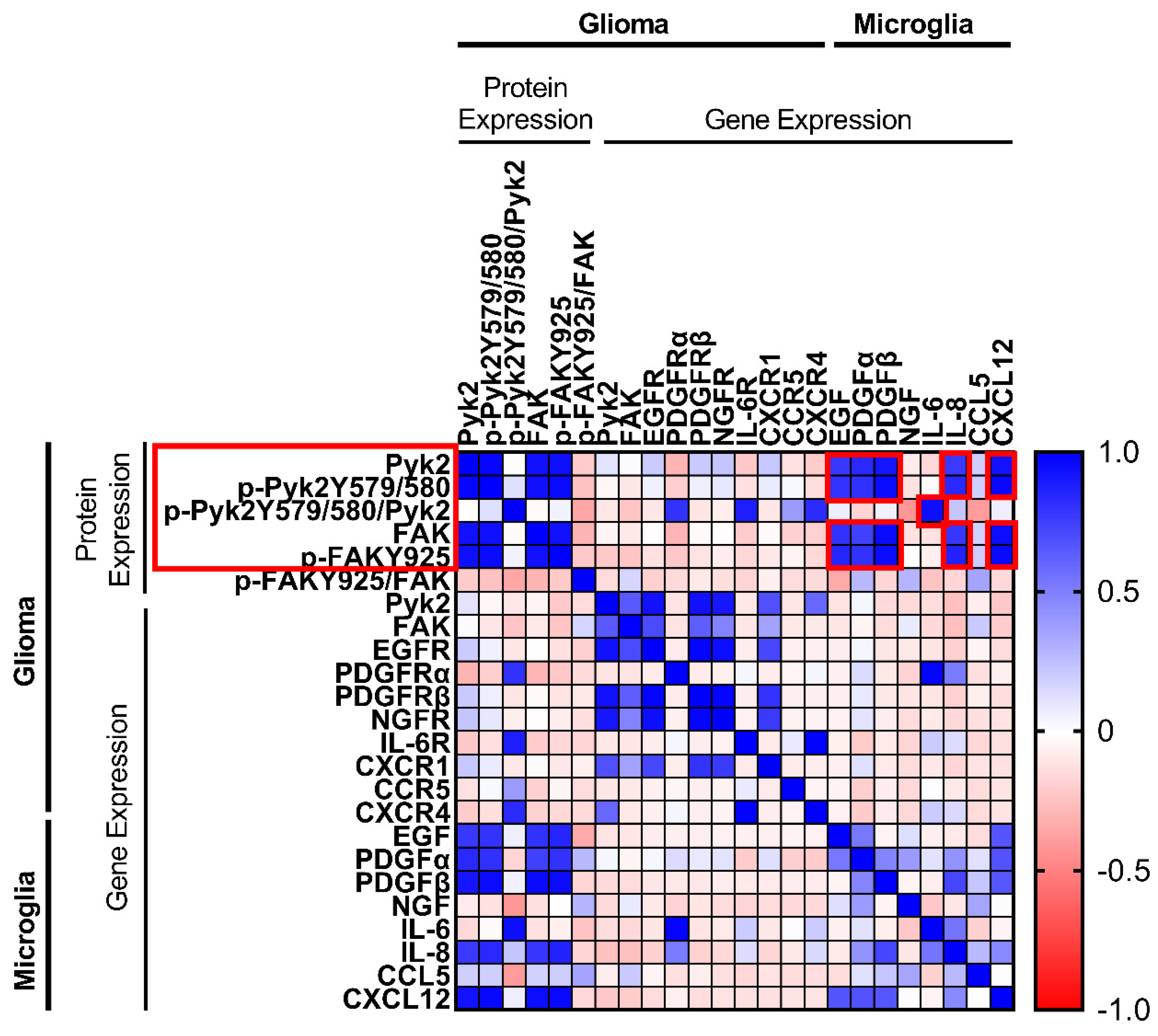
3.1. Correlation between Gene Expression of Cytokines in Microglia and Pyk2 and FAK Protein Expression in Glioma Cells from GBM Tumor Samples
Recent studies [
10,
25,
27] suggest that microglial-derived factors drive the expression of Pyk2 and FAK in glioma cells to promote their proliferation and invasiveness. To determine the association between the gene expression of microglial cytokines/chemokines and Pyk2 and FAK gene and protein expression in glioma cells, a Pearson’s correlation test was performed. Using RT-PCR, the Percoll-purified microglia fraction from 20 human GBM specimens was analyzed for the gene expression of key cytokines and chemokines (encoding EGF, PDGFα, PDGFβ, NGF, IL-6, IL-8, IL-10, CXCR12, and CCL5). The glioma cell tumor fraction was analyzed for the level of gene expression of corresponding receptors, as well as Pyk2 and FAK. The protein expression of Pyk2 and FAK and the phosphorylation levels of Pyk2 (Y579/Y580) and FAK (Y925) were evaluated by western blotting. Pierson correlation analysis was performed, and cytokine pairs with a strong correlation (0.6–1.0) were selected for further study (
Figure 1).
A strong positive correlation (r > 0.6) between Pyk2 and FAK gene expression and cytokine/chemokine receptors was observed in glioma fractions. Statistically significant correlations of gene expression of Pyk2 with that of EGFR (r = 0.93, p < 0.05), PDGFβR (r = 0.99, p < 0.05), NGFR (r = 0.91, p < 0.05), CXCR1 (also known as the IL-8 receptor, r = 0.68, p < 0.05), and CXCR4 (also known as SDF-1αR, r = 0.6, p < 0.05) were observed. Similar results were obtained when comparing FAK with EGFR and PDGFβR (r = 0.71 and r = 0.63, respectively, p < 0.05). Despite the fact that no significant correlation between Pyk2 and FAK gene expression in glioma and the corresponding cytokines in microglia was found, a strong positive correlation for phosphorylated and total Pyk2 and FAK protein expression in glioma cell fractions and cytokine expression in microglia was observed. Phosphorylated and total Pyk2 protein expression demonstrated a strong correlation with EGF (r = 0.8, p < 0.05), PDGFα (r ≥ 0.81, p < 0.05), PDFGβ (r > 0.92, p < 0.05), IL-8 (r > 0.78, p < 0.05), and SDF-1α (r > 0.93, p < 0.05). Similar results were obtained when FAK was evaluated. Of note, no correlation was found between listed microglial cytokines/chemokines and glioma Pyk2 and FAK intensity of phosphorylation, calculated as a proportion of phosphorylated protein to total protein expression. This result might indicate that the observed correlation between microglial factors and the level of phosphorylated Pyk2 and FAK in glioma cells is due to an impact on protein expression, but not phosphorylation. By contrast, IL-6 expression in microglia did not demonstrate a correlation with Pyk2/FAK phosphorylated and total protein expression; however, it was correlated with the intensity of Pyk2 phosphorylation (Pyk2 (Y579/Y580))/total Pyk2), indicating the possible regulation of Pyk2 phosphorylation by this cytokine.
No correlation was observed between the genes encoding Pyk2 and FAK and their phosphorylated and total protein expression. Taken together with the lack of correlation between expression of the genes encoding Pyk2 and FAK and microglial factors, this finding suggests a microglial-dependent regulation of Pyk2 and FAK signaling in glioma only at the protein expression level. In addition, cytokine/chemokine gene expression in microglia and their respective receptors in glioma were not significantly correlated.
