
Genome-Wide Profiling Reveals HPV Integration Pattern and Activated Carcinogenic Pathways in Penile Squamous Cell Carcinoma

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. PSCC Patients and Samples
2.2. Library Preparation and HIVID
2.3. Bioinformatic Analysis for HPV Integration
2.4. Immunohistochemistry
2.5. Cell Culture, siRNA/Plasmid Transfection, and Cell Proliferation Assay
2.6. Migration and Invasion Assay
2.7. RNA Extraction and qPCR
2.8. Statistical Analysis
3. Results
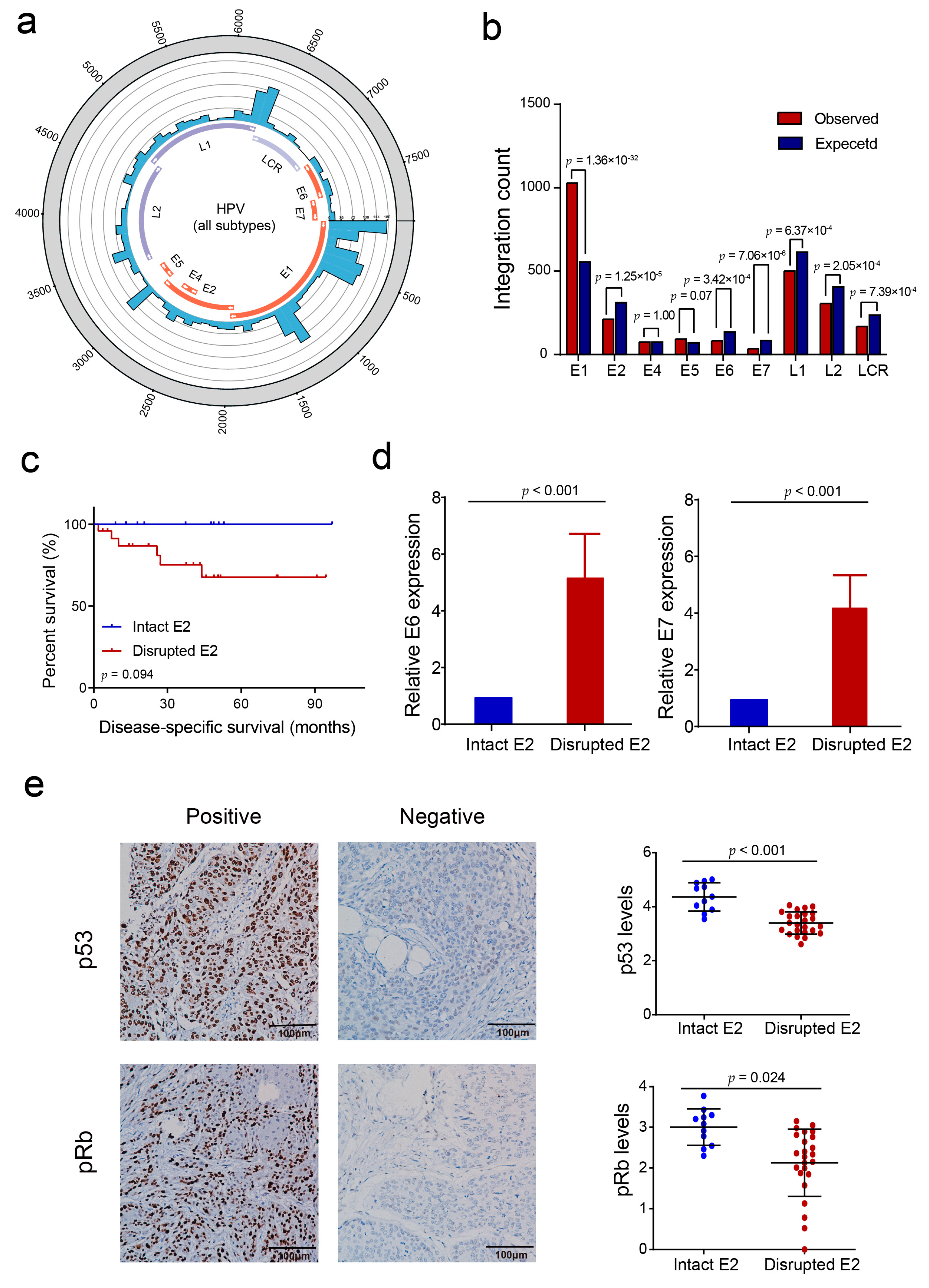
3.1. HPV Integration Status and HPV Subtypes in PSCC
3.2. HPV Integration Pattern with Respect to the Human PSCC Genome
3.3. HPV E2 Disruption by Viral DNA Integration Is Not a Necessary Event in HPV-Induced Penile Carcinogenesis
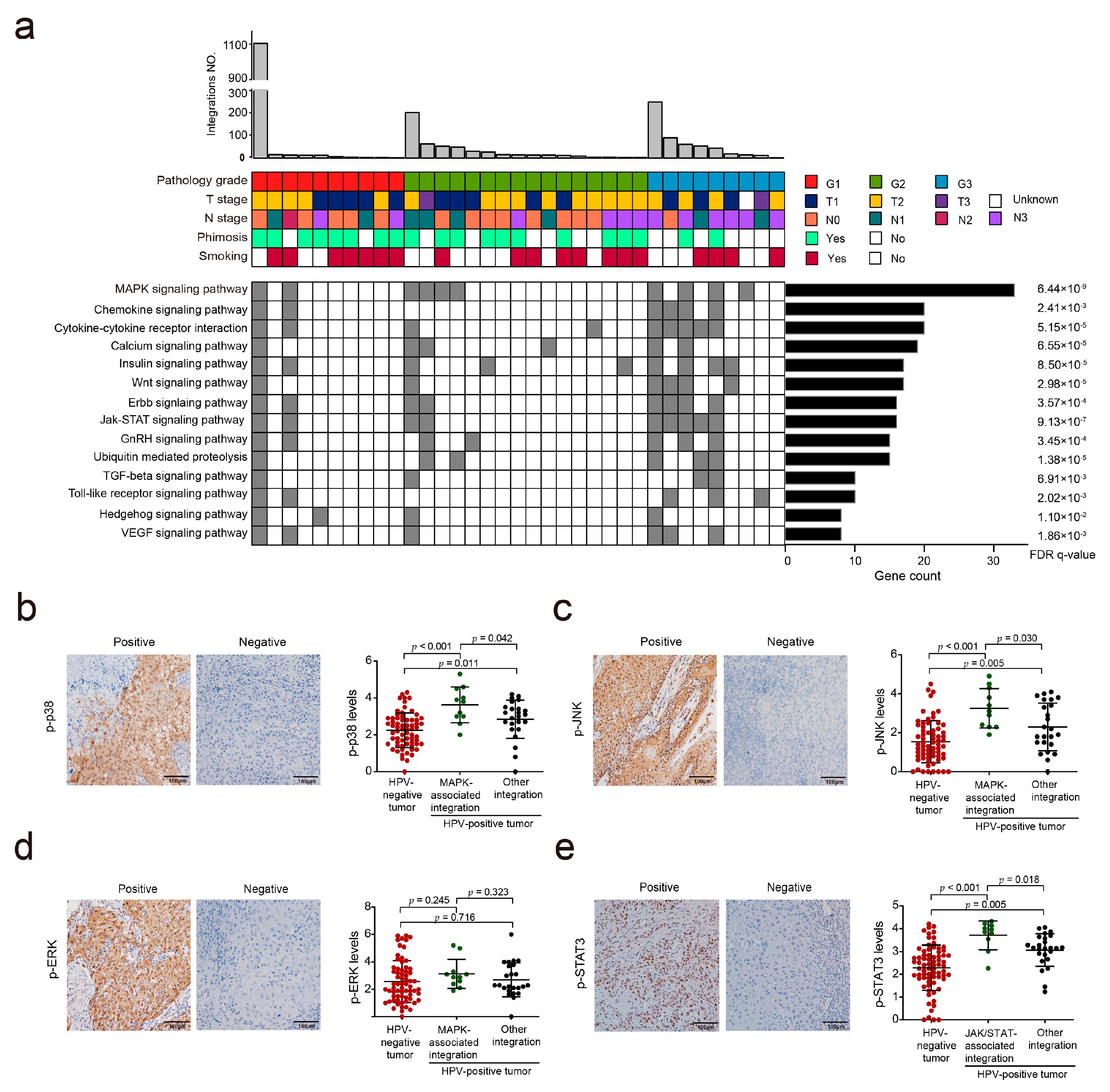
3.4. HPV Integration May Affect the MAPK and JAK/STAT Signaling Pathways in PSCC
3.5. Hotspot Genes Affected by HPV Integration in PSCC
3.6. CADM2 Inhibited the Proliferation and Invasion of PSCC Cells In Vitro
3.7. KLF5 Promoted the Proliferation and Invasion of PSCC Cells In Vitro
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hakenberg, O.W.; Comperat, E.M.; Minhas, S.; Necchi, A.; Protzel, C.; Watkin, N. Eau guidelines on penile cancer: 2014 update. Eur. Urol. 2015, 67, 142–150. Available online: http://www.ncbi.nlm.nih.gov/pubmed/25457021 (accessed on 1 October 2021). [CrossRef]
- Klaes, R.; Woerner, S.M.; Ridder, R.; Wentzensen, N.; Duerst, M.; Schneider, A.; Lotz, B.; Melsheimer, P.; von Knebel Doeberitz, M. Detection of high-risk cervical intraepithelial neoplasia and cervical cancer by amplification of transcripts derived from integrated papillomavirus oncogenes. Cancer Res. 1999, 59, 6132–6136. [Google Scholar]
- Collins, S.I.; Constandinou-Williams, C.; Wen, K.; Young, L.S.; Roberts, S.; Murray, P.G.; Woodman, C.B. Disruption of the e2 gene is a common and early event in the natural history of cervical human papillomavirus infection: A longitudinal cohort study. Cancer Res. 2009, 69, 3828–3832. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19401452 (accessed on 5 October 2021). [CrossRef] [Green Version]
