lajous, M.; Shields, J.A.; Shields, C.L. The association between host susceptibility factors and uveal melanoma: A meta-analysis. Arch. Ophthalmol. 2006, 124, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Shah, C.P.; Weis, E.; Lajous, M.; Shields, J.A.; Shields, C.L. Intermittent and chronic ultraviolet light exposure and uveal melanoma: A meta-analysis. Ophthalmology 2005, 112, 1599–1607. [Google Scholar] [CrossRef]
- Shields, C.L.; Kaliki, S.; Livesey, M.; Walker, B.; Garoon, R.; Bucci, M.; Feinstein, E.; Pesch, A.; Gonzalez, C.; Lally, S.E.; et al. Association of ocular and oculodermal melanocytosis with the rate of uveal melanoma metastasis: Analysis of 7872 consecutive eyes. JAMA Ophthalmol. 2013, 131, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Hammer, H.; Oláh, J.; Tóth-Molnár, E. Dysplastic nevi are a risk factor for uveal melanoma. Eur. J. Ophthalmol. 1996, 6, 472–474. [Google Scholar] [CrossRef]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.D.; Bergman, L.; Seregard, S. Uveal melanoma: Epidemiologic aspects. Ophthalmol Clin N. Am 2005, 18, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Arnesen, K. The neural crest origin of uveal melanomas. Int. Ophthalmol. 1985, 7, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Damato, B. Ocular treatment of choroidal melanoma in relation to the prevention of metastatic death-A personal view. Prog. Retin. Eye Res. 2018, 66, 187–199. [Google Scholar] [CrossRef]
- Collaborative Ocular Melanoma Study Group. Assessment of metastatic disease status at death in 435 patients with large choroidal melanoma in the Collaborative Ocular Melanoma Study (COMS): COMS report no. 15. Arch. Ophthalmol. 2001, 119, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Kujala, E.; Mäkitie, T.; Kivelä, T. Very long-term prognosis of patients with malignant uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4651–4659. [Google Scholar] [CrossRef] [Green Version]
- Diener-West, M.; Reynolds, S.M.; Agugliaro, D.J.; Caldwell, R.; Cumming, K.; Earle, J.D.; Hawkins, B.S.; Hayman, J.A.; Jaiyesimi, I.; Jampol, L.M.; et al. Development of metastatic disease after enrollment in the COMS trials for treatment of choroidal melanoma: Collaborative Ocular Melanoma Study Group Report No. 26. Arch. Ophthalmol. 2005, 123, 1639–1643. [Google Scholar] [PubMed]
- Eskelin, S.; Pyrhönen, S.; Summanen, P.; Hahka-Kemppinen, M.; Kivelä, T. Tumor doubling times in metastatic malignant melanoma of the uvea: Tumor progression before and after treatment. Ophthalmology 2000, 107, 1443–1449. [Google Scholar] [CrossRef]
- Callejo, S.A.; Antecka, E.; Blanco, P.L.; Edelstein, C.; Burnier, M.N. Identification of circulating malignant cells and its correlation with prognostic factors and treatment in uveal melanoma. A prospective longitudinal study. Eye 2007, 21, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Ossowski, L.; Aguirre-Ghiso, J.A. Dormancy of metastatic melanoma. Pigment Cell Melanoma Res. 2010, 23, 41–56. [Google Scholar] [CrossRef]
- Singh, A.D.; Turell, M.E.; Topham, A.K. Uveal melanoma: Trends in incidence, treatment, and survival. Ophthalmology 2011, 118, 1881–1885. [Google Scholar] [CrossRef]
- Park, J.J.; Diefenbach, R.J.; Joshua, A.M.; Kefford, R.F.; Carlino, M.S.; Rizos, H. Oncogenic signaling in uveal melanoma. Pigment Cell Melanoma Res. 2018, 31, 661–672. [Google Scholar] [CrossRef]
- Yang, J.; Manson, D.K.; Marr, B.P.; Carvajal, R.D. Treatment of uveal melanoma: Where are we now? Ther. Adv. Med. Oncol. 2018, 10, 1758834018757175. [Google Scholar] [CrossRef]
- Rantala, E.S.; Hernberg, M.; Kivelä, T.T. Overall survival after treatment for metastatic uveal melanoma: A systematic review and meta-analysis. Melanoma Res. 2019, 29, 561–568. [Google Scholar] [CrossRef]
- Khoja, L.; Atenafu, E.G.; Suciu, S.; Leyvraz, S.; Sato, T.; Marshall, E.; Keilholz, U.; Zimmer, L.; Patel, S.P.; Piperno-Neumann, S.; et al. Meta-analysis in metastatic uveal melanoma to determine progression free and overall survival benchmarks: An international rare cancers initiative (IRCI) ocular melanoma study. Ann. Oncol. 2019, 30, 1370–1380. [Google Scholar] [CrossRef]
- Spagnolo, F.; Caltabiano, G.; Queirolo, P. Uveal melanoma. Cancer Treat. Rev. 2012, 38, 549–553. [Google Scholar] [CrossRef]
- Spagnolo, F.; Grosso, M.; Picasso, V.; Tornari, E.; Pesce, M.; Queirolo, P. Treatment of metastatic uveal melanoma with intravenous fotemustine. Melanoma Res. 2013, 23, 196–198. [Google Scholar] [CrossRef]
