
Organ Specific Copy Number Variations in Visceral Metastases of Human Melanoma

 , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Metastatic Melanoma Cohort
2.2. CNV Landscape of Primary Tumors and Metastases of Skin Melanoma
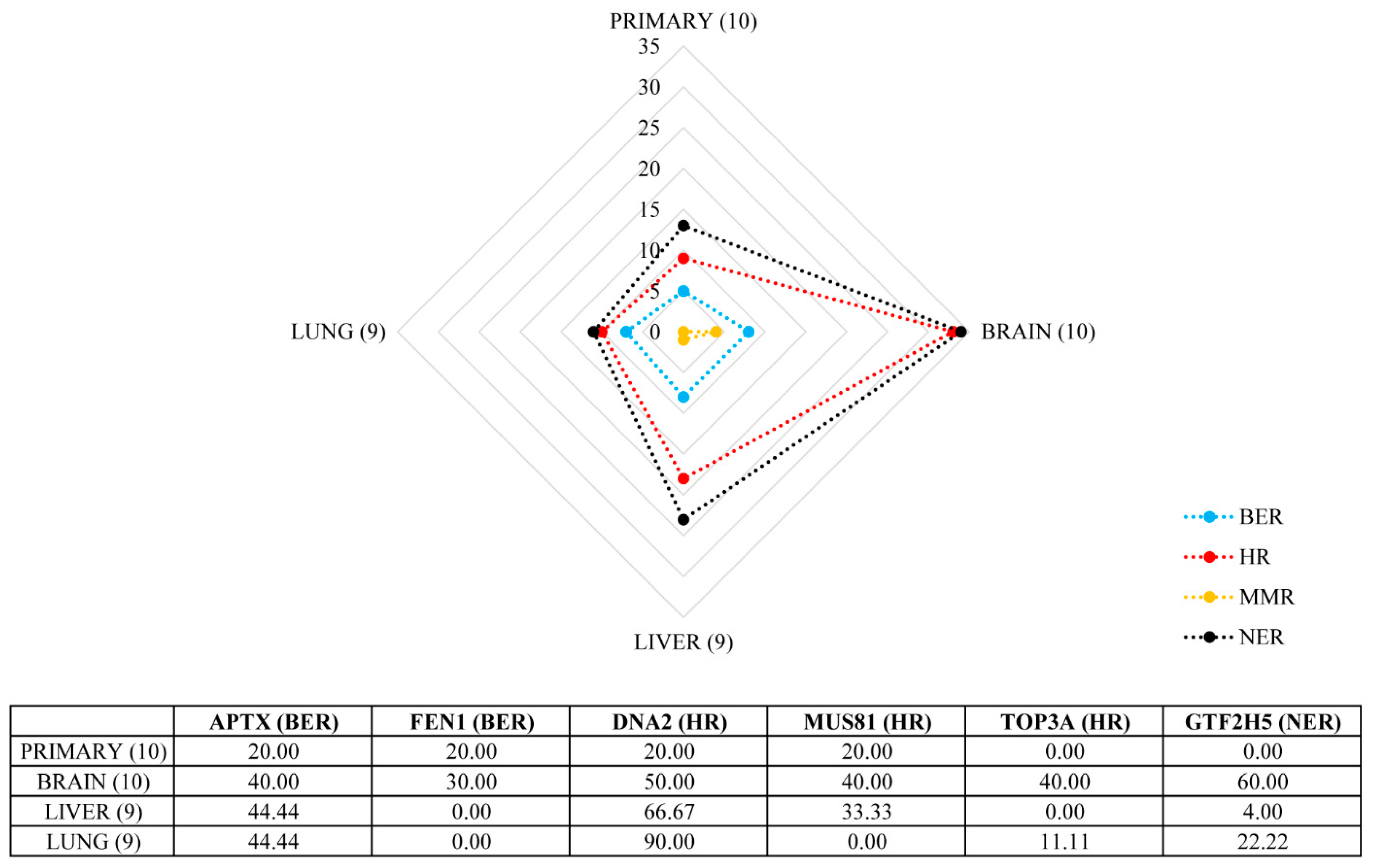
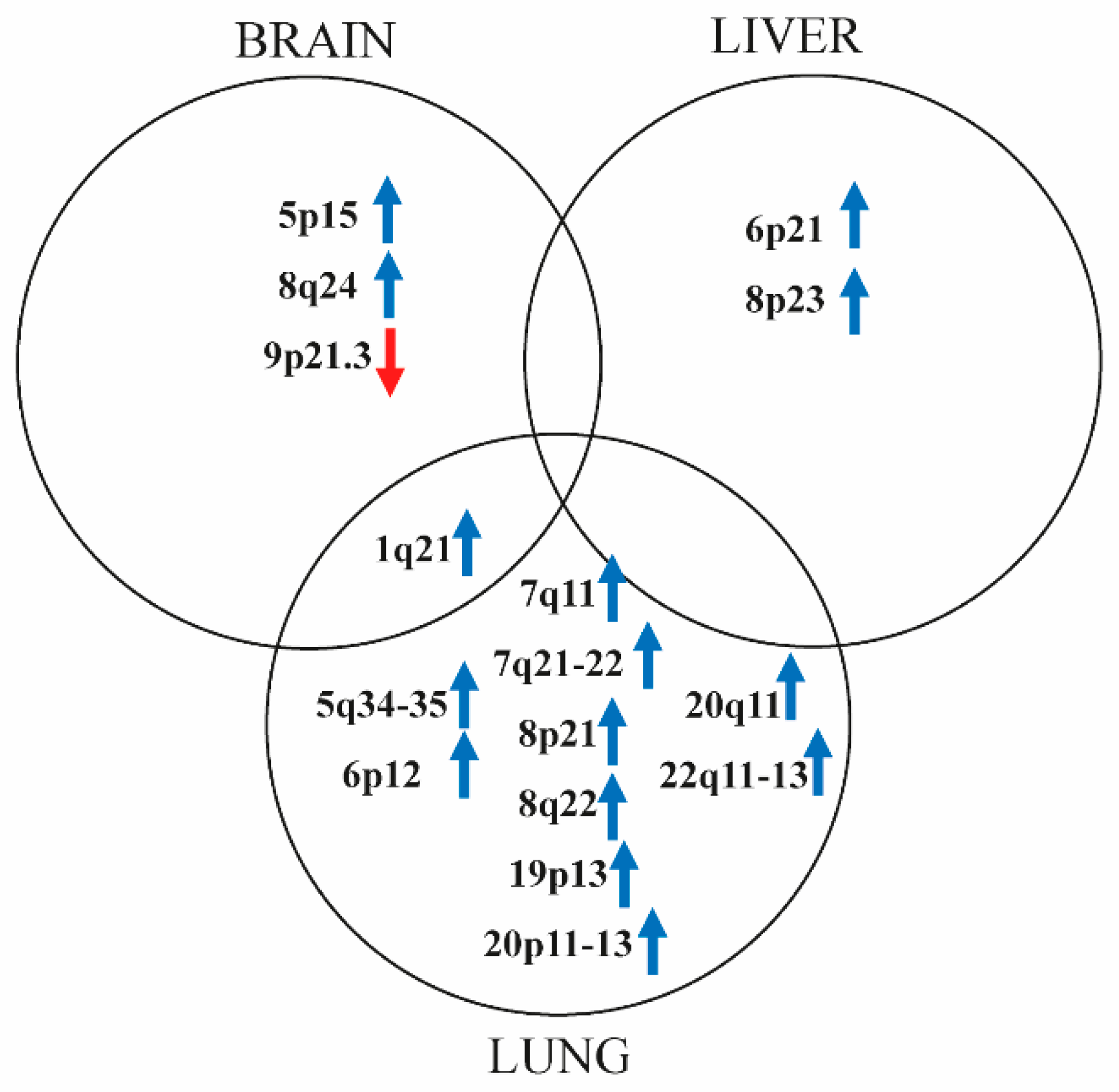
2.3. Marked Genetic Differences between Visceral Metastases of Melanoma
2.4. Expression Profiles of the Immunogenic Mimicry Related Genes
2.5. Mutant Allele Frequency (MAF) Changes of Driver Oncogenes during Progression
3. Discussion
4. Materials and Methods
4.1. Primary Tumors and Visceral Metastases
4.2. DNA Extraction, Quality Control, and Microarray Hybridization
4.3. aCGH Experiments and Analysis
4.4. Driver Mutation Analysis and Defining Mutant Allele Frequency
4.5. Gene Expression Analyses of the IGM Related Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Makohon-Moore, A.P.; Lipson, E.J.; Hooper, J.E.; Zucker, A.; Hong, J.; Bielski, C.M.; Hayashi, A.; Tokheim, C.; Baez, P.; Kappagantula, R.; et al. The Genetic Evolution of Treatment-Resistant Cutaneous, Acral, and Uveal Melanomas. Clin. Cancer Res. 2021, 27, 1516–1525. [Google Scholar] [CrossRef]
- Liu, L.; Bai, X.; Wang, J.; Tang, X.-R.; Wu, D.-H.; Du, S.-S.; Du, X.-J.; Zhang, Y.-W.; Zhu, H.-B.; Fang, Y.; et al. Combination of TMB and CNA Stratifies Prognostic and Predictive Responses to Immunotherapy Across Metastatic Cancer. Clin. Cancer Res. 2019, 25, 7413–7423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimann, J.D.R.; Salim, S.; Velazquez, E.F.; Wang, L.; Williams, K.M.; Flejter, W.L.; Brooke, L.; Sunder, S.; Busam, K.J. Comparison of melanoma gene expression score with histopathology, fluorescence in situ hybridization, and SNP array for the classification of melanocytic neoplasms. Mod. Pathol. 2018, 31, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.; Garraway, L.A.; Fisher, D.E. Malignant melanoma: Genetics and therapeutics in the genomic era. Genes Dev. 2006, 20, 2149–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rákosy, Z.; Vízkeleti, L.; Ecsedi, S.; Vokó, Z.; Bégány, A.; Barok, M.; Krekk, Z.; Gallai, M.; Szentirmay, Z.; Adány, R.; et al. EGFR gene copy number alterations in primary cutaneous malignant melanomas are associated with poor prognosis. Int. J. Cancer 2007, 121, 1729–1737. [Google Scholar] [CrossRef]