- Ojesina, A.I.; Lichtenstein, L.; Freeman, S.S.; Pedamallu, C.S.; Imaz-Rosshandler, I.; Pugh, T.J.; Cherniack, A.D.; Ambrogio, L.; Cibulskis, K.; Bertelsen, B.; et al. Landscape of genomic alterations in cervical carcinomas. Nature 2014, 506, 371–375. Available online: https://www.ncbi.nlm.nih.gov/pubmed/24390348 (accessed on 12 November 2021). [CrossRef]
- Rusan, M.; Li, Y.Y.; Hammerman, P.S. Genomic landscape of human papillomavirus-associated cancers. Clin. Cancer Res. 2015, 21, 2009–2019. [Google Scholar] [CrossRef] [Green Version]
- Spiess, P.E.; Dhillon, J.; Baumgarten, A.S.; Johnstone, P.A.; Giuliano, A.R. Pathophysiological basis of human papillomavirus in penile cancer: Key to prevention and delivery of more effective therapies. CA Cancer J. Clin. 2016, 66, 481–495. [Google Scholar] [CrossRef]
- Durst, M.; Croce, C.M.; Gissmann, L.; Schwarz, E.; Huebner, K. Papillomavirus sequences integrate near cellular oncogenes in some cervical carcinomas. Proc. Natl. Acad. Sci. USA 1987, 84, 1070–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodelon, C.; Untereiner, M.E.; Machiela, M.J.; Vinokurova, S.; Wentzensen, N. Genomic characterization of viral integration sites in hpv-related cancers. Int. J. Cancer 2016, 139, 2001–2011. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhu, D.; Wang, W.; Li, W.; Jia, W.; Zeng, X.; Ding, W.; Yu, L.; Wang, X.; Wang, L.; et al. Genome-wide profiling of hpv integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat. Genet. 2015, 47, 158–163. Available online: http://www.nature.com/articles/ng.3178 (accessed on 5 July 2021). [CrossRef] [PubMed]
- Busso-Lopes, A.F.; Marchi, F.A.; Kuasne, H.; Scapulatempo-Neto, C.; Trindade-Filho, J.C.; De Jesus, C.M.; Lopes, A.; Guimarães, G.C.; Rogatto, S.R. Genomic profiling of human penile carcinoma predicts worse prognosis and survival. Cancer Prev. Res. (Phila.) 2015, 8, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Feber, A.; Worth, D.C.; Chakravarthy, A.; de Winter, P.; Shah, K.; Arya, M.; Saqib, M.; Nigam, R.; Malone, P.R.; Tan, W.S.; et al. Csn1 somatic mutations in penile squamous cell carcinoma. Cancer Res. 2016, 76, 4720–4727. [Google Scholar] [CrossRef] [Green Version]
- Feber, A.; Arya, M.; de Winter, P.; Saqib, M.; Nigam, R.; Malone, P.R.; Tan, W.S.; Rodney, S.; Lechner, M.; Freeman, A.; et al. Epigenetics markers of metastasis and hpv-induced tumorigenesis in penile cancer. Clin. Cancer Res. 2015, 21, 1196–1206. [Google Scholar] [CrossRef] [Green Version]