Based on these results, EGF, PDGFα, PDFGβ, IL-6, IL-8, and SDF-1α were selected as candidates for the microglial regulation of Pyk2 and FAK signaling in glioma cells. The effects of these factors on Pyk2- and FAK-related glioma cell proliferation and dispersal were further investigated in three primary human GBM cell lines developed from the analyzed specimens. The three selected cell lines represent several basal levels of Pyk2 and FAK expression. The patient information and cell line characterizations are provided in
Figure S1. Cell line 1 (CL1) is characterized by high expression of total Pyk2 (
Figure S1d) and a low expression level of FAK (
Figure S1f) with moderate phosphorylation of Pyk2 (
Figure S1c) and high phosphorylation of FAK (
Figure S1e). Cell line 2 (CL2) is characterized by low total expression of Pyk2 (
Figure S1d) with moderate phosphorylation (
Figure S1c) and high total FAK expression (
Figure S1f) with low phosphorylation (
Figure S1e). Cell line 3 (CL3) is characterized by moderate total Pyk2 expression (
Figure S1d) with moderate phosphorylation (
Figure S1c) and high total FAK expression (
Figure S1f) with low phosphorylation (
Figure S1e).
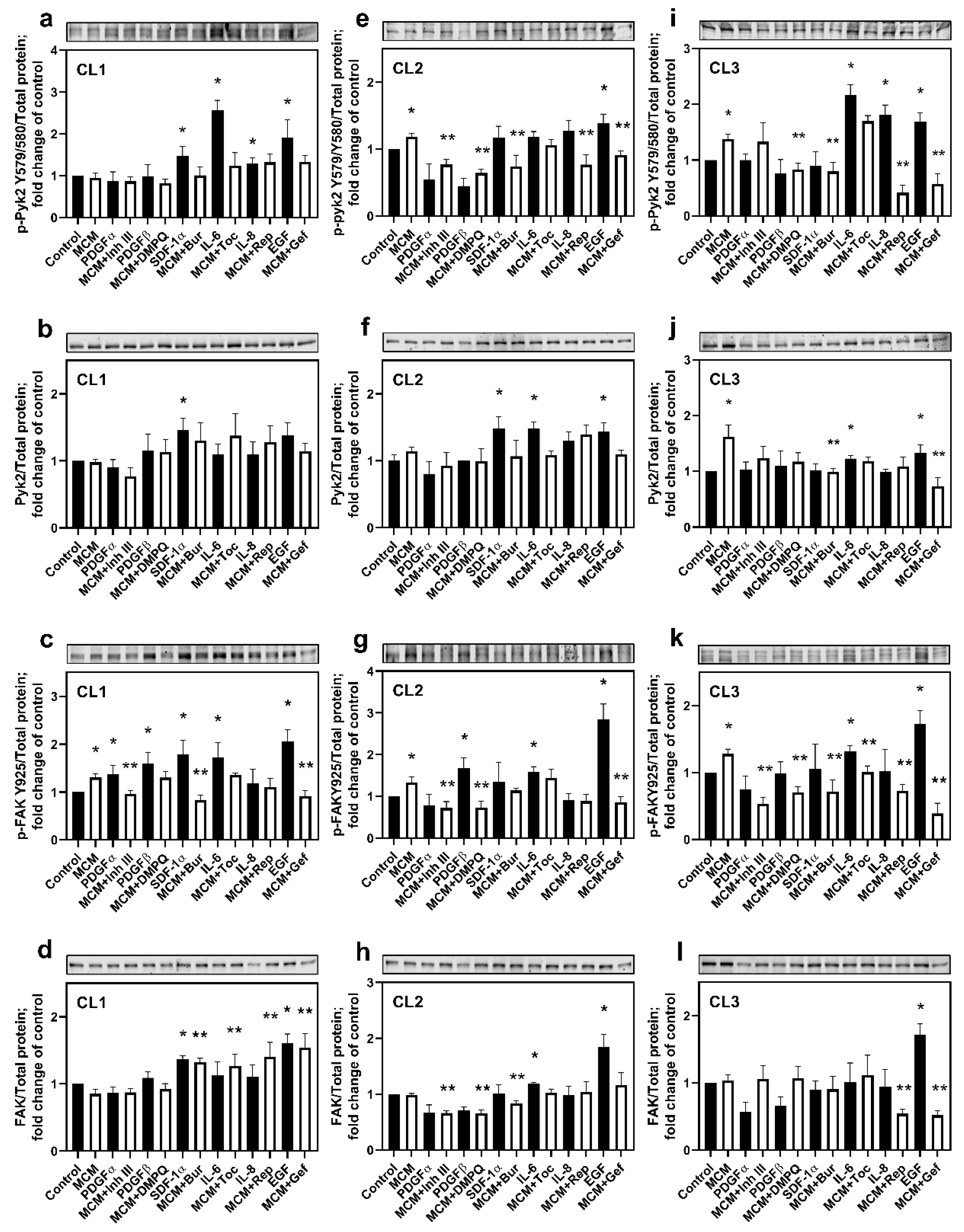
3.2. Cytokines Released by Microglia Activate Pyk2 and FAK Signaling Pathways in Human Glioma Cells
To identify the microglial factors activating Pyk2 and FAK signaling in glioma cells, the phosphorylated (p-Pyk2 (Y579/Y580) and p-FAK (Y925)) and total protein levels of these kinases were evaluated upon cell treatment with MCM or with each cytokine/chemokine identified above (EGF, PDGFα, PDGFβ, SDF-1α, IL-6, and IL-8). Additionally, the effect of treatment with MCM in combination with any of the inhibitors for cytokine/chemokine receptors was evaluated. Because of inter-individual variability, we chose to present the results of these experiments for each cell line individually.
CL1:
Figure 2a shows that SDF-1α, IL-8, IL-6, and EGF significantly upregulated Pyk2 phosphorylation compared with control (≥29% for all,
p < 0.05). From this group, IL-6 induced the strongest activation of Pyk2 phosphorylation (260%,
p < 0.05). The total protein levels of Pyk2 (
Figure 2b) were not significantly increased, except for SDF-1α treatment (47%,
p < 0.05), indicating that the observed increase in phosphorylated Pyk2 in the presence of SDF-1α is due to the increase in total Pyk2 protein and not to increased phosphorylation. EGF also appears to promote an increase in total protein levels of Pyk2, although this effect did not reach statistical significance (38%,
p = 0.07). MCM did not affect the activation of Pyk2 in CL1. Correspondingly, blockers of SDF-1α, IL-6, IL-8, and EGF receptors (burixafor, tocilizumab, reparixin, and gefitinib, respectively) in the presence of MCM did not affect Pyk2 phosphorylation. A possible explanation of this effect is the extremely high autocrine gene expression of SDF-1α, IL-8, and EGF by CL1 glioma cells (
Figure S2b) and, consequently, a high basal level of Pyk2 expression and phosphorylation in this cell line (
Figure S1c). Therefore, microglial-derived cytokines/chemokines did not provide an additive effect for the activation of Pyk2, which was basally supported by autocrine mechanisms. For this reason, cells treated with chemokine/cytokine receptor blockers individually did not significantly affect the activation of Pyk2 due to the strong stimulatory effect of other cytokines released by glioma cells.
In contrast to the insensitivity of p-Pyk2 to MCM, FAK was activated by MCM by 31% compared with control (
p < 0.05,
Figure 2c). Additionally, all cytokines evaluated, except for IL-8, enhanced the phosphorylation of FAK (by ≥38% fold,
p < 0.05 for each). Treatments with inhibitor III, burixafor, and gefitinib reduced the stimulatory effect of MCM (by between 21 and 69%,
p < 0.05 for each), supporting the roles of PDGFα, SDF-1α, and EGF in the MCM-mediated activation of FAK. Despite the stimulatory effect of PDGFβ and IL-6 on FAK phosphorylation, their corresponding inhibitors, DMPQ and tocilizumab, did not reverse MCM-dependent phosphorylation of FAK in this cell line, probably due to the strong stimulatory effect of other chemokines, such as SDF-1α and EGF, that are present at high concentrations in MCM (a cytokine expression profile of microglia treated with medium conditioned from each of the investigated cell lines is provided in
Figure S2a). Changes in the total expression of FAK were observed with SDF-1α and EGF (
Figure 2d, 37 and 60% increase, respectively,
p < 0.05 for each), which were the same as those observed for the total expression of Pyk2 protein (
Figure 2b). Of note, treatment with burixafor, tocilizumab, reparixin, and gefitinib increased total FAK in the presence of MCM by between 55 and 81% (
p < 0.05, for each). These results suggest roles for SDF-1α, IL-6, and EGF in upregulation of both Pyk2 and FAK phosphorylated proteins, while PDGFα and PDGFβ are involved only in FAK regulation when it is related to CL1.