- Augsburger, J.J.; Corrêa, Z.M.; Shaikh, A.H. Effectiveness of treatments for metastatic uveal melanoma. Am. J. Ophthalmol. 2009, 148, 119–127. [Google Scholar] [CrossRef]
- Schmittel, A.; Schmidt-Hieber, M.; Martus, P.; Bechrakis, N.E.; Schuster, R.; Siehl, J.M.; Foerster, M.H.; Thiel, E.; Keilholz, U. A randomized phase II trial of gemcitabine plus treosulfan versus treosulfan alone in patients with metastatic uveal melanoma. Ann. Oncol. 2006, 17, 1826–1829. [Google Scholar] [CrossRef]
- Homsi, J.; Bedikian, A.Y.; Papadopoulos, N.E.; Kim, K.B.; Hwu, W.-J.; Mahoney, S.L.; Hwu, P. Phase 2 open-label study of weekly docosahexaenoic acid-paclitaxel in patients with metastatic uveal melanoma. Melanoma Res. 2010, 20, 507–510. [Google Scholar] [CrossRef]
- Kelderman, S.; van der Kooij, M.K.; van den Eertwegh, A.J.M.; Soetekouw, P.M.M.B.; Jansen, R.L.H.; van den Brom, R.R.H.; Hospers, G.A.P.; Haanen, J.B.A.G.; Kapiteijn, E.; Blank, C.U. Ipilimumab in pretreated metastastic uveal melanoma patients. Results of the Dutch Working group on Immunotherapy of Oncology (WIN-O). Acta Oncol. (Madr.) 2013, 52, 1786–1788. [Google Scholar] [CrossRef] [Green Version]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 2017, 32, 204–220.e15. [Google Scholar] [CrossRef] [Green Version]
- Heppt, M.V.; Steeb, T.; Schlager, J.G.; Rosumeck, S.; Dressler, C.; Ruzicka, T.; Nast, A.; Berking, C. Immune checkpoint blockade for unresectable or metastatic uveal melanoma: A systematic review. Cancer Treat. Rev. 2017, 60, 44–52. [Google Scholar] [CrossRef]
- Mignard, C.; Deschamps Huvier, A.; Gillibert, A.; Duval Modeste, A.B.; Dutriaux, C.; Khammari, A.; Avril, M.-F.; Kramkimel, N.; Mortier, L.; Marcant, P.; et al. Efficacy of Immunotherapy in Patients with Metastatic Mucosal or Uveal Melanoma. J. Oncol. 2018, 2018, 1908065. [Google Scholar] [CrossRef] [Green Version]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rimoldi, D.; Salvi, S.; Liénard, D.; Lejeune, F.J.; Speiser, D.; Zografos, L.; Cerottini, J.-C. Lack of BRAF mutations in uveal melanoma. Cancer Res. 2003, 63, 5712–5715. [Google Scholar] [PubMed]
- Onken, M.D.; Worley, L.A.; Long, M.D.; Duan, S.; Council, M.L.; Bowcock, A.M.; Harbour, J.W. Oncogenic mutations in GNAQ occur early in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5230–5234. [Google Scholar] [CrossRef] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Saldanha, G.; Purnell, D.; Fletcher, A.; Potter, L.; Gillies, A.; Pringle, J.H. High BRAF mutation frequency does not characterize all melanocytic tumor types. Int. J. Cancer 2004, 111, 705–710. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Johansson, P.; Aoude, L.G.; Wadt, K.; Glasson, W.J.; Warrier, S.K.; Hewitt, A.W.; Kiilgaard, J.F.; Heegaard, S.; Isaacs, T.; Franchina, M.; et al. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget 2016, 7, 4624–4631. [Google Scholar] [CrossRef] [Green Version]
- Furney, S.J.; Pedersen, M.; Gentien, D.; Dumont, A.G.; Rapinat, A.; Desjardins, L.; Turajlic, S.; Piperno-Neumann, S.; de la Grange, P.; Roman-Roman, S.; et al. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 2013, 3, 1122–1129. [Google Scholar] [CrossRef] [Green Version]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef] [Green Version]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.-X.; Luo, J.; Mo, J.-S.; Liu, G.; Kim, Y.C.; Meng, Z.; Zhao, L.; Peyman, G.; Ouyang, H.; Jiang, W.; et al. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell 2014, 25, 822–830. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Degese, M.S.; Iglesias-Bartolome, R.; Vaque, J.P.; Molinolo, A.A.; Rodrigues, M.; Zaidi, M.R.; Ksander, B.R.; Merlino, G.; Sodhi, A.; et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell 2014, 25, 831–845. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.R.; Ceraudo, E.; Sher, J.J.; Guan, Y.; Shoushtari, A.N.; Chang, M.T.; Zhang, J.Q.; Walczak, E.G.; Kazmi, M.A.; Taylor, B.S.; et al. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat. Genet. 2016, 48, 675–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceraudo, E.; Horioka, M.; Mattheisen, J.M.; Hitchman, T.D.; Moore, A.R.; Kazmi, M.A.; Chi, P.; Chen, Y.; Sakmar, T.P.; Huber, T. Direct evidence that the GPCR CysLTR2 mutant causative of uveal melanoma is constitutively active with highly biased signaling. J. Biol. Chem. 2021, 296, 100163. [Google Scholar] [CrossRef]
- Offermanns, S. G-proteins as transducers in transmembrane signalling. Prog. Biophys. Mol. Biol. 2003, 83, 101–130. [Google Scholar] [CrossRef]