- Tímár, J.; Vizkeleti, L.; Doma, V.; Barbai, T.; Rásó, E. Genetic progression of malignant melanoma. Cancer Metastasis Rev. 2016, 35, 93–107. [Google Scholar] [CrossRef]
- Turner, N.; Ware, O.; Bosenberg, M. Genetics of metastasis: Melanoma and other cancers. Clin. Exp. Metastasis 2018, 35, 379–391. [Google Scholar] [CrossRef]
- Boi, S.; Tebaldi, T.; Re, A.; Cantaloni, C.; Adami, V.; Barbareschi, M.; Cristofolini, M.; Pasini, L.; Quattrone, A. Increased frequency of minimal homozygous deletions is associated with poor prognosis in primary malignant melanoma patients. Genes. Chromosomes Cancer 2014, 53, 487–496. [Google Scholar] [CrossRef]
- Gandolfi, G.; Longo, C.; Moscarella, E.; Zalaudek, I.; Sancisi, V.; Raucci, M.; Manzotti, G.; Gugnoni, M.; Piana, S.; Argenziano, G.; et al. The extent of whole-genome copy number alterations predicts aggressive features in primary melanomas. Pigment. Cell Melanoma Res. 2016, 29, 163–175. [Google Scholar] [CrossRef]
- Chiu, C.G.; Nakamura, Y.; Chong, K.K.; Huang, S.K.; Kawas, N.P.; Triche, T.; Elashoff, D.; Kiyohara, E.; Irie, R.F.; Morton, D.L.; et al. Genome-wide characterization of circulating tumor cells identifies novel prognostic genomic alterations in systemic melanoma metastasis. Clin. Chem. 2014, 60, 873–885. [Google Scholar] [CrossRef] [Green Version]
- Marzese, D.M.; Scolyer, R.A.; Roqué, M.; Vargas-Roig, L.M.; Huynh, J.L.; Wilmott, J.S.; Murali, R.; Buckland, M.E.; Barkhoudarian, G.; Thompson, J.F.; et al. DNA methylation and gene deletion analysis of brain metastases in melanoma patients identifies mutually exclusive molecular alterations. Neuro. Oncol. 2014, 16, 1499–1509. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, S.D.; Zheng, S.; Xiu, J.; Zhou, S.; Khasraw, M.; Brastianos, P.K.; Kesari, S.; Hu, J.; Rudnick, J.; Salacz, M.E.; et al. Profiles of brain metastases: Prioritization of therapeutic targets. Int. J. Cancer 2018, 143, 3019–3026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doma, V.; Kárpáti, S.; Rásó, E.; Barbai, T.; Tímár, J. Dynamic and unpredictable changes in mutant allele fractions of BRAF and NRAS during visceral progression of cutaneous malignant melanoma. BMC Cancer 2019, 19, 853. [Google Scholar] [CrossRef]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Betancourt, L.H.; Gil, J.; Kim, Y.; Doma, V.; Çakır, U.; Sanchez, A.; Murillo, J.R.; Kuras, M.; Parada, I.P.; Sugihara, Y.; et al. The human melanoma proteome atlas—Defining the molecular pathology. Clin. Transl. Med. 2021, 11, 11. [Google Scholar] [CrossRef]