CL2:
Figure 2e shows that MCM increased phosphorylated Pyk2 by 19% compared with control (
p < 0.05). Of all the cytokines and chemokines evaluated, only EGF significantly upregulated Pyk2 phosphorylation (by 39%,
p < 0.05), despite non-significant upregulation of p-Pyk (Y579/Y580), which was also observed for SDF-1α, IL-6, and IL-8. However, inhibitor III, DMPQ, burixafor, reparixin, and gefitinib inhibited the activation of Pyk2 in the presence of MCM by between 23 and 46% (
p < 0.05). This non-significant upregulation of Pyk2 phosphorylation by SDF-1α, IL-6, and IL-8, together with the strong inhibitory effect of the corresponding receptors’ blockers, can be explained by the high autocrine expression of SDF-1α, IL-6, and IL-8 by CL2 (
Figure S2b). The lack of stimulatory effect of PDGFα and PDGFβ on Pyk2 phosphorylation, together with the inhibitory effect of their corresponding blockers on the MCM-dependent upregulation of p-Pyk2 (Y579/Y580), can be explained by the complex recognition between PDGFs and PDGFRs and the multitude of possible PDGF–PDGFR interactions. PDGFRα are activated by homodimeric PDGF-αα, PDGF-ββ, and heterodimeric PDGF-αβ. Additionally, the heterodimerization of PDGFRαβ can be induced by the PDGF-ββ homodimer or the PDGF-αβ heterodimer [
32]. Taking in account this complexity, the application of single PDGFα or PDGFβ may not result in the activation of Pyk2. However, inhibition of either PDGFRα or PDGFRβ may affect PDGFR dimerization and signaling. Deep studies directed at the elucidation of the mechanisms of PDGFR signaling on the activation of Pyk2 are needed. In a frame of the current study, our findings support the involvement of PDFGα, PDGFβ, SDF-1α, IL-6, IL8, and EGF in the activation of Pyk2 by MCM. Moreover, SDF-1α, IL-6, and EGF promoted the elevation of the expression of total Pyk2 (
Figure 2f) to a similar extent (by between 44 and 48%,
p < 0.05).
Figure 2g demonstrates that MCM also upregulated FAK phosphorylation in CL2 compared with control (by 34%,
p < 0.05). Similarly, PDFGβ, IL-6, and EGF increased p-FAK (Y925) expression by 69%, 58%, and 285%, respectively (
p < 0.05). Inhibitor III, DMPQ, and gefitinib partially reversed the effect of MCM on FAK phosphorylation by between 35% and 46% (
p < 0.05), while an inhibitory effect of the IL-6R blocker tocilizumab was not observed. Taking into account the high levels of expression of PDGFβ and EGF, together with the low expression of IL-6 in microglia/CL2-conditioned medium (
Figure S2a), we propose that an inhibitory effect of tocilizumab on FAK phosphorylation was masked by a strong activation effect of PDGFβ and EGF in MCM. These results suggest that FAK is activated by microglia-derived PDGFβ, IL-6, and EGF in CL2.
Figure 2h indicates an increase in total FAK expression induced by IL-6 and EGF compared with control (by 19% and 84%, respectively,
p < 0.05). Treatment with inhibitor III, DMPQ, or burixafor decreased total expression of the kinase (by 33% for inhibitor III or DMPQ and by 15% for burixafor,
p < 0.05), which is consistent with FAK phosphorylated protein modifications in MCM-treated cells. These findings support the role for EGF, PDGFβ, SDF-1α, IL6, and IL-8 in the activation of Pyk2 and FAK signaling in CL2.
CL3:
Figure 2i illustrates that, as with CL2, p-Pyk2 (Y579/Y580) in CL3 was significantly upregulated by MCM compared with control (by 37%,
p < 0.05). Consistent with the results in CL1, activation of p-Pyk2 (Y579/Y580) was observed in the presence of IL-6 (217%), IL-8 (81%), and EGF (68%,
p < 0.05 for each). The MCM-dependent increase in phosphorylated Pyk2 was significantly decreased by DMPQ, burixafor, reparixin, and gefitinib (≥40% for all,
p < 0.05), supporting the involvement of PDGFβ and SDF-1α in the activation of Pyk2, in addition to IL-6, IL-8, and EGF. The lack of inhibitory effect of the IL-6R blocker tocilizumab in the presence of MCM, together with the strong Pyk2 (Y579/Y580) upregulation in response to IL-6, can be explained by the high expression level of EGF and IL-8 in microglia/CL3 conditioned medium (
Figure S2a). The lack of effect of PDGFβ and SDF-1α on Pyk2 phosphorylation, together with the inhibitory effect of their corresponding receptors’ blockers, DMPQ and burixafor, might be due to the low expression level of PDGFRβ and CXCR12, together with their autocrine activation, which may exceed the receptor activation capacity (
Figures S1b and S2b). However, significant downregulation of Pyk2 phosphorylation by DMPQ and burixafor indicates the important role of PDGFβ and SDF-1α in the regulation of Pyk2 signaling.
In contrast to CL1 and CL2, a significant increase in total Pyk2 was observed in MCM-treated cells compared with control (by 62%,
p < 0.05,
Figure 2j). Both IL-6 and EGF enhanced this parameter by 23% and 34%, respectively (
p < 0.05). Treatment with burixafor and gefitinib reduced the total expression of Pyk2 in MCM-treated cells (by 39% and 55%, respectively,
p < 0.05).
Treatment with MCM also upregulated FAK phosphorylation by 30% (
p < 0.05,
Figure 2k). Similar to observations in CL1 and CL2, IL-6 and EGF activated FAK by 32% and 73%, respectively (
p < 0.05). Of note, the activating effect of MCM was reversed by inhibitor III, DMPQ, burixafor, reparixin, and gefitinib (by ≥21% for all,
p < 0.05), suggesting a role for PDGFα, PDGFβ, SDF-1α, and IL-8 in the activation of FAK signaling. Total FAK expression was increased by EGF by 72% compared with control (
Figure 2l). No effect of MCM on this parameter was observed. However, reparixin and gefitinib reversed the effect of MCM on total FAK expression (by 50% for both,
p < 0.05).
Original uncropped western blot images and loading controls with REVERT Total Protein Stain are presented in
Figures S3 and S4.
These results suggest that the activation of Pyk2 and FAK signaling is mostly IL-6- and EGF-dependent in CL3, while PDGFβ, SDF-1α, and IL-8 also contribute to Pyk2 and FAK phosphorylation. Altogether, these findings indicate that, in all three evaluated specimens, Pyk2 and FAK signaling pathway activation significantly depend on microglia-derived EGF, PDGFβ, SDF-1α, IL-6, and IL-8, although the extent of their regulatory contribution differs from patient to patient.