- Johnston, C.A.; Siderovski, D.P. Receptor-mediated activation of heterotrimeric G-proteins: Current structural insights. Mol. Pharmacol. 2007, 72, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Milligan, G.; Kostenis, E. Heterotrimeric G-proteins: A short history. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S46–S55. [Google Scholar] [CrossRef] [Green Version]
- Oldham, W.M.; Hamm, H.E. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 2008, 9, 60–71. [Google Scholar] [CrossRef]
- Yoo, J.H.; Shi, D.S.; Grossmann, A.H.; Sorensen, L.K.; Tong, Z.; Mleynek, T.M.; Rogers, A.; Zhu, W.; Richards, J.R.; Winter, J.M.; et al. ARF6 Is an Actionable Node that Orchestrates Oncogenic GNAQ Signaling in Uveal Melanoma. Cancer Cell 2016, 29, 889–904. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.Q.; Lee, C.H.; Rhee, S.G.; Simon, M.I. Activation of phospholipase C by the alpha subunits of the Gq and G11 proteins in transfected Cos-7 cells. J. Biol. Chem. 1992, 267, 1811–1817. [Google Scholar] [CrossRef]
- Ross, E.M.; Wilkie, T.M. GTPase-activating proteins for heterotrimeric G proteins: Regulators of G protein signaling (RGS) and RGS-like proteins. Annu. Rev. Biochem. 2000, 69, 795–827. [Google Scholar] [CrossRef] [Green Version]
- Kimple, A.J.; Bosch, D.E.; Giguère, P.M.; Siderovski, D.P. Regulators of G-protein signaling and their Gα substrates: Promises and challenges in their use as drug discovery targets. Pharmacol. Rev. 2011, 63, 728–749. [Google Scholar] [CrossRef] [Green Version]
- O’Hayre, M.; Vázquez-Prado, J.; Kufareva, I.; Stawiski, E.W.; Handel, T.M.; Seshagiri, S.; Gutkind, J.S. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat. Rev. Cancer 2013, 13, 412–424. [Google Scholar] [CrossRef]
- Van Eps, N.; Preininger, A.M.; Alexander, N.; Kaya, A.I.; Meier, S.; Meiler, J.; Hamm, H.E.; Hubbell, W.L. Interaction of a G protein with an activated receptor opens the interdomain interface in the alpha subunit. Proc. Natl. Acad. Sci. USA 2011, 108, 9420–9424. [Google Scholar] [CrossRef] [Green Version]
- Kleuss, C.; Raw, A.S.; Lee, E.; Sprang, S.R.; Gilman, A.G. Mechanism of GTP hydrolysis by G-protein alpha subunits. Proc. Natl. Acad. Sci. USA 1994, 91, 9828–9831. [Google Scholar] [CrossRef] [Green Version]
- Takasaki, J.; Saito, T.; Taniguchi, M.; Kawasaki, T.; Moritani, Y.; Hayashi, K.; Kobori, M. A novel Galphaq/11-selective inhibitor. J. Biol. Chem. 2004, 279, 47438–47445. [Google Scholar] [CrossRef] [Green Version]
- Weber, A.; Hengge, U.R.; Urbanik, D.; Markwart, A.; Mirmohammadsaegh, A.; Reichel, M.B.; Wittekind, C.; Wiedemann, P.; Tannapfel, A. Absence of mutations of the BRAF gene and constitutive activation of extracellular-regulated kinase in malignant melanomas of the uvea. Lab. Investig. 2003, 83, 1771–1776. [Google Scholar] [CrossRef] [Green Version]
- Zuidervaart, W.; van Nieuwpoort, F.; Stark, M.; Dijkman, R.; Packer, L.; Borgstein, A.M.; Pavey, S.; van der Velden, P.; Out, C.; Jager, M.J.; et al. Activation of the MAPK pathway is a common event in uveal melanomas although it rarely occurs through mutation of BRAF or RAS. Br. J. Cancer 2005, 92, 2032–2038. [Google Scholar] [CrossRef] [Green Version]
- Edmunds, S.C.; Cree, I.A.; Dí Nícolantonío, F.; Hungerford, J.L.; Hurren, J.S.; Kelsell, D.P. Absence of BRAF gene mutations in uveal melanomas in contrast to cutaneous melanomas. Br. J. Cancer 2003, 88, 1403–1405. [Google Scholar] [CrossRef] [Green Version]
- Vaqué, J.P.; Dorsam, R.T.; Feng, X.; Iglesias-Bartolome, R.; Forsthoefel, D.J.; Chen, Q.; Debant, A.; Seeger, M.A.; Ksander, B.R.; Teramoto, H.; et al. A genome-wide RNAi screen reveals a Trio-regulated Rho GTPase circuitry transducing mitogenic signals initiated by G protein-coupled receptors. Mol. Cell 2013, 49, 94–108. [Google Scholar] [CrossRef] [Green Version]
- Babchia, N.; Calipel, A.; Mouriaux, F.; Faussat, A.-M.; Mascarelli, F. The PI3K/Akt and mTOR/P70S6K signaling pathways in human uveal melanoma cells: Interaction with B-Raf/ERK. Investig. Ophthalmol. Vis. Sci. 2010, 51, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Chua, V.; Lapadula, D.; Randolph, C.; Benovic, J.L.; Wedegaertner, P.B.; Aplin, A.E. Dysregulated GPCR signaling and therapeutic options in uveal melanoma. Mol. Cancer Res. 2017, 15, 501–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Li, J.; Zhu, M.; Fletcher, J.A.; Hodi, F.S. Protein kinase C inhibitor AEB071 targets ocular melanoma harboring GNAQ mutations via effects on the PKC/Erk1/2 and PKC/NF-κB pathways. Mol. Cancer Ther. 2012, 11, 1905–1914. [Google Scholar] [CrossRef] [Green Version]