- Betancourt, L.H.; Gil, J.; Sanchez, A.; Doma, V.; Kuras, M.; Murillo, J.R.; Velasquez, E.; Çakır, U.; Kim, Y.; Sugihara, Y.; et al. The Human Melanoma Proteome Atlas—Complementing the melanoma transcriptome. Clin. Transl. Med. 2021, 11, e451. [Google Scholar] [CrossRef] [PubMed]
- Toh, M.; Ngeow, J. Homologous Recombination Deficiency: Cancer Predispositions and Treatment Implications. Oncologist 2021, 26, e1526–e1537. [Google Scholar] [CrossRef]
- Diossy, M.; Reiniger, L.; Sztupinszki, Z.; Krzystanek, M.; Timms, K.M.; Neff, C.; Solimeno, C.; Pruss, D.; Eklund, A.C.; Tóth, E.; et al. Breast cancer brain metastases show increased levels of genomic aberration-based homologous recombination deficiency scores relative to their corresponding primary tumors. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2018, 29, 1948–1954. [Google Scholar] [CrossRef]
- Kim, K.B.; Soroceanu, L.; de Semir, D.; Millis, S.Z.; Ross, J.; Vosoughi, E.; Dar, A.A.; Nosrati, M.; Desprez, P.-Y.; Ice, R.; et al. Prevalence of Homologous Recombination Pathway Gene Mutations in Melanoma: Rationale for a New Targeted Therapeutic Approach. J. Invest. Dermatol. 2021, 141, 2028–2036.e2. [Google Scholar] [CrossRef]
- Adhikari, S.; Nice, E.C.; Deutsch, E.W.; Lane, L.; Omenn, G.S.; Pennington, S.R.; Paik, Y.-K.; Overall, C.M.; Corrales, F.J.; Cristea, I.M.; et al. A high-stringency blueprint of the human proteome. Nat. Commun. 2020, 11, 5301. [Google Scholar] [CrossRef] [PubMed]
- Uruga, H.; Mino-Kenudson, M. Predictive biomarkers for response to immune checkpoint inhibitors in lung cancer: PD-L1 and beyond. Virchows Arch. 2021, 478, 31–44. [Google Scholar] [CrossRef]
- Majidpoor, J.; Mortezaee, K. The efficacy of PD-1/PD-L1 blockade in cold cancers and future perspectives. Clin. Immunol. 2021, 226, 108707. [Google Scholar] [CrossRef]