- Macedo, J.; Silva, E.; Nogueira, L.; Coelho, R.; da Silva, J.; Dos Santos, A.; Teixeira-Júnior, A.A.; Belfort, M.; Silva, G.; Khayat, A.; et al. Genomic profiling reveals the pivotal role of hrhpv driving copy number and gene expression alterations, including mrna downregulation of tp53 and rb1 in penile cancer. Mol. Carcinog. 2020, 59, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 who classification of tumours of the urinary system and male genital organs-part a: Renal, penile, and testicular tumours. Eur. Urol. 2016, 70, 93–105. [Google Scholar] [CrossRef]
- Li, W.; Zeng, X.I.; Lee, N.P.; Liu, X.; Chen, S.; Guo, B.; Yi, S.; Zhuang, X.; Chen, F.; Wang, G.; et al. Hivid: An efficient method to detect hbv integration using low coverage sequencing. Genomics 2013, 102, 338–344. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeitouni, B.; Boeva, V.; Janoueix-Lerosey, I.; Loeillet, S.; Legoix-Né, P.; Nicolas, A.; Delattre, O.; Barillot, E. Svdetect: A tool to identify genomic structural variations from paired-end and mate-pair sequencing data. Bioinformatics 2010, 26, 1895–1896. [Google Scholar] [CrossRef]
- Wang, J.; Mullighan, C.G.; Easton, J.; Roberts, S.; Heatley, S.L.; Ma, J.; Rusch, M.C.; Chen, K.; Harris, C.C.; Ding, L.; et al. Crest maps somatic structural variation in cancer genomes with base-pair resolution. Nat. Methods 2011, 8, 652–654. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. Annovar: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. Pgc-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267. Available online: https://www.nature.com/articles/ng1180#supplementary-information (accessed on 5 October 2021). [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Chen, H.; Chen, S.; Mo, X.; Li, T.; Xiao, B.; Yu, R.; Guo, J. Circular rna 0000096 affects cell growth and migration in gastric cancer. Br. J. Cancer 2017, 116, 626–633. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28081541 (accessed on 5 October 2021). [CrossRef] [Green Version]