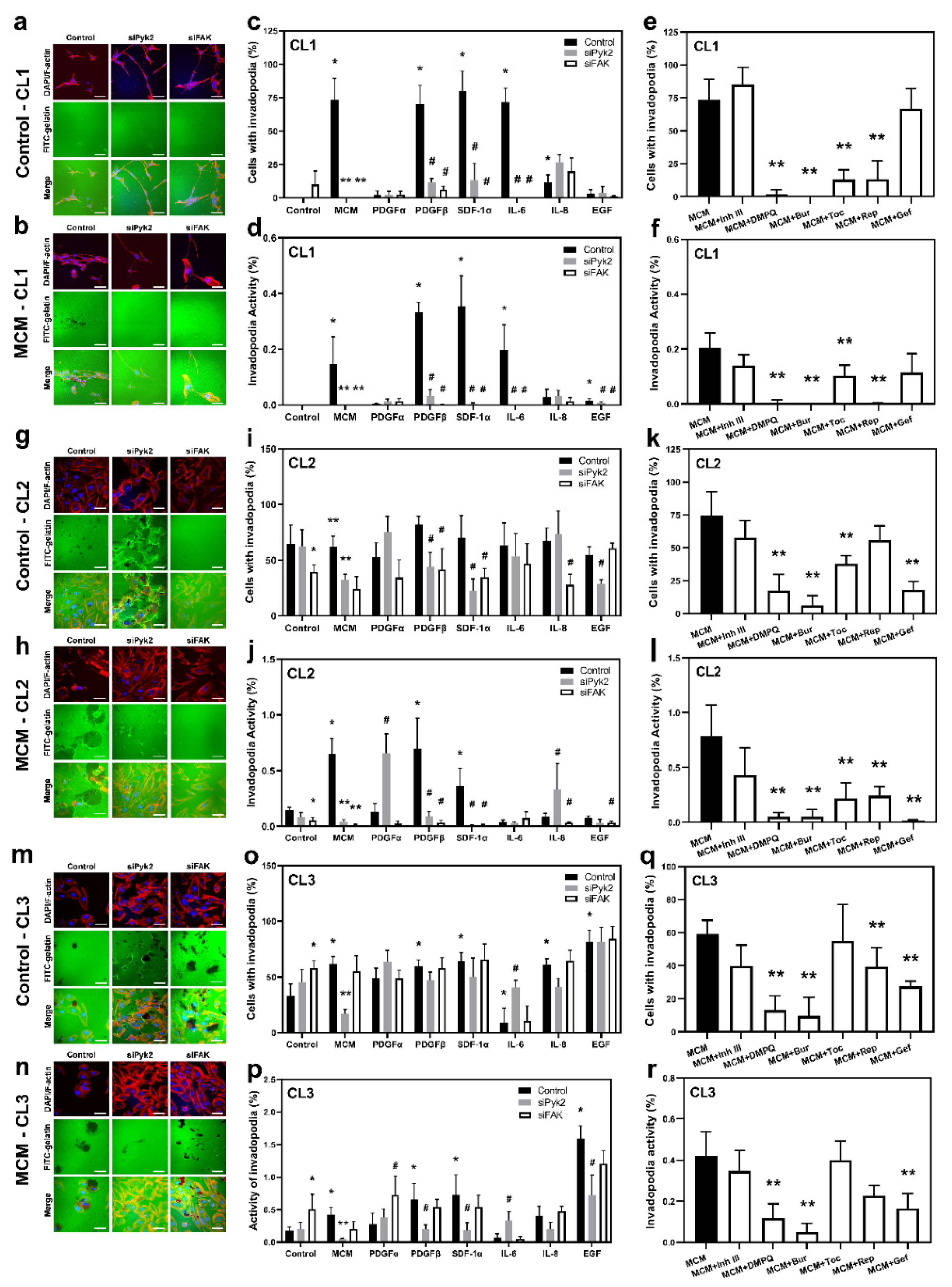
3.3. Cytokines/Chemokines Released by Microglia Promote Glioma Cell Extracellular Matrix Degradation through Pyk2 and FAK Signaling
Glioma cell invasion is a complex process composed of extracellular matrix degradation and migration of the cell. As different intracellular and extracellular signaling pathways can be involved, in this study, the regulation of matrix degradation and migration were investigated independently.
Invadopodia are actin-rich membrane protrusions that serve as mediators of cell invasion [
33]. To evaluate the role of the cytokines and chemokines investigated in the Pyk2- and FAK-dependent formation of functional invadopodia and extracellular matrix degradation, a FITC-conjugated gelatin-degradation assay was performed. The number of cells that form invadopodia (invadopodia formation, IF) and the portion of gelatin matrix degradation (invadopodia activity, IA) were evaluated for the CL1, CL2, and CL3 cell lines. siRNA knockdown against Pyk2 or FAK, together with mock control cells, were treated with MCM or any one of the cytokines investigated (EGF, IL-6, IL-8, SDF-1α, PDGFα, or PDGFβ), as well as with MCM in combination with the corresponding inhibitors of cytokine receptors (gefitinib, tocilizumab, reparixin, burixafor, inhibitor III, or DMPQ). The efficacy of Pyk2 and FAK knockdown is presented in
Figure S5).
CL1: Glioma cells from CL1 did not exhibit IF and extracellular matrix degradation under control conditions (
Figure 3c,d). However, treatment with MCM induced both IF and IA, with 73% of the cells forming invadopodia and 0.15% degradation of extracellular matrix (
p < 0.05). siPyk2 and siFAK knockdown abolished this effect of MCM (
Figure 3b–d). These results demonstrate a stimulatory effect of MCM on functional invadopodia formation in glioma cells that is Pyk2 and FAK dependent.
The results presented in
Figure 3c demonstrate that PDGFβ, SDF-1α, and IL-6 increased IF in the mock control experimental group to a similar extent as MCM treatment (70%, 83%, and 91% of cells formed invadopodia, respectively,
p < 0.05,
Figure 3b), and there was an increase in IA to 0.32%, 0.35%, and 0.2% of degraded matrix, respectively, compared with the complete lack of matrix degradation in control (
Figure 3d). IL-8 slightly increased both parameters (by 12% IF and by 0.05% IA), although it did not reach statistical significance (
p = 0.37). The stimulatory effect of PDGFβ, SDF-1α, and IL-6 on IF and IA was reversed by either siPyk2 or siFAK knockdown. Inhibitors of the corresponding receptors, DMPQ, burixafor, tocilizumab, and reparixin, eliminated the stimulatory effect of MCM on IF and IA (
Figure 3e,f), supporting the involvement of PDGFβ, SDF-1α, IL-6, and IL8 in MCM-induced CL1 glioma cell invasion. These findings suggest that Pyk2 and FAK are key regulators of IF and IA in CL1 through PDGFβ, SDF-1α, and IL-6.
CL2: In contrast to CL1, CL2 presented strong IF and AI under basal conditions, which are FAK dependent (
Figure 3g,i,j). Indeed, siFAK knockdown reduced the IF by 39% (
p < 0.05) compared with the control group, together with a 60% reduction in AI. Despite the fact that Pyk2 knockdown did not significantly affect IF and IA compared with the control, it increased stress-fiber tension, inducing cells to pull and tear the extracellular matrix (
Figure 3g). The total number of cells with invadopodia was not affected by MCM (
Figure 3h,i); however, AI resulted in an increase in extracellular matrix degradation by 430% (
p < 0.05) compared with the control group in a manner that is Pyk2- and FAK-dependent (a 94% decrease,
p < 0.05,
Figure 3j). Similarly, siPyk2 and siFAK reduced IF by 47 and 61%, respectively, following MCM treatment (
p < 0.05). Of note, siRNA knockdown against Pyk2 increased the tension of cell attachment to the extracellular matrix in MCM treatment in the same way as in the control group, although to a lesser extent. Therefore, Pyk2 could be involved in the regulation of cell detachment, which is important for the cell migration process. In addition, the reduced number of cells forming invadopodia after siPyk2 and siFAK knockdown indicates the relevance of Pyk2 and FAK in MCM-induced glioma invasion.