- Kumar, S.; Sharawat, S.K. Epigenetic regulators of programmed death-ligand 1 expression in human cancers. Transl. Res. 2018, 202, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.; Deschoolmeester, V.; Zwaenepoel, K.; Rolfo, C.; Silence, K.; Rottey, S.; Lardon, F.; Smits, E.; Pauwels, P. CD70: An emerging target in cancer immunotherapy. Pharmacol. Ther. 2015, 155, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Zhang, R.; Li, S.; Liu, J. IDO1: An important immunotherapy target in cancer treatment. Int. Immunopharmacol. 2017, 47, 70–77. [Google Scholar] [CrossRef]
- Soto-Pantoja, D.R.; Terabe, M.; Ghosh, A.; Ridnour, L.A.; DeGraff, W.G.; Wink, D.A.; Berzofsky, J.A.; Roberts, D.D. CD47 in the tumor microenvironment limits cooperation between antitumor T-cell immunity and radiotherapy. Cancer Res. 2014, 74, 6771–6783. [Google Scholar] [CrossRef] [Green Version]
- Ring, N.G.; Herndler-Brandstetter, D.; Weiskopf, K.; Shan, L.; Volkmer, J.-P.; George, B.M.; Lietzenmayer, M.; McKenna, K.M.; Naik, T.J.; McCarty, A.; et al. Anti-SIRPα antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc. Natl. Acad. Sci. USA 2017, 114, E10578–E10585. [Google Scholar] [CrossRef] [Green Version]
- Aloia, A.; Müllhaupt, D.; Chabbert, C.D.; Eberhart, T.; Flückiger-Mangual, S.; Vukolic, A.; Eichhoff, O.; Irmisch, A.; Alexander, L.T.; Scibona, E.; et al. A Fatty Acid Oxidation-dependent Metabolic Shift Regulates the Adaptation of BRAF-mutated Melanoma to MAPK Inhibitors. Clin. Cancer Res. 2019, 25, 6852–6867. [Google Scholar] [CrossRef]
- Amato, C.M.; Hintzsche, J.D.; Wells, K.; Applegate, A.; Gorden, N.T.; Vorwald, V.M.; Tobin, R.P.; Nassar, K.; Shellman, Y.G.; Kim, J.; et al. Pre-Treatment Mutational and Transcriptomic Landscape of Responding Metastatic Melanoma Patients to Anti-PD1 Immunotherapy. Cancers 2020, 12, 1943. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Czyz, M. HGF/c-MET Signaling in Melanocytes and Melanoma. Int. J. Mol. Sci. 2018, 19, 3844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adachi, E.; Sakai, K.; Nishiuchi, T.; Imamura, R.; Sato, H.; Matsumoto, K. Different growth and metastatic phenotypes associated with a cell-intrinsic change of Met in metastatic melanoma. Oncotarget 2016, 7, 70779–70793. [Google Scholar] [CrossRef] [PubMed]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iranzo, J.; Martincorena, I.; Koonin, E.V. Cancer-mutation network and the number and specificity of driver mutations. Proc. Natl. Acad. Sci. USA 2018, 115, E6010–E6019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moosavi, F.; Giovannetti, E.; Saso, L.; Firuzi, O. HGF/MET pathway aberrations as diagnostic, prognostic, and predictive biomarkers in human cancers. Crit. Rev. Clin. Lab. Sci. 2019, 56, 533–566. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Alexander, S. Cancer Invasion and the Microenvironment: Plasticity and Reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orgaz, J.L.; Sanz-Moreno, V. Emerging molecular targets in melanoma invasion and metastasis. Pigment. Cell Melanoma Res. 2013, 26, 39–57. [Google Scholar] [CrossRef]
- Sahai, E.; Marshall, C.J. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 2003, 5, 711–719. [Google Scholar] [CrossRef]
- Yang, X.; Liang, R.; Liu, C.; Liu, J.A.; Cheung, M.P.L.; Liu, X.; Man, O.Y.; Guan, X.-Y.; Lung, H.L.; Cheung, M. SOX9 is a dose-dependent metastatic fate determinant in melanoma. J. Exp. Clin. Cancer Res. 2019, 38, 17. [Google Scholar] [CrossRef] [PubMed]
- Gaggioli, C.; Sahai, E. Melanoma invasion—Current knowledge and future directions. Pigment. Cell Res. 2007, 20, 161–172. [Google Scholar] [CrossRef]
- Rákosy, Z.; Vízkeleti, L.; Ecsedi, S.; Bégány, Á.; Emri, G.; Ádány, R.; Balázs, M. Characterization of 9p21 copy number alterations in human melanoma by fluorescence in situ hybridization. Cancer Genet. Cytogenet. 2008, 182, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.J.; Flaherty, K. MAP kinase signaling and inhibition in melanoma. Oncogene 2013, 32, 2373–2379. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.B.; Menzies, A.M.; Zimmer, L.; Eroglu, Z.; Ye, F.; Zhao, S.; Rizos, H.; Sucker, A.; Scolyer, R.A.; Gutzmer, R.; et al. Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur. J. Cancer 2015, 51, 2792–2799. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Fung, C.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Hyman, J.; Shahheydari, H.; Tembe, V.; Thompson, J.F.; Saw, R.P.; et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat. Commun. 2014, 5, 5694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathanson, K.L.; Martin, A.-M.; Wubbenhorst, B.; Greshock, J.; Letrero, R.; D’Andrea, K.; O’Day, S.; Infante, J.R.; Falchook, G.S.; Arkenau, H.