- Piperno-Neumann, S.; Kapiteijn, E.; Larkin, J.M.G.; Carvajal, R.D.; Luke, J.J.; Seifert, H.; Roozen, I.; Zoubir, M.; Yang, L.; Choudhury, S.; et al. Phase I dose-escalation study of the protein kinase C (PKC) inhibitor AEB071 in patients with metastatic uveal melanoma. J. Clin. Oncol. 2014, 32, 9030. [Google Scholar] [CrossRef]
- Chen, X.; Wu, Q.; Tan, L.; Porter, D.; Jager, M.J.; Emery, C.; Bastian, B.C. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene 2014, 33, 4724–4734. [Google Scholar] [CrossRef] [Green Version]
- Ambrosini, G.; Pratilas, C.A.; Qin, L.-X.; Tadi, M.; Surriga, O.; Carvajal, R.D.; Schwartz, G.K. Identification of unique MEK-dependent genes in GNAQ mutant uveal melanoma involved in cell growth, tumor cell invasion, and MEK resistance. Clin. Cancer Res. 2012, 18, 3552–3561. [Google Scholar] [CrossRef] [Green Version]
- Carvajal, R.D.; Sosman, J.A.; Quevedo, J.F.; Milhem, M.M.; Joshua, A.M.; Kudchadkar, R.R.; Linette, G.P.; Gajewski, T.F.; Lutzky, J.; Lawson, D.H.; et al. Effect of selumetinib vs. chemotherapy on progression-free survival in uveal melanoma: A randomized clinical trial. JAMA 2014, 311, 2397–2405. [Google Scholar] [CrossRef]
- Adjei, A.A.; LoRusso, P.; Ribas, A.; Sosman, J.A.; Pavlick, A.C.; Dy, G.K.; Zhou, X.; Gangolli, E.A.; Walker, R.M.; Kneissl, M.; et al. Phase I, dose-escalation study of the investigational drug TAK-733, an oral MEK inhibitor, in patients (pts) with advanced solid tumors. JCO 2013, 31, 2528. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Piperno-Neumann, S.; Kapiteijn, E.; Chapman, P.B.; Frank, S.; Joshua, A.M.; Piulats, J.M.; Wolter, P.; Cocquyt, V.; Chmielowski, B.; et al. Selumetinib in combination with dacarbazine in patients with metastatic uveal melanoma: A phase III, multicenter, randomized trial (SUMIT). J. Clin. Oncol. 2018, 36, 1232–1239. [Google Scholar] [CrossRef] [Green Version]
- Kirkwood, J.M.; Bastholt, L.; Robert, C.; Sosman, J.; Larkin, J.; Hersey, P.; Middleton, M.; Cantarini, M.; Zazulina, V.; Kemsley, K.; et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin. Cancer Res. 2012, 18, 555–567. [Google Scholar] [CrossRef] [Green Version]
- Shoushtari, A.N.; Kudchadkar, R.R.; Panageas, K.; Murthy, R.K.; Jung, M.; Shah, R.; O’Donnell, B.; Khawaja, T.T.; Shames, Y.; Prempeh-Keteku, N.A.; et al. A randomized phase 2 study of trametinib with or without GSK2141795 in patients with advanced uveal melanoma. J. Clin. Oncol. 2016, 34, 9511. [Google Scholar] [CrossRef]
- Falchook, G.S.; Lewis, K.D.; Infante, J.R.; Gordon, M.S.; Vogelzang, N.J.; DeMarini, D.J.; Sun, P.; Moy, C.; Szabo, S.A.; Roadcap, L.T.; et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 782–789. [Google Scholar] [CrossRef] [Green Version]
- Steeb, T.; Wessely, A.; Ruzicka, T.; Heppt, M.V.; Berking, C. How to MEK the best of uveal melanoma: A systematic review on the efficacy and safety of MEK inhibitors in metastatic or unresectable uveal melanoma. Eur. J. Cancer 2018, 103, 41–51. [Google Scholar] [CrossRef]
- Cheng, H.; Chua, V.; Liao, C.; Purwin, T.J.; Terai, M.; Kageyama, K.; Davies, M.A.; Sato, T.; Aplin, A.E. Co-targeting HGF/cMET Signaling with MEK Inhibitors in Metastatic Uveal Melanoma. Mol. Cancer Ther. 2017, 16, 516–528. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Terai, M.; Kageyama, K.; Ozaki, S.; McCue, P.A.; Sato, T.; Aplin, A.E. Paracrine effect of NRG1 and HGF drives resistance to MEK inhibitors in metastatic uveal melanoma. Cancer Res. 2015, 75, 2737–2748. [Google Scholar] [CrossRef] [Green Version]
- Decaudin, D.; El Botty, R.; Diallo, B.; Massonnet, G.; Fleury, J.; Naguez, A.; Raymondie, C.; Davies, E.; Smith, A.; Wilson, J.; et al. Selumetinib-based therapy in uveal melanoma patient-derived xenografts. Oncotarget 2018, 9, 21674–21686. [Google Scholar] [CrossRef] [Green Version]
- Truong, A.; Yoo, J.H.; Scherzer, M.T.; Sanchez, J.M.S.; Dale, K.J.; Kinsey, C.G.; Richards, J.R.; Shin, D.; Ghazi, P.C.; Onken, M.D.; et al. Chloroquine Sensitizes GNAQ/11-mutated Melanoma to MEK1/2 Inhibition. Clin. Cancer Res. 2020, 26, 6374–6386. [Google Scholar] [CrossRef]