- Zhou, Q.H.; Deng, C.Z.; Li, Z.S.; Chen, J.P.; Yao, K.; Huang, K.B.; Liu, T.Y.; Liu, Z.W.; Qin, Z.K.; Zhou, F.J.; et al. Molecular characterization and integrative genomic analysis of a panel of newly established penile cancer cell lines. Cell Death Dis. 2018, 9, 684. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.H.; Deng, C.Z.; Chen, J.P.; Huang, K.B.; Liu, T.Y.; Yao, K.; Liu, Z.W.; Qin, Z.K.; Li, Y.H.; Guo, S.J.; et al. Elevated serum lamc2 is associated with lymph node metastasis and predicts poor prognosis in penile squamous cell carcinoma. Cancer Manag. Res. 2018, 10, 2983–2995. [Google Scholar] [CrossRef] [Green Version]
- Du, W.Y.; Lu, Z.H.; Ye, W.; Fu, X.; Zhou, Y.; Kuang, C.M.; Wu, J.X.; Pan, Z.Z.; Chen, S.; Liu, R.Y.; et al. The loss-of-function mutations and down-regulated expression of asb3 gene promote the growth and metastasis of colorectal cancer cells. Chin. J. Cancer 2017, 36, 11. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28088228 (accessed on 5 October 2021). [CrossRef] [PubMed] [Green Version]
- Liu, H.; Zhang, H.; Wu, X.; Ma, D.; Wu, J.; Wang, L.; Jiang, Y.; Fei, Y.; Zhu, C.; Tan, R.; et al. Nuclear cgas suppresses DNA repair and promotes tumorigenesis. Nature 2018, 563, 131–136. [Google Scholar] [CrossRef]
- Atkinson, C.J.; Kawamata, F.; Liu, C.; Ham, S.; Győrffy, B.; Munn, A.L.; Wei, M.Q.; Möller, A.; Whitehall, V.; Wiegmans, A.P. Egfr and prion protein promote signaling via foxo3a-klf5 resulting in clinical resistance to platinum agents in colorectal cancer. Mol. Oncol. 2019, 13, 725–737. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30478887 (accessed on 5 October 2021). [CrossRef] [PubMed] [Green Version]
- Groves, I.J.; Coleman, N. Human papillomavirus genome integration in squamous carcinogenesis: What have next-generation sequencing studies taught us? J. Pathol. 2018, 245, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Balaji, H.; Demers, I.; Wuerdemann, N.; Schrijnder, J.; Kremer, B.; Klussmann, J.P.; Huebbers, C.U.; Speel, E.J. Causes and consequences of hpv integration in head and neck squamous cell carcinomas: State of the art. Cancers (Basel) 2021, 13, 4089. [Google Scholar] [CrossRef]
- Lindel, K.; Beer, K.T.; Laissue, J.; Greiner, R.H.; Aebersold, D.M. Human papillomavirus positive squamous cell carcinoma of the oropharynx: A radiosensitive subgroup of head and neck carcinoma. Cancer 2001, 92, 805–813. [Google Scholar] [CrossRef]