-T.; et al. Tumor Genetic Analyses of Patients with Metastatic Melanoma Treated with the BRAF Inhibitor Dabrafenib (GSK2118436). Clin. Cancer Res. 2013, 19, 4868–4878. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Moriceau, G.; Kong, X.; Lee, M.-K.; Lee, H.; Koya, R.C.; Ng, C.; Chodon, T.; Scolyer, R.A.; Dahlman, K.B.; et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat. Commun. 2012, 3, 724. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Tumor | N = 10 (100%) |
|---|---|
| Breslow thickness: range+SEM | 4.44 (1.0–9.25) |
| <1.0 | 1 (10) |
| 1.1–2.0 | 2 (20) |
| 2.1–4.0 | 2 (20) |
| >4.1 | 5 (50) |
| Histological types | |
| SSM | 3 (30) |
| NM | 2 (20) |
| LMM | 1 (10) |
| unclassified | 4 (40) |
| Anatomical location | |
| trunk | 2 (20) |
| head and neck | 2 (20) |
| extremities | 6 (60) |
| Mutational status | |
| BRAFV600K/E | 6 (60) |
| NRASQ61L/R | 3 (30) |
| BRAF/NRAS/KIT wild type | 1 (10) |
| Gender | |
| male | 6 (60) |
| female | 4 (40) |
| Age (years) | 47.6 + 18.1 |
| Overall survival (months): range + SD | 69.3 + 39.6 |
| Treatment | |
| IFN2α | 7 (70) |
| BRAFi | 3 (30) |
| DTIC | 2 (20) |
| cisplatinum | 2 (20) |
| Metastasis | N = 28 (100%) |
| brain | 10 (36) |
| liver | 9 (32) |
| lung | 9 (32) |
| Gene | Locus | BRAIN | LIVER | LUNG | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean CN | Type of Change | Frequency (%) | Mean CN | Type of Change | Frequency (%) | Mean CN | Type of Change | Frequency (%) | ||
| CDKN2A | 9p21.3 | 0 | hoCNL | 20 | - | - | - | 0 | hoCNL | 22 |
| CDKN2B | 0 | hoCNL | 10 | - | - | - | 0 | hoCNL | 11 | |
| TERT | 5p15.33 | 4.5 | amp. | 20 | - | - | - | - | - | - |
| MAPK15 | 8q24.3 | 5 | amp. | 10 | 4.5 | amp. | 22 | 4.66 | amp. | 33 |
| TWIST1 | 7p21.1 | 5 | amp. | 10 | 5 | amp. | 11 | 4.75 | amp. | 44 |
| TWIST2 | 2q37.3 | - | - | - | - | - | - | 4 | amp. | 11 |
| SNAI1 | 20q13.13 | - | - | - | - | - | - | 4 | amp. | 11 |
| SNAI2 | - | - | - | 5 | amp. | 22 | 4.66 | amp. | 22 | |
| S100A9 | 1q21.3 | - | - | - | 6 | amp. | 11 | 4.66 | amp. | 33 |
| S100A10 | - | - | - | 5 | amp. | 11 | 4.66 | amp. | 33 | |
| S100A11 | - | - | - | 5 | amp. | 11 | 4.66 | amp. | 33 | |
| S100A12 | - | - | - | 6 | amp. | 11 | 4.66 | amp. | 33 | |
| NEDD9 | 6p24.2 | - | - | - | - | - | - | 4 | amp. | 22 |
| TEAD1 | 11p15.3 | - | - | - | - | - | - | 4 | amp. | 11 |
| TEAD2 | 19q13.33 | - | - | - | - | - | - | 4 | amp. | 22 |