- Chen, X.; Wu, Q.; Depeille, P.; Chen, P.; Thornton, S.; Kalirai, H.; Coupland, S.E.; Roose, J.P.; Bastian, B.C. Rasgrp3 mediates MAPK pathway activation in GNAQ mutant uveal melanoma. Cancer Cell 2017, 31, 685–696.e6. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.R.; Ran, L.; Guan, Y.; Sher, J.J.; Hitchman, T.D.; Zhang, J.Q.; Hwang, C.; Walzak, E.G.; Shoushtari, A.N.; Monette, S.; et al. GNA11 Q209L mouse model reveals RasGRP3 as an essential signaling node in uveal melanoma. Cell Rep. 2018, 22, 2455–2468. [Google Scholar] [CrossRef] [Green Version]
- Khalili, J.S.; Yu, X.; Wang, J.; Hayes, B.C.; Davies, M.A.; Lizee, G.; Esmaeli, B.; Woodman, S.E. Combination small molecule MEK and PI3K inhibition enhances uveal melanoma cell death in a mutant GNAQ- and GNA11-dependent manner. Clin. Cancer Res. 2012, 18, 4345–4355. [Google Scholar] [CrossRef] [Green Version]
- Musi, E.; Ambrosini, G.; de Stanchina, E.; Schwartz, G.K. The phosphoinositide 3-kinase α selective inhibitor BYL719 enhances the effect of the protein kinase C inhibitor AEB071 in GNAQ/GNA11-mutant uveal melanoma cells. Mol. Cancer Ther. 2014, 13, 1044–1053. [Google Scholar] [CrossRef] [Green Version]
- Ambrosini, G.; Musi, E.; Ho, A.L.; de Stanchina, E.; Schwartz, G.K. Inhibition of mutant GNAQ signaling in uveal melanoma induces AMPK-dependent autophagic cell death. Mol. Cancer Ther. 2013, 12, 768–776. [Google Scholar] [CrossRef] [Green Version]
- Shoushtari, A.N.; Ong, L.T.; Schoder, H.; Singh-Kandah, S.; Abbate, K.T.; Postow, M.A.; Callahan, M.K.; Wolchok, J.; Chapman, P.B.; Panageas, K.S.; et al. A phase 2 trial of everolimus and pasireotide long-acting release in patients with metastatic uveal melanoma. Melanoma Res. 2016, 26, 272–277. [Google Scholar] [CrossRef] [Green Version]
- Amirouchene-Angelozzi, N.; Frisch-Dit-Leitz, E.; Carita, G.; Dahmani, A.; Raymondie, C.; Liot, G.; Gentien, D.; Némati, F.; Decaudin, D.; Roman-Roman, S.; et al. The mTOR inhibitor Everolimus synergizes with the PI3K inhibitor GDC0941 to enhance anti-tumor efficacy in uveal melanoma. Oncotarget 2016, 7, 23633–23646. [Google Scholar] [CrossRef] [Green Version]
- Carita, G.; Frisch-Dit-Leitz, E.; Dahmani, A.; Raymondie, C.; Cassoux, N.; Piperno-Neumann, S.; Némati, F.; Laurent, C.; De Koning, L.; Halilovic, E.; et al. Dual inhibition of protein kinase C and p53-MDM2 or PKC and mTORC1 are novel efficient therapeutic approaches for uveal melanoma. Oncotarget 2016, 7, 33542–33556. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Qi, H.-X.; Hu, Z.-M.; Chang, Y.-N.; Shi, Z.-M.; Han, X.-H.; Han, Y.-W.; Zhang, R.-X.; Zhang, Z.; Chen, T.; et al. YAP and TAZ take center stage in cancer. Biochemistry 2015, 54, 6555–6566. [Google Scholar] [CrossRef]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.-J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [Green Version]
- Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [Green Version]
- Ramos, A.; Camargo, F.D. The Hippo signaling pathway and stem cell biology. Trends Cell Biol. 2012, 22, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.-X.; Guan, K.-L. The Hippo pathway: Regulators and regulations. Genes Dev. 2013, 27, 355–371. [Google Scholar] [CrossRef] [Green Version]
- Lyubasyuk, V.; Ouyang, H.; Yu, F.-X.; Guan, K.-L.; Zhang, K. YAP inhibition blocks uveal melanogenesis driven by GNAQ or GNA11 mutations. Mol. Cell. Oncol. 2015, 2, e970957. [Google Scholar] [CrossRef]
- Kim, Y.J.; Lee, S.C.; Kim, S.E.; Kim, S.H.; Kim, S.K.; Lee, C.S. YAP Activity is Not Associated with Survival of Uveal Melanoma Patients and Cell Lines. Sci. Rep. 2020, 10, 6209. [Google Scholar] [CrossRef] [Green Version]
- Brouwer, N.J.; Konstantinou, E.K.; Gragoudas, E.S.; Marinkovic, M.; Luyten, G.P.M.; Kim, I.K.; Jager, M.J.; Vavvas, D.G. Targeting the YAP/TAZ pathway in uveal and conjunctival melanoma with verteporfin. Investig. Ophthalmol. Vis. Sci. 2021, 62, 3. [Google Scholar] [CrossRef]
- Feng, X.; Arang, N.; Rigiracciolo, D.C.; Lee, J.S.; Yeerna, H.; Wang, Z.; Lubrano, S.; Kishore, A.; Pachter, J.A.; König, G.M.; et al. A Platform of Synthetic Lethal Gene Interaction Networks Reveals that the GNAQ Uveal Melanoma Oncogene Controls the Hippo Pathway through FAK. Cancer Cell 2019, 35, 457–472.e5. [Google Scholar] [CrossRef] [Green Version]
- Paradis, J.S.; Acosta, M.; Saddawi-Konefka, R.; Kishore, A.; Gomes, F.; Arang, N.; Tiago, M.; Coma, S.; Lubrano, S.; Wu, X.; et al. Synthetic Lethal Screens Reveal Cotargeting FAK and MEK as a Multimodal Precision Therapy for GNAQ-Driven Uveal Melanoma. Clin. Cancer Res. 2021, 27, 3190–3200. [Google Scholar] [CrossRef]
- Ma, J.; Weng, L.; Bastian, B.C.; Chen, X. Functional characterization of uveal melanoma oncogenes. Oncogene 2021, 40, 806–820. [Google Scholar] [CrossRef]