Figure 3i demonstrates that none of the cytokines evaluated in CL2 significantly affected IF compared with controls. However, PDGFβ and SDF-1α increased the IA by 4.7- and 2.5-fold, respectively, compared with control (
p < 0.05,
Figure 3f). This stimulatory effect was abolished by siPyk2 and siFAK (by ≥87% for both kinases,
p < 0.05), indicating that they are parts of the PDGFβ and SDF-α signaling pathways controlling the activity of invadopodia. No significant changes were observed in IF and in AI in the presence of EGF, PDGFα, IL-6, and IL-8. For these treatments, siPyk2 knockdown resulted in increased tension to the matrix, the same as observed in the siPyk2 control group, indicating that these factors are not involved in the regulation of invadopodia in CL2.
Interesting results were obtained from cell treatment with MCM in combination with cytokine/chemokine blockers (
Figure 3k,l). While an increase in AI was detected for PDGFβ and SDF-1α treatments only (
Figure 3j), the MCM effect on IA was abrogated by their corresponding receptor blockers, DMPQ and burixafor, and also by inhibitors of EGFR, IL-6R, and IL-8R (gefitinib, tocilizumab, and reparixin, respectively; by ≥50% for all,
p < 0.05;
Figure 3l), indicating the autocrine regulation of IF and IA. These findings suggest that both Pyk2 and FAK signaling, mediated by PDGFβ and SDF-1α, are involved in the regulation of IF and IA (with possible autocrine regulation by EGF, IL-6, and IL-8) in CL2. In addition, the regulation of extracellular matrix detachment could also be mediated by Pyk2.
CL3: IF and AI under basal conditions in CL3 were similar to what was observed for CL2 (
Figure 3m,o,p). In contrast to CL2, silencing of FAK under control conditions increased both IF by 69% (
p < 0.05) and AI (by 280%,
p < 0.05), suggesting an anti-matrix degradation role of this kinase in CL3. Pyk2 knockdown led to increased matrix attachment and stress-fiber tension, resulting in folding and tearing of the matrix, similar to CL2 (
Figure 3m). As shown in CL1, treatment with MCM increased IF and IA (by 78% and 83%, respectively,
p < 0.05) in CL3, and this activation was reversed by Pyk2 knockdown, but not by FAK knockdown (
Figure 3n–p). Indeed, siPyk2 decreased the stimulatory effect of MCM on IF by 4-fold, and IA by 10-fold (
p < 0.05). These findings indicate that in CL3 both Pyk2 and FAK are involved in the regulation of extracellular matrix degradation, with Pyk2 acting as a booster of matrix degradation and cell mobility, and FAK reducing the activity of matrix degradation.
Figure 3o demonstrates that EGF, PDGFβ, SDF-1α, and IL-8 increased the number of cells with invadopodia (by ≥170% for all,
p < 0.05). A stimulatory effect of EGF, PDGFβ, and SDF-1α was also observed in IA (by 890%, 370%, and 400%, respectively,
p < 0.05 for each;
Figure 3p). This effect was inhibited by siPyk2 (by ≥ 55% for all,
p < 0.05), indicating that Pyk2 is part of the EGF, PDGFβ, and SDF-1α signaling pathways in the promotion of cell invasion. Additionally, the inhibitors of EGFR, PDGFβR, and SDF-1αR (gefitinib, DMPQ, and burixafor) and, to a lesser extent, the inhibitor of IL-8R (reparixin), significantly reduced FI and AI in the presence of MCM (by ≥300% for gefitinib, DMPQ, and burixafor, and by 30% for reparixin,
p < 0.05;
Figure 3q,r), supporting an important role for microglia-derived EGF, PDGFβ, SDF-1α, and IL-8 in extracellular matrix degradation in CL3. By contrast, IL-6 reduced IF by 73% compared with control (
p < 0.05). IL-6 in the presence of Pyk2 knockdown resulted in increased damage to the extracellular matrix caused by stress-fiber tension. This observed effect does not allow us to determine whether Pyk2 signaling in CL3 is mediated by IL-6. However, considering the lack of effect by tocilizumab in MCM-stimulated IF and IA, the role of IL-6 in microglia-driven extracellular matrix degradation in CL3 is negligible.
Altogether, these findings suggest that in the three evaluated cell lines, both Pyk2 and FAK signaling are involved in the regulation of extracellular matrix degradation, with microglia-derived PDGFβ and SDF-1α being common upstream regulators of Pyk2 and FAK for all three cell lines. EGF, IL-6, and IL-8 are also involved in Pyk2- and FAK-dependent extracellular matrix degradation; however, the extent of their involvement is patient dependent.
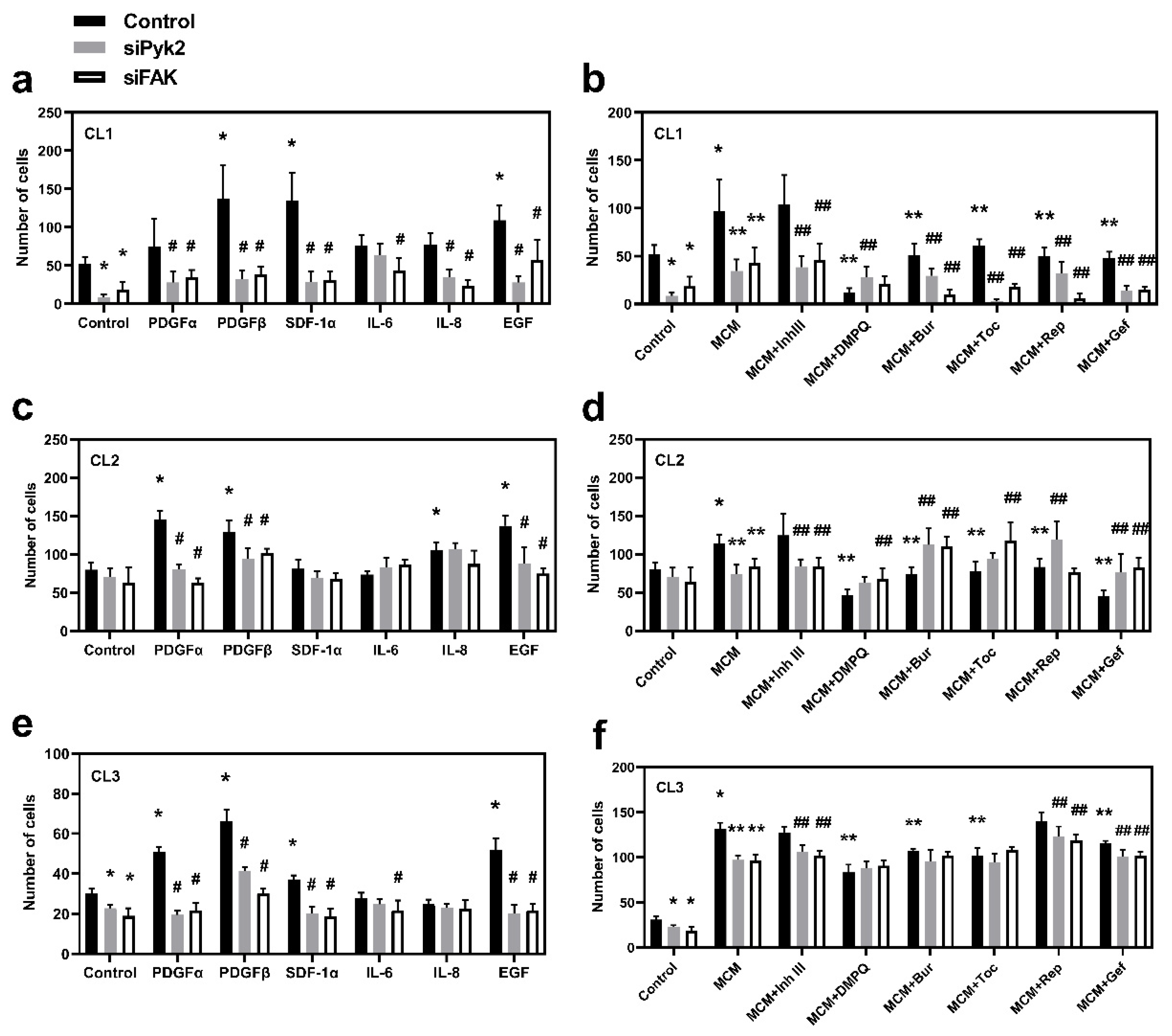
3.4. Cytokines/Chemokines Released by Microglia Promote Glioma Cell Migration through Pyk2 and FAK Signaling
Transwell migration assays were performed for all three cell lines investigated in order to evaluate the role of Pyk2 and FAK, as well as to identify their extracellular upstream regulators, which lead to increased migration capacity of glioma cells. Mock cells and cells that were siRNA-transfected against Pyk2 or FAK were used for the assay with PDGFα, PDGFβ, SDF1α, EGF, IL6, IL8, or MCM or MCM in combination with corresponding chemokine/cytokine receptor inhibitors in the lower compartment. The migration times were found to be 23 h for CL1 and 8 h for both CL2 and CL3.