| TEAD4 | 12p13.33 | - | - | - | - | - | - | 4 | amp. | 22 |
| CDK6 | 7q21.2 | - | - | - | - | - | - | 4 | amp. | 44 |
| MAPK1 | 22q11.22 | - | - | - | - | - | - | 4 | amp. | 22 |
| ABL2 | 1q25.2 | - | - | - | - | - | - | 4.66 | amp. | 33 |
| HGF | 7q21.11 | 6 | amp. | 10 | 8.33 | amp. | 33 | |||
| MET | 7q31 | 6 | amp. | 11 | 9 | amp. | 11 | |||
| Gene | Locus | BRAIN | LIVER | LUNG | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean CN | Type of Change | Frequency (%) | Mean CN | Type of Change | Frequency (%) | Mean CN | Type of Change | Frequency (%) | ||
| CD160 | 1q21.1 | 4 | amp. | 10 | - | - | - | - | - | - |
| CD40 | 20q13.12 | - | - | - | 5.5 | amp. | 22 | 6 | amp. | 22 |
| CD83 | 6p23 | - | - | - | 5 | amp. | 11 | 4 | amp. | 22 |
| CD172 | 20p13 | - | - | - | 5 | amp. | 11 | 4.5 | amp. | 22 |
| CD1A-E | 1q23.1 | - | - | - | - | - | - | 4.66 | amp. | 33 |
| CD48 | 1q23.3 | - | - | - | - | - | - | 4.66 | amp. | 33 |
| CD84 | - | - | - | - | - | - | 4.66 | amp. | 33 | |
| CD244 | - | - | - | - | - | - | 4.66 | amp. | 33 | |
| CD247 | 1q24.2 | - | - | - | - | - | - | 4.66 | amp. | 33 |
| IDO1 | 8p11.21 | - | - | - | - | - | - | 5 | amp. | 22 |
| IDO2 | - | - | - | - | - | - | 5 | amp. | 22 | |
| BCR | 22q11.23 | - | - | - | - | - | - | 4 | amp. | 33 |
| IL17R | 22q11.1 | - | - | - | - | - | - | 4 | amp. | 22 |
| CD36 | 7q21.11 | - | - | - | - | - | - | 4.25 | amp. | 44 |
| CD37 | 19q13.33 | - | - | - | - | - | - | 4 | amp. | 22 |
| CD70 | 19p13.3 | - | - | - | - | - | - | 4 | amp. | 11 |
| CD93 | 20p11.21 | - | - | - | - | - | - | 4.5 | amp. | 22 |
| CD209 | 19p13.2 | - | - | - | - | - | - | 4 | amp. | 11 |
| CD320 | - | - | - | - | - | - | 5 | amp. | 11 | |
| Scheme. | Site | Driver Mutation in DM | % of Mutant Cells * (P **) | % of Mutant Cells * (DM ***) | DM/P Ratio | Alteration of Driver Mutant Gene in Primary Tumor | Alteration of Driver Mutant Gene in Metastasis |
|---|---|---|---|---|---|---|---|
| BRAF primary | |||||||
| 17 | liver | BRAF | 29.1 | 69.1 | 2.37 | n.d. | CN 4 + LOH |
| lung | BRAF | 77.9 | 2.68 | CN 3 + LOH | |||
| 19 | liver | BRAF | 40.8 | 50.4 | 1.24 | CN 3 | CN 3 |
| lung | BRAF | 55.3 | 1.36 | CN 3 | |||
| 24 | brain | BRAF | 38.4 | 28.6 | 0.74 | no change | no change |
| brain | BRAF | 24.0 | 0.63 | no change | |||