- Shain, A.H.; Bagger, M.M.; Yu, R.; Chang, D.; Liu, S.; Vemula, S.; Weier, J.F.; Wadt, K.; Heegaard, S.; Bastian, B.C.; et al. The genetic evolution of metastatic uveal melanoma. Nat. Genet. 2019, 51, 1123–1130. [Google Scholar] [CrossRef]
- Zhang, H.; Nielsen, A.L.; Strømgaard, K. Recent achievements in developing selective Gq inhibitors. Med. Res. Rev. 2020, 40, 135–157. [Google Scholar] [CrossRef]
- Kostenis, E.; Pfeil, E.M.; Annala, S. Heterotrimeric Gq proteins as therapeutic targets? J. Biol. Chem. 2020, 295, 5206–5215. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, M.; Nagai, K.; Arao, N.; Kawasaki, T.; Saito, T.; Moritani, Y.; Takasaki, J.; Hayashi, K.; Fujita, S.; Suzuki, K.; et al. YM-254890, a novel platelet aggregation inhibitor produced by Chromobacterium sp. QS3666. J. Antibiot. 2003, 56, 358–363. [Google Scholar] [CrossRef] [Green Version]
- Uemura, T.; Kawasaki, T.; Taniguchi, M.; Moritani, Y.; Hayashi, K.; Saito, T.; Takasaki, J.; Uchida, W.; Miyata, K. Biological properties of a specific Galpha q/11 inhibitor, YM-254890, on platelet functions and thrombus formation under high-shear stress. Br. J. Pharmacol. 2006, 148, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Taniguchi, M.; Moritani, Y.; Uemura, T.; Shigenaga, T.; Takamatsu, H.; Hayashi, K.; Takasaki, J.; Saito, T.; Nagai, K. Pharmacological properties of YM-254890, a specific G(alpha)q/11 inhibitor, on thrombosis and neointima formation in mice. Thromb. Haemost. 2005, 94, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Taniguchi, M.; Moritani, Y.; Hayashi, K.; Saito, T.; Takasaki, J.; Nagai, K.; Inagaki, O.; Shikama, H. Antithrombotic and thrombolytic efficacy of YM-254890, a G q/11 inhibitor, in a rat model of arterial thrombosis. Thromb. Haemost. 2003, 90, 406–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, X.-F.; Zhang, H.; Underwood, C.R.; Harpsøe, K.; Gardella, T.J.; Wöldike, M.F.; Mannstadt, M.; Gloriam, D.E.; Bräuner-Osborne, H.; Strømgaard, K. Total synthesis and structure-activity relationship studies of a series of selective G protein inhibitors. Nat. Chem. 2016, 8, 1035–1041. [Google Scholar] [CrossRef] [Green Version]
- Fujioka, M.; Koda, S.; Morimoto, Y.; Biemann, K. Structure of FR900359, a cyclic depsipeptide from Ardisia crenata sims. J. Org. Chem. 1988, 53, 2820–2825. [Google Scholar] [CrossRef]
- Carlier, A.; Fehr, L.; Pinto-Carbó, M.; Schäberle, T.; Reher, R.; Dessein, S.; König, G.; Eberl, L. The genome analysis of Candidatus Burkholderia crenata reveals that secondary metabolism may be a key function of the Ardisia crenata leaf nodule symbiosis. Environ. Microbiol. 2016, 18, 2507–2522. [Google Scholar] [CrossRef]
- Inamdar, V.; Patel, A.; Manne, B.K.; Dangelmaier, C.; Kunapuli, S.P. Characterization of UBO-QIC as a Gαq inhibitor in platelets. Platelets 2015, 26, 771–778. [Google Scholar] [CrossRef]
- Schrage, R.; Schmitz, A.-L.; Gaffal, E.; Annala, S.; Kehraus, S.; Wenzel, D.; Büllesbach, K.M.; Bald, T.; Inoue, A.; Shinjo, Y.; et al. The experimental power of FR900359 to study Gq-regulated biological processes. Nat. Commun. 2015, 6, 10156. [Google Scholar] [CrossRef] [Green Version]
- Zaima, K.; Deguchi, J.; Matsuno, Y.; Kaneda, T.; Hirasawa, Y.; Morita, H. Vasorelaxant effect of FR900359 from Ardisia crenata on rat aortic artery. J. Nat. Med. 2013, 67, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Carr, R.; Koziol-White, C.; Zhang, J.; Lam, H.; An, S.S.; Tall, G.G.; Panettieri, R.A.; Benovic, J.L. Interdicting Gq Activation in Airway Disease by Receptor-Dependent and Receptor-Independent Mechanisms. Mol. Pharmacol. 2016, 89, 94–104. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.-G.; Jacobson, K.A. On the selectivity of the Gαq inhibitor UBO-QIC: A comparison with the Gαi inhibitor pertussis toxin. Biochem. Pharmacol. 2016, 107, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Pfeil, E.M.; Brands, J.; Merten, N.; Vögtle, T.; Vescovo, M.; Rick, U.; Albrecht, I.-M.; Heycke, N.; Kawakami, K.; Ono, Y.; et al. Heterotrimeric G Protein Subunit Gαq Is a Master Switch for Gβγ-Mediated Calcium Mobilization by Gi-Coupled GPCRs. Mol. Cell 2020, 80, 940–954.e6. [Google Scholar] [CrossRef] [PubMed]
- Crüsemann, M.; Reher, R.; Schamari, I.; Brachmann, A.O.; Ohbayashi, T.; Kuschak, M.; Malfacini, D.; Seidinger, A.; Pinto-Carbó, M.; Richarz, R.; et al. Heterologous Expression, Biosynthetic Studies, and Ecological Function of the Selective Gq-Signaling Inhibitor FR900359. Angew. Chem. Int. Ed. Engl. 2018, 57, 836–840. [Google Scholar] [CrossRef] [PubMed]