- Li, D.; Zhang, Y.; Zhang, H.; Zhan, C.; Li, X.; Ba, T.; Qiu, Z.; Fang, E.; Lv, G.; Zou, C.; et al. Cadm2, as a new target of mir-10b, promotes tumor metastasis through fak/akt pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 46. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Yang, C.; Bai, W.; Wang, Z.; Wang, X.; Johnson, M.; Wang, W.; Zhang, P.; Yang, H.; Liu, H.; et al. Cadm2 inhibits human glioma proliferation, migration and invasion. Oncol. Rep. 2019, 41, 2273–2280. [Google Scholar] [CrossRef]
- Jacob, J.M.; Ferry, E.K.; Gay, L.M.; Elvin, J.A.; Vergilio, J.A.; Ramkissoon, S.; Severson, E.; Necchi, A.; Killian, J.K.; Ali, S.M.; et al. Comparative genomic profiling of refractory and metastatic penile and nonpenile cutaneous squamous cell carcinoma: Implications for selection of systemic therapy. J. Urol. 2019, 201, 541–548. [Google Scholar] [CrossRef]
- Ferrándiz-Pulido, C.; Hernández-Losa, J.; Masferrer, E.; Vivancos, A.; Somoza, R.; Marés, R.; Valverde, C.; Salvador, C.; Placer, J.; Morote, J.; et al. Identification of somatic gene mutations in penile squamous cell carcinoma. Genes Chromosomes Cancer 2015, 54, 629–637. Available online: http://www.ncbi.nlm.nih.gov/pubmed/26216163 (accessed on 8 October 2021). [CrossRef] [PubMed]
- McDaniel, A.S.; Hovelson, D.H.; Cani, A.K.; Liu, C.J.; Zhai, Y.; Zhang, Y.; Weizer, A.Z.; Mehra, R.; Feng, F.Y.; Alva, A.S.; et al. Genomic profiling of penile squamous cell carcinoma reveals new opportunities for targeted therapy. Cancer Res. 2015, 75, 5219–5227. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.M.; Pal, S.K.; Wang, K.; Palma, N.A.; Sanford, E.; Bailey, M.; He, J.; Elvin, J.A.; Chmielecki, J.; Squillace, R.; et al. Comprehensive genomic profiling of advanced penile carcinoma suggests a high frequency of clinically relevant genomic alterations. Oncologist 2016, 21, 33–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, K.; Chen, Y.; Zhou, J.; Liang, Y.; Yang, X.; Li, X.; Cao, Y.; Wang, D.; Luo, L.; et al. Mutational landscape of penile squamous cell carcinoma in a chinese population. Int. J. Cancer 2019, 145, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Jadli, M.; Thakur, K.; Shishodia, G.; Mahata, S.; Basir, S.F.; Das, B.C.; Bharti, A.C. Level of phospho-stat3 (tyr705) correlates with copy number and physical state of human papillomavirus 16 genome in cervical precancer and cancer lesions. PLoS ONE 2019, 14, e0222089. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | No. of Cases | No. of HPV-Negative Tumors (%) | No. of HPV-Positive Tumors with Integration (%) | p-Value 2 | Median Integrations in Positive Tumors | p-Value 3 | |
|---|---|---|---|---|---|---|---|
| Total | 102 | 67 (65.7) | 35 (34.3) | ||||
| Age (years) | <55 ≧55 | 48 54 | 31 (64.6) 36 (66.7) | 17 (35.4) 18 (33.3) | 0.825 | 14.0 15.0 | 0.613 |
| Phimosis | No Yes | 44 58 | 29 (65.9) 38 (65.5) | 15 (34.1) 20 (34.5) | 0.967 | 14.0 14.0 | 0.705 |
| Smoking history | No Yes | 38 64 | 22 (57.9) 45 (70.3) | 16 (42.1) 19 (39.7) | 0.266 | 30.0 9.0 | 0.002 |
| T stage | T1 T2 or higher | 40 62 | 27 (67.5) 40 (64.5) | 13 (32.5) 22 (35.5) | 0.757 | 14.0 14.5 | 0.960 |
| N stage | N0 N+ | 52 50 | 38 (73.1) 29 (58.0) | 14 (26.9) 21 (42.0) | 0.109 | 13.5 15.0 | 0.987 |
| Histologic grade | G1 G2 G3 | 48 38 16 | 38 (79.2) 22 (57.9) 7 (43.8) | 10 (20.8) 16 (42.1) 9 (56.2) | 0.016 | 10.0 14.5 46.0 | 0.105 |
| Histologic subtype 1 | Non-HPV-related HPV-related | 79 23 | 62 (78.5) 5 (21.7) | 17 (21.5) 18 (78.3) | <0.001 | 14.0 15.0 | 0.732 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, K.-B.; Guo, S.-J.; Li, Y.-H.; Zhang, X.-K.; Chen, D.; Spiess, P.E.; Li, Z.-S.; Deng, C.-Z.; Chen, J.-P.; Zhou, Q.-H.; et al. Genome-Wide Profiling Reveals HPV Integration Pattern and Activated Carcinogenic Pathways in Penile Squamous Cell Carcinoma. Cancers 2021, 13, 6104. https://doi.org/10.3390/cancers13236104
Huang K-B, Guo S-J, Li Y-H, Zhang X-K, Chen D, Spiess PE, Li Z-S, Deng C-Z, Chen J-P, Zhou Q-H, et al. Genome-Wide Profiling Reveals HPV Integration Pattern and Activated Carcinogenic Pathways in Penile Squamous Cell Carcinoma. Cancers. 2021; 13(23):6104. https://doi.org/10.3390/cancers13236104
Chicago/Turabian StyleHuang, Kang-Bo, Sheng-Jie Guo, Yong-Hong Li, Xin-Ke Zhang, Dong Chen, Philippe E. Spiess, Zai-Shang Li, Chuang-Zhong Deng, Jie-Ping Chen, Qiang-Hua Zhou, and et al. 2021. "Genome-Wide Profiling Reveals HPV Integration Pattern and Activated Carcinogenic Pathways in Penile Squamous Cell Carcinoma" Cancers 13, no. 23: 6104. https://doi.org/10.3390/cancers13236104