| 28 | brain | BRAF | 21.2 | 35.7 | 1.68 | n.d. | CN 3 |
| brain | BRAF | 32.8 | 1.55 | no change | |||
| lung | BRAF | 37.5 | 1.77 | CN 3 | |||
| 31 | liver | WT | 27.4 | 8.3 | 0.30 | n.d. | no change |
| lung | BRAF | 29.5 | 1.08 | no change | |||
| 32 | liver | BRAF | 4.0 | 14.9 | 3.73 | no change | SNP |
| lung | BRAF | 33.1 | 8.28 | CN 5 | |||
| 36 | brain | BRAF | 23.7 | 68.6 | 2.89 | n.d. | CN 3 |
| 48 | brain | BRAF | 40.3 | 50.9 | 1.26 | no change | no change |
| 54 | lung | BRAF | 2.2 | 80.0 | 36.36 | n.d. | CN 3 + LOH |
| 56 | liver | BRAF | 4.7 | 14.0 | 2.98 | CN 3 | CN 3 |
| lung | BRAF | 54.1 | 11.51 | CN 4 | |||
| 57 | brain | BRAF | 11.0 | 24.8 | 2.25 | no change | CN 3 |
| liver | BRAF | 28.0 | 2.55 | CN 3 | |||
| lung | BRAF | 37.8 | 3.44 | CN 3 | |||
| 59 | lung | BRAF | 4.3 | 75.4 | 17.53 | no change | CN 5 |
| NRAS primary | |||||||
| 20 | liver | NRAS | 4.6 | 29.2 | 6.35 | − | |
| 53 | brain | NRAS | 15.8 | 49.4 | 3.13 | no change | LOH |
| 55 | brain | NRAS | 35.4 | 22.8 | 0.64 | LOH | no change |
| liver | NRAS | 38.0 | 1.07 | no change | |||
| WT primary **** | |||||||
| 33 | brain | WT | 0.0 | 0.0 | − | n.r. | n.r. |
| BRAF | Copy Number State (N) | |||
|---|---|---|---|---|
| CN2 (7) | CN3 (11) | CN4 (2) | CN5 (2) | |
| MAF * (mean ± SE) | 27.00 ± 5.16 | 46.36 ± 6.63 | 61.60 ± 7.50 | 52.25 ± 21.25 |
| heterozygous | 1 | 2 | 1 | 0 |
| homozygous | 0 | 3 | 1 | 1 |
| subclonal | 6 | 6 | 0 | 1 |
| Correlation coefficient ** (p-value) | 0.540 (0.009) | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papp, O.; Doma, V.; Gil, J.; Markó-Varga, G.; Kárpáti, S.; Tímár, J.; Vízkeleti, L. Organ Specific Copy Number Variations in Visceral Metastases of Human Melanoma. Cancers 2021, 13, 5984. https://doi.org/10.3390/cancers13235984
Papp O, Doma V, Gil J, Markó-Varga G, Kárpáti S, Tímár J, Vízkeleti L. Organ Specific Copy Number Variations in Visceral Metastases of Human Melanoma. Cancers. 2021; 13(23):5984. https://doi.org/10.3390/cancers13235984
Chicago/Turabian StylePapp, Orsolya, Viktória Doma, Jeovanis Gil, György Markó-Varga, Sarolta Kárpáti, József Tímár, and Laura Vízkeleti. 2021. "Organ Specific Copy Number Variations in Visceral Metastases of Human Melanoma" Cancers 13, no. 23: 5984. https://doi.org/10.3390/cancers13235984