A significant reduction in migration in Pyk2- and FAK-knockdown cells under basal conditions was detected in CL1 and CL3 (
Figure 4a,e). A reduction in migration was also observed in CL2; however, this did not reach statistical significance. Of note, in all the investigated cell lines, stimulation with PDGFβ or EGF drastically increased migration, and this effect was eliminated by either Pyk2 or FAK knockdown (
Figure 4a,c,e). In addition, PDGFα induced the Pyk2- and FAK-dependent activation of migration in CL2 and CL3 (
Figure 4c,e), while SDF-1α induced the Pyk2- and FAK-dependent activation of migration in CL1 and CL3 (
Figure 4a,e).
MCM resulted in 30–300% upregulation of migration in all cell lines (
Figure 4b,d,f;
p < 0.05). Knockout of either Pyk2 or FAK reduced this effect. Inhibitors of PDGFRβ, EGFR, or SDF-1αR (DMPQ, gefitinib, and burixafor, respectively) significantly reduced the stimulatory effect of MCM in all cell lines, confirming the key role of microglia-derived PDGFβ, EGF, and SDF-1α in glioma cell migration (
Figure 4b,d,f). No additive effects of Pyk2 and FAK knockdown in DMPQ treatment were detected in any of the cell lines, indicating that PDGFβ stimulates migration through both Pyk2 and FAK. The additive effect of Pyk2 and FAK knockdown in gefitinib and burixafor treatment was detected in CL1 (
Figure 4b), but not in other cell lines, indicating that pathways other than the Pyk2 and FAK pathways may be involved in EGF- and SDF-1α-mediated activation of migration in CL1. The lack of effect of SDF-1α in CL2, together with the strong inhibitory effect of burixafor (
Figure 4d), could be a consequence of the significant autocrine expression of SDF-1α in CL2, which was confirmed by PCR analysis (
Figure S2b). Therefore, SDF1α, together with EGF and PDGFβ, can be classified as a key microglia-derived stimulator of migration in all cell lines.
Despite the fact that PDGFα significantly stimulated migration in CL2 and CL3 (
Figure 4a,e), inhibitor III (blocker of PDGFRα) did not reduce the effect of MCM in any of the investigated cell lines (
Figure 4b,d,f). RT-PCR analysis of cytokine/chemokine expression in microglia pre-activated with medium conditioned from glioma cells, did not detect expression of PDGFα in microglia pre-activated by CL2 and CL3 but detected trace expression of PDGFα in microglia pre-activated by CL1 (
Figure S2a). PDGFα gene expression was also not detected in any of the glioma cell lines investigated (
Figure S2b). These results suggest that, despite the findings that PDGFα can be actively involved in the regulation of glioma cell migration and that this regulation is carried out through Pyk2 and FAK signaling, the absence or low expression of PDGFα in the glioma–microglia microenvironment reduces the role of this factor in microglia-driven glioma cell migration.
3.5. Microglial-Derived EGF, SDF1α, and IL-8 Stimulate Glioma Cell Viability
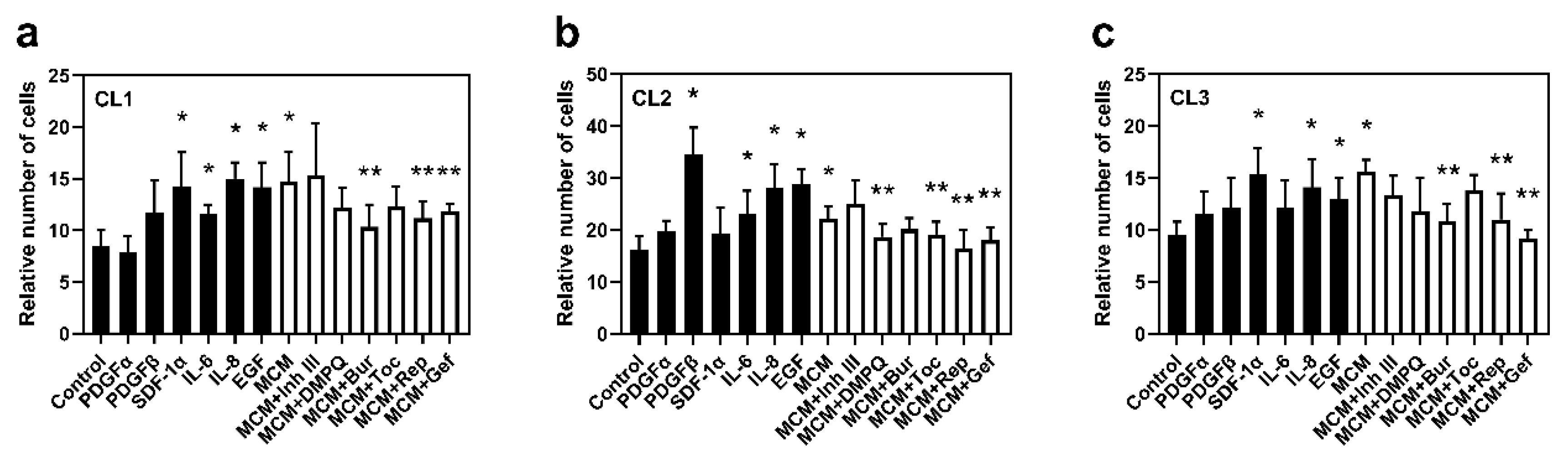
To determine the role of microglia-derived factors on glioma cell viability, the trypan blue exclusion test was used. Cells were incubated for 72 h with the following treatments: control, PDGFα, PDGFβ, EGF, SDF-1α, IL-6, IL-8, MCM, and MCM in combination with inhibitors of the corresponding receptors.
A significant increase in the number of viable cells after 72 h of incubation with MCM compared with control was observed in all investigated cell lines (by 74% in CL1, 37% in CL2, and 64% in CL3,
p < 0.05 for each,
Figure 5). Incubation with EGF or IL-8 resulted in a significant increase in the number of cells at 72 h compared with control in all cell lines (by 37–78%,
p < 0.05). The EGFR and IL-8R blockers, gefitinib and reparixin, respectively, abolished the MCM-enhanced viability effect, reducing the number of viable cells under each treatment in all cell lines (by 19–41%,
p < 0.05).
SDF1α increased viability in CL1 and CL3 (by 68 and 62%, respectively,
p < 0.05;
Figure 5a,c), while burixafor reduced the stimulatory effect of MCM in all investigated cell lines. Considering the strong autocrine gene expression of SDF-1α in CL2 (
Figure S2b), the absence of an effect of SDF1α on cell viability in this cell line could be due to the saturation of CXCR4 (also known as SDF-1αR) receptor activation. Therefore, SDF1α, together with EGF and IL-8, are key regulators of cell viability in the glioma–microglia microenvironment, in all cell lines.