- Pistorius, D.; Buntin, K.; Bouquet, C.; Richard, E.; Weber, E.; Wollbrett, S. Genetic Engineering of Chromobacterium vaccinii DSM 25150 for Improved Production of FR900359; ChemRxiv; Cambridge Open Engage: Cambridge, UK, 2021; This content is a preprint and has not been peer-reviewed. [Google Scholar] [CrossRef]
- Nishimura, A.; Kitano, K.; Takasaki, J.; Taniguchi, M.; Mizuno, N.; Tago, K.; Hakoshima, T.; Itoh, H. Structural basis for the specific inhibition of heterotrimeric Gq protein by a small molecule. Proc. Natl. Acad. Sci. USA 2010, 107, 13666–13671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malfacini, D.; Patt, J.; Annala, S.; Harpsøe, K.; Eryilmaz, F.; Reher, R.; Crüsemann, M.; Hanke, W.; Zhang, H.; Tietze, D.; et al. Rational design of a heterotrimeric G protein α subunit with artificial inhibitor sensitivity. J. Biol. Chem. 2019, 294, 5747–5758. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Nielsen, A.L.; Boesgaard, M.W.; Harpsøe, K.; Daly, N.L.; Xiong, X.-F.; Underwood, C.R.; Haugaard-Kedström, L.M.; Bräuner-Osborne, H.; Gloriam, D.E.; et al. Structure-activity relationship and conformational studies of the natural product cyclic depsipeptides YM-254890 and FR900359. Eur. J. Med. Chem. 2018, 156, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Kuschak, M.; Namasivayam, V.; Rafehi, M.; Voss, J.H.; Garg, J.; Schlegel, J.G.; Abdelrahman, A.; Kehraus, S.; Reher, R.; Küppers, J.; et al. Cell-permeable high-affinity tracers for Gq proteins provide structural insights, reveal distinct binding kinetics and identify small molecule inhibitors. Br. J. Pharmacol. 2020, 177, 1898–1916. [Google Scholar] [CrossRef] [Green Version]
- Lapadula, D.; Farias, E.; Randolph, C.E.; Purwin, T.J.; McGrath, D.; Charpentier, T.H.; Zhang, L.; Wu, S.; Terai, M.; Sato, T.; et al. Effects of oncogenic Gαq and Gα11 inhibition by FR900359 in uveal melanoma. Mol. Cancer Res. 2019, 17, 963–973. [Google Scholar] [CrossRef]
- Onken, M.D.; Makepeace, C.M.; Kaltenbronn, K.M.; Kanai, S.M.; Todd, T.D.; Wang, S.; Broekelmann, T.J.; Rao, P.K.; Cooper, J.A.; Blumer, K.J. Targeting nucleotide exchange to inhibit constitutively active G protein α subunits in cancer cells. Sci. Signal. 2018, 11, 546. [Google Scholar] [CrossRef] [Green Version]
- Annala, S.; Feng, X.; Shridhar, N.; Eryilmaz, F.; Patt, J.; Yang, J.; Pfeil, E.M.; Cervantes-Villagrana, R.D.; Inoue, A.; Häberlein, F.; et al. Direct targeting of Gαq and Gα11 oncoproteins in cancer cells. Sci. Signal. 2019, 12, 573. [Google Scholar] [CrossRef]
- Onken, M.D.; Makepeace, C.M.; Kaltenbronn, K.M.; Choi, J.; Hernandez-Aya, L.; Weilbaecher, K.N.; Piggott, K.D.; Rao, P.K.; Yuede, C.M.; Dixon, A.J.; et al. Targeting primary and metastatic uveal melanoma with a Gprotein inhibitor. J. Biol. Chem. 2021, 296, 100403. [Google Scholar] [CrossRef]
- Hitchman, T.D.; Bayshtok, G.; Ceraudo, E.; Moore, A.R.; Lee, C.; Jia, R.; Wang, N.; Pachai, M.R.; Shoushtari, A.N.; Francis, J.H.; et al. Combined inhibition of Gαq and MEK enhances therapeutic efficacy in uveal melanoma. Clin. Cancer Res. 2021, 27, 1476–1490. [Google Scholar] [CrossRef]
- Schlegel, J.G.; Tahoun, M.; Seidinger, A.; Voss, J.H.; Kuschak, M.; Kehraus, S.; Schneider, M.; Matthey, M.; Fleischmann, B.K.; König, G.M.; et al. Macrocyclic Gq Protein Inhibitors FR900359 and/or YM-254890-Fit for Translation? ACS Pharmacol. Transl. Sci. 2021, 4, 888–897. [Google Scholar] [CrossRef] [PubMed]
- Onken, M.D.; Worley, L.A.; Ehlers, J.P.; Harbour, J.W. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004, 64, 7205–7209. [Google Scholar] [CrossRef] [Green Version]
- Bauer, J.; Kilic, E.; Vaarwater, J.; Bastian, B.C.; Garbe, C.; de Klein, A. Oncogenic GNAQ mutations are not correlated with disease-free survival in uveal melanoma. Br. J. Cancer 2009, 101, 813–815. [Google Scholar] [CrossRef] [Green Version]
- Offermanns, S.; Hashimoto, K.; Watanabe, M.; Sun, W.; Kurihara, H.; Thompson, R.F.; Inoue, Y.; Kano, M.; Simon, M.I. Impaired motor coordination and persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking Galphaq. Proc. Natl. Acad. Sci. USA 1997, 94, 14089–14094. [Google Scholar] [CrossRef] [Green Version]