IL-6 upregulated cell viability in CL1 and CL2. This result is correlated with the reduction in the MCM effect by the inhibitor of IL-6R, tocilizumab, in CL1 and CL2 (by 16 and 13%, respectively, p < 0.05) but not in CL3, indicating that cell viability in CL3 is IL-6-independent.
PDGFβ significantly increased cell viability only in CL2, promoting a 2.1-fold increase (p < 0.05). In CL1 and CL3, a non-significant increase in cell viability was observed under PDGFβ stimulation. Correspondingly, DMPQ significantly reduced the effect of MCM in CL2 only (a 16% decrease, p < 0.05) along with a non-significant reduction in CL1 and CL3, suggesting that PDGFβ could be involved in microglia-driven glioma cell viability. However, the extent of this regulation is patient dependent.
3.6. Microglia-Derived EGF and IL-6 Induce Mitosis through Pyk2 and FAK Signaling
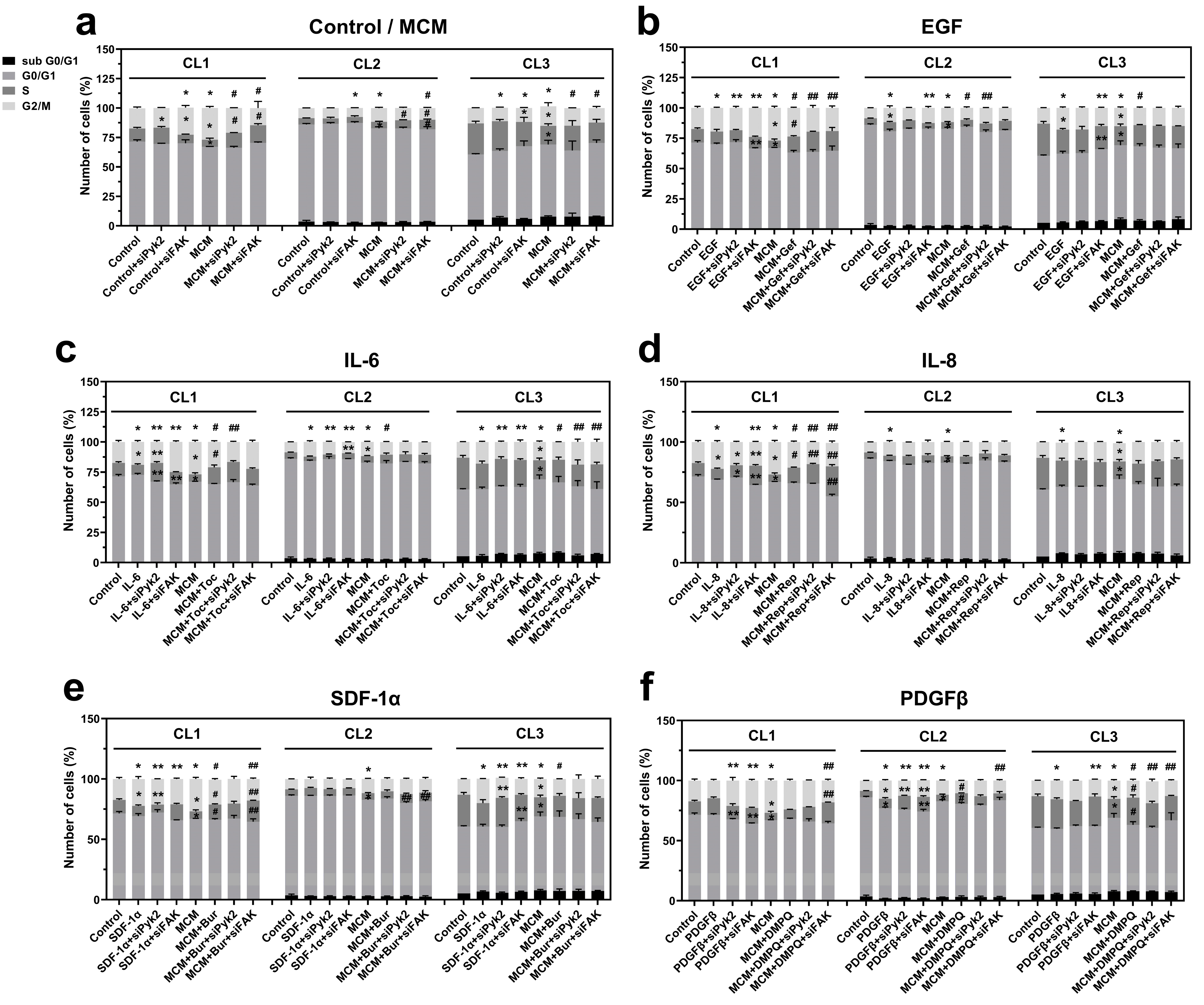
In order to further investigate the role of Pyk2 and FAK in microglia-driven glioma cell proliferation and to identify the upstream signaling mechanisms, cell cycle analysis was performed for mock control and siRNA knockdown against Pyk2 and FAK cells. Cells were incubated for 24 h with the following treatments: control, PDGFα, PDGFβ, EGF, SDF-1α, IL-6, IL-8, MCM, and MCM in combination with each of the inhibitors of the corresponding receptors. The cell distribution at defined cell cycle phases was analyzed by flow cytometry.
Cell cycle analysis of glioma cells with knockdown of Pyk2 or FAK revealed that the involvement of Pyk2 and FAK in cell cycle regulation is patient dependent. In CL1, FAK knockdown resulted in significant accumulation of cells in G2/M (35%,
p < 0.05) and a strong reduction in cells in the S phase (35%,
p < 0.05;
Figure 6a), indicating cell cycle arrest at G2/M. In CL2, FAK silencing decreased the percentage of cells in the G2/M phase by 12% (
p < 0.05) and increased the accumulation of cells in G1 (3%,
n.s.
p = 0.58), indicating that, in CL1 and CL2, FAK is involved in the regulation of cell cycle progression and stimulates cell proliferation. The dramatic effect of FAK silencing in CL1 compared with CL2 can be explained by the two-fold stronger activity of FAK signaling in CL1 compared with CL2 (
Figure S1e).
Silencing of Pyk2 did not affect the cell cycle in CL1 and CL2. However, in CL3, siPyk2 reduced the number of cells in G2/M by 9% (
p < 0.05), while FAK silencing resulted in an accumulation of cells in G1, a reduction in cells in the S phase, and a nonsignificant reduction in cells in G2/M (
Figure 6a). This result indicates that in CL3, Pyk2 has a dominant role over FAK in the stimulation of cell division, and this can be related to the strong (three-fold) activation of Pyk2 phosphorylation in CL3 compared with CL1 and CL2, together with low activation of FAK. These results suggest that both Pyk2 and FAK may be involved in modulation of the cell cycle in glioma cells under basal conditions, leading to the activation of proliferation. The extent of this modulation and the prevalence of one signaling pathway over the other is patient dependent.
MCM increased the percentage of cells in the G2/M phase compared with control in all evaluated patients (by between 25 and 52%,
p < 0.05;
Figure 6a). The reduction in cells in the G1 and S phases and the increase in viability in response to MCM treatment (
Figure 5) confirms the stimulatory effect of MCM on glioma cell proliferation. This effect was reversed by silencing of FAK in all investigated cell lines and also partially by siPyk2 in CL1 and CL3. These results demonstrate that MCM promotes cell proliferation by stimulating cell cycle progression through both the FAK and Pyk2 signaling pathways.
To identify the microglial-released factors involved in the stimulation of cell proliferation through FAK and Pyk2 signaling, glioma cells were treated with previously selected cytokines/chemokines, including EGF, IL-6, IL-8, SDF-1α, PDGFα, PDGFβ, or MCM in combination with one of their respective blockers (gefitinib, tocilizumab, reparixin, burixafor, inhibitor III, or DMPQ, respectively). As demonstrated in
Figure 6b–e, administration of EGF, IL-6, IL-8, and SDF-1α led to an increased number of cells in the G2/M phase compared with control in all evaluated cell lines, which is consistent with the cell viability results. PDGFα increased mitosis in only CL3 (
Figure S6), and PDGFβ increased mitosis in CL2 and slightly in CL3 (
Figure 6f). These results are consistent with the significant increase in viability observed in response to these factors (
Figure 5b,c).
The stimulatory effect of EGF on mitosis (
Figure 6b) was reversed by Pyk2 silencing in CL2 (decreased by 16%,
p < 0.05) and by FAK silencing in CL3 (decreased by 14%,
p < 0.05). In CL1, FAK knockdown in the presence of EGF resulted in G2/M arrest, similar to what was observed under basal conditions (
Figure 6a). Treatment with gefitinib inhibited the stimulatory effect of the G2/M phase by MCM (decreased by 7% in CL1, 17% in CL2, and 9% in CL3,
p < 0.05 for each), suggesting a significant contribution of the EGF stimulation of G2/M by MCM. Silencing of Pyk2 and FAK did not significantly add to the inhibitory effect of gefitinib in MCM in CL2 and CL3, indicating that in these cell lines, EGF is a key regulator of Pyk2 and FAK signaling, leading to cell cycle modulation. However, in CL1, both Pyk2 and FAK silencing provided an additive effect to gefitinib inhibition, indicating that in this cell line, factors other than EGF are involved in the Pyk2- and FAK-dependent regulation of the cell cycle. Therefore, these results suggest that the proliferative effect of EGF is mediated by both FAK and Pyk2 in all patients, but to different extents.
As depicted in
Figure 6c, IL-6 increased the number of cells in the G2/M phase in all evaluated patients (by between 10 and 45%,
p < 0.05). This effect was inhibited by siPyk2 in all patients and by siFAK in CL2 and CL3, while in CL1, siFAK resulted in G2/M arrest, as shown under basal conditions (
Figure 6a). These results suggest that Pyk2 and FAK are part of the proliferative mechanism of IL-6. Treatment with tocilizumab blocked the MCM-dependent stimulation of the G2/M phase in all evaluated patients, indicating that the release of this cytokine by microglia is critical for stimulation of the cell cycle. Under MCM + tocilizumab conditions, siPyk2 induced a greater inhibition of the G2/M phase compared with tocilizumab alone (by 21%,
p < 0.05) in CL1, indicating that IL-6 is involved in cell cycle regulation through Pyk2-dependent and -independent signaling mechanisms. siFAK in CL1 and CL2, as well as Pyk2 silencing in CL2, did not have additive effects in the presence of tocilizumab, suggesting that, in CL2, the main route for IL6′s regulation of the cell cycle is through Pyk2 and FAK. An interesting result was observed in CL3, in which siFAK and siPyk2 reduced the inhibitory effect of tocilizumab by 21% (
p < 0.05), probably as a result of over-inhibition. These results suggest that IL6 is an important regulator of glioma cell proliferation, but the extent of involvement of FAK and Pyk2 signaling in the process is patient dependent.
The stimulatory effect of IL-8 was slightly reduced by both Pyk2 and FAK silencing (decreased by 13%,
p < 0.05) in CL1, while in CL2 and CL3, Pyk2 and FAK silencing had no effect on IL-8-driven mitosis (
Figure 6d), suggesting that these kinases have little or no involvement in the mechanisms of the IL8-dependent activation of mitosis. A role for IL-8 in MCM-driven cell proliferation in CL1 was demonstrated by the inhibitory effect of reparaxin in the G2/M phase compared with MCM alone (decreased by 19%,
p < 0.05). However, under MCM + reparaxin conditions, the silencing of FAK and Pyk2 induced a greater inhibition of the G2/M phase (decreased by 5% and 15%, respectively,
p < 0.05), suggesting that IL8 is just a minor contributor to the Pyk2/FAK regulation of the cell cycle.
As shown in the
Figure 6e, SDF-1α increased the number of cells in G2/M by 33 and 64% in CL1 and CL3, respectively, compared with control (
p < 0.05, for each), and did not significantly affect CL2. siFAK and siPyk2 reversed the effect of SDF-1α in CL1 and CL3, suggesting a critical role for these kinases in the promotion of cell proliferation by SDF-1α. In the CL1 and CL3 cell lines, burixafor abolished MCM-induced mitosis. siFAK in the presence of MCM + burixafor resulted in a reduction in G2/M by 16% (
p < 0.05) while increasing the number of cells in the S phase by 33% (
p < 0.05) in CL1, suggesting that in this cell line, SDF-1α can affect mitosis through FAK-dependent and -independent signaling pathways. However, no significant additive effects were observed with siPyK and siFAK in CL3 or siPyk2 in CL1 compared with MCM + burixafor. These results suggest that in two of three cell lines, SDF-1α serves as a key regulator of mitosis. In addition, Pyk2 and FAK could serve as a main route of SDF-1α-driven proliferation, and other signaling pathways could also be involved.
As shown in
Figure 6f, PDFGβ increased the number of cells in the G2/M phase in CL2 and CL3 compared with control (by 75% and 25%, respectively,
p < 0.05). The effects of PDFGβ appear to be mediated by FAK and Pyk2 in CL2, because siFAK and siPyk2 decreased the number of cells in the G2/M phase compared with PDGFβ alone (decreased by 9% and 17%, respectively,
p < 0.05). However, in CL3, the PDGFβ-mediated proliferative effect seems to be Pyk2-independent, because siPyk2 did not diminish the effect of PDGFβ. Treatment with DMPQ reduced the number of cells in the G2/M phase by 10% (
p < 0.05), while arresting the cells in the S phase (increased by 13% and 36%, for CL2 and CL3, respectively,
p < 0.05). Under conditions of MCM + DMPQ, siFAK did not show an additive inhibitory effect in CL2, suggesting that this is the main route for PDGFβ in the regulation of mitosis, while siPyk2 increased the G2/M phase in CL2, suggesting an over-inhibition effect. In CL3, siFAK in combination with MCM + DMPQ treatment decreased the G2/M phase further than MCM + DMPQ treatment alone (decreased by 10%,
p < 0.05), suggesting that other pathways may be involved in this process.
PDGFα induced cell proliferation only in CL3, which is consistent with the significant increase in viability in this cell line (
Figure 5c). Indeed, the G2/M phase was increased 52% by PDGFα compared with control (
p < 0.05,
Figure S4). This effect appears to be FAK/Pyk2 dependent because the silencing of both kinases decreased the G2/M phase by 14% and 20%, respectively (
p < 0.05 for both). Inhibitor III, in the presence of MCM, decreased the number of cells in the G2/M phase by 9% and arrested cells in the S phase (38% increase,
p < 0.05) compared with MCM alone. Under conditions of MCM + inhibitor III, siPyk2 further decreased the number of cells in the G2/M phase (decreased by 9%,
p < 0.05), suggesting the involvement of signaling pathways other than Pyk2. By contrast, siFAK stimulated mitosis by 9%, (
p < 0.05) compared with the MCM + inhibitor III group, probably by over-inhibition.
In conclusion, although there is heterogenicity in the cytokine-dependent pathways involved in cell cycle regulation in glioma, our results support a pivotal role of FAK and Pyk2 in this process. In contrast to PDGFα, PDGFβ, and SDF-1α, whose functions appear to be patient-specific, EGF and IL-6 are implicated in Pyk2- and FAK-mediated glioma cell proliferation in all subjects evaluated.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