- Offermanns, S.; Zhao, L.P.; Gohla, A.; Sarosi, I.; Simon, M.I.; Wilkie, T.M. Embryonic cardiomyocyte hypoplasia and craniofacial defects in G alpha q/G alpha 11-mutant mice. EMBO J. 1998, 17, 4304–4312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frederick, A.L.; Saborido, T.P.; Stanwood, G.D. Neurobehavioral phenotyping of Gαq knockout mice reveals impairments in motor functions and spatial working memory without changes in anxiety or behavioral despair. Front. Behav. Neurosci. 2012, 6, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesen, K.; Kaiser, E.; Schröder, L.; Scholz, A.; Ruppenthal, S.; Reil, J.-C.; Backes, C.; Meese, E.; Meier, C.; Bogdanova, A.; et al. Cardiac remodeling in Gαq and Gα11 knockout mice. Int. J. Cardiol. 2016, 202, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Rensing, D.T.; Uppal, S.; Blumer, K.J.; Moeller, K.D. Toward the Selective Inhibition of G Proteins: Total Synthesis of a Simplified YM-254890 Analog. Org. Lett. 2015, 17, 2270–2273. [Google Scholar] [CrossRef]
- Kaur, H.; Harris, P.W.R.; Little, P.J.; Brimble, M.A. Total synthesis of the cyclic depsipeptide YM-280193, a platelet aggregation inhibitor. Org. Lett. 2015, 17, 492–495. [Google Scholar] [CrossRef]
- Zhang, H.; Xiong, X.-F.; Boesgaard, M.W.; Underwood, C.R.; Bräuner-Osborne, H.; Strømgaard, K. Structure-Activity Relationship Studies of the Cyclic Depsipeptide Natural Product YM-254890, Targeting the Gq Protein. Chem. Med. Chem. 2017, 12, 830–834. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.-F.; Zhang, H.; Boesgaard, M.W.; Underwood, C.R.; Bräuner-Osborne, H.; Strømgaard, K. Structure-Activity Relationship Studies of the Natural Product Gq/11 Protein Inhibitor YM-254890. Chem. Med. Chem. 2019, 14, 865–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reher, R.; Kühl, T.; Annala, S.; Benkel, T.; Kaufmann, D.; Nubbemeyer, B.; Odhiambo, J.P.; Heimer, P.; Bäuml, C.A.; Kehraus, S.; et al. Deciphering Specificity Determinants for FR900359-Derived Gq α Inhibitors Based on Computational and Structure-Activity Studies. Chem. Med. Chem. 2018, 13, 1634–1643. [Google Scholar] [CrossRef] [PubMed]
- Reher, R.; Kuschak, M.; Heycke, N.; Annala, S.; Kehraus, S.; Dai, H.-F.; Müller, C.E.; Kostenis, E.; König, G.M.; Crüsemann, M. Applying molecular networking for the detection of natural sources and analogues of the selective Gq protein inhibitor FR900359. J. Nat. Prod. 2018, 81, 1628–1635. [Google Scholar] [CrossRef]
- Hermes, C.; Richarz, R.; Wirtz, D.A.; Patt, J.; Hanke, W.; Kehraus, S.; Voß, J.H.; Küppers, J.; Ohbayashi, T.; Namasivayam, V.; et al. Thioesterase-mediated side chain transesterification generates potent Gq signaling inhibitor FR900359. Nat. Commun. 2021, 12, 144. [Google Scholar] [CrossRef]
- Hanke, W.; Patt, J.; Alenfelder, J.; Voss, J.H.; Zdouc, M.M.; Kehraus, S.; Kim, J.B.; Grujičić, G.V.; Namasivayam, V.; Reher, R.; et al. Feature-Based Molecular Networking for the Targeted Identification of Gq-Inhibiting FR900359 Derivatives. J. Nat. Prod. 2021, 84, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Shi, S.; Deng, J.-J.; Chen, X.-P.; Song, Z.; Liu, L.; Lou, L.; Zhang, X.; Xiong, X.-F. Design, synthesis, and evaluation of small molecule Gαq/11 protein inhibitors for the treatment of uveal melanoma. J. Med. Chem. 2021, 64, 3131–3152. [Google Scholar] [CrossRef]
Figure 1.
Signaling Pathways in Uveal Melanoma. Activating mutations in GPCR CysLTR2, Gαq/11, and PLCβ4 stimulate downstream signaling pathways such as ARF6, MAPK, FAK/YAP, and AKT/MTOR, all of which can lead to UM cell proliferation and survival.
Figure 1.
Signaling Pathways in Uveal Melanoma. Activating mutations in GPCR CysLTR2, Gαq/11, and PLCβ4 stimulate downstream signaling pathways such as ARF6, MAPK, FAK/YAP, and AKT/MTOR, all of which can lead to UM cell proliferation and survival.

Figure 2.
Chemical structures of Gq/11 depsipeptide inhibitors YM-254890 and FR900359. Highlighted areas show the regions of YM-254890 and FR900359 that differ.
Figure 2.
Chemical structures of Gq/11 depsipeptide inhibitors YM-254890 and FR900359. Highlighted areas show the regions of YM-254890 and FR900359 that differ.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lapadula, D.; Benovic, J.L. Targeting Oncogenic Gαq/11 in Uveal Melanoma. Cancers 2021, 13, 6195. https://doi.org/10.3390/cancers13246195
AMA Style
Lapadula D, Benovic JL. Targeting Oncogenic Gαq/11 in Uveal Melanoma. Cancers. 2021; 13(24):6195. https://doi.org/10.3390/cancers13246195
Chicago/Turabian StyleLapadula, Dominic, and Jeffrey L. Benovic. 2021. "Targeting Oncogenic Gαq/11 in Uveal Melanoma" Cancers 13, no. 24: 6195. https://doi.org/10.3390/cancers13246195
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.