
Clinical Value of NGS Genomic Studies for Clinical Management of Pediatric and Young Adult Bone Sarcomas

 ,
,  , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. Extraction of DNA/RNA from Tissues
2.3. NGS Library Preparation and Sequencing
2.4. Data Analysis
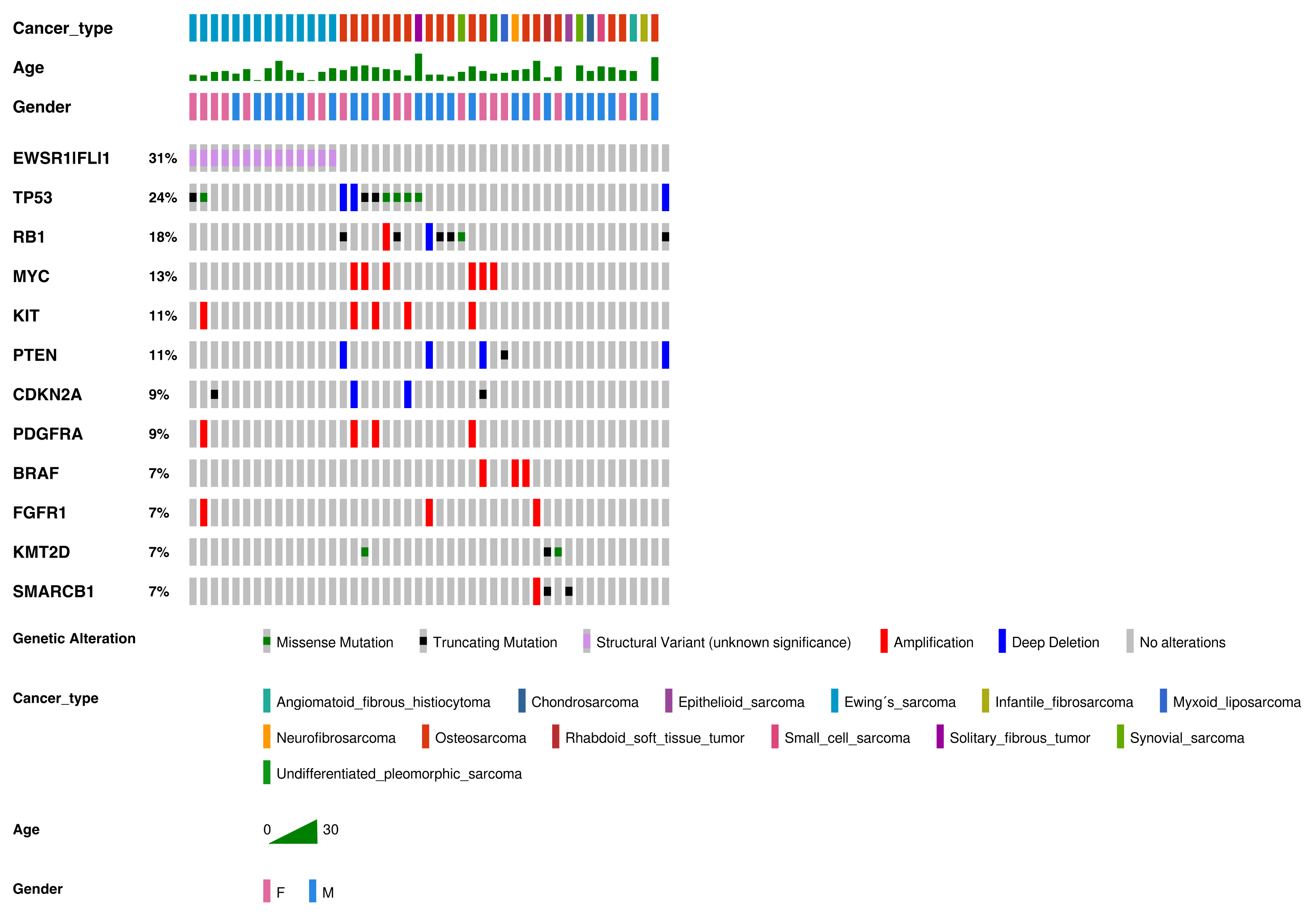
3. Results
3.1. Patient and Sample Characteristics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Number (%) |
|---|---|
| Median age at diagnosis (range) | 11.8 (0–30.8) |
| Gender | |
| Male | 30 (56.6) |
| Female | 23 (43.4) |
| Ethnic origin | |
| European | 47 (88.6) |
| Latin | 3 (5.7) |
| African | 3 (5.7) |
| Classification of the sarcoma | |
| Osteosarcoma | 25 (47.2) |
| Ewing´s sarcoma | 16 (30.2) |
| Other | 12 (22.6) |
| Pathological subtype | |
| Osteosarcoma; n = 25 | |
| Osteoblastic | 12 (48) |
| Unknown | 5 (20) |
| Chondroblastic | 4 (16) |
| Osteo-chondroblastic | 3 (12) |
| Parostal | 1 (4) |
| Ewing’s sarcoma; n = 16 | |
| Bone | 10 (62.5) |
| Askin tumor | 3 (18.75) |
| Extraskeletal | 2 (12.5) |
| Ewing-like | 1 (6.25) |
| Other sarcomas; n = 12 | |
| Undifferentiated pleomorphic sarcoma | 2 (16.7) |
| Chondrosarcoma | 1 (8.3, each) |
| Infantile fibrosarcoma | |
| Angiomatoid fibrous histiocytoma | |
| Neurofibrosarcoma | |
| Synovial sarcoma | |
| Epitheloid sarcoma | |
| Desmoplastic small round cell tumor | |
| Solitary fibrous tumor | |
| Rhabdoid soft tissue tumor | |
| Myxoid liposarcoma | |
| Location of the primary tumor | |
| Lower extremity | 22 (41) |
| Upper extremity | 11 (20.8) |
| Trunk | 16 (30.2) |
| Other | 4 (7.5) |
| Disease stage at diagnosis | |
| Localized | 43 (81.1) |
| Metastatic | 10 (19.9) |
| Treatment regimen | |
| Neoadjuvant chemotherapy | 43 (81.1) |
| Surgery | 49 (92.5) |
| Adjuvant chemotherapy | 47 (88.7) |
| Radiotherapy | 29 (54.7) |
| Targeted therapy | 6 (11.3) |
| Immunotherapy | 6 (11.3) |
| Stem cell transplant | 4 (7.5) |
| Current status | |
| Alive | 34 (62.1) |
| Dead | 16 (30.2) |
| Unknown | 3 (5.7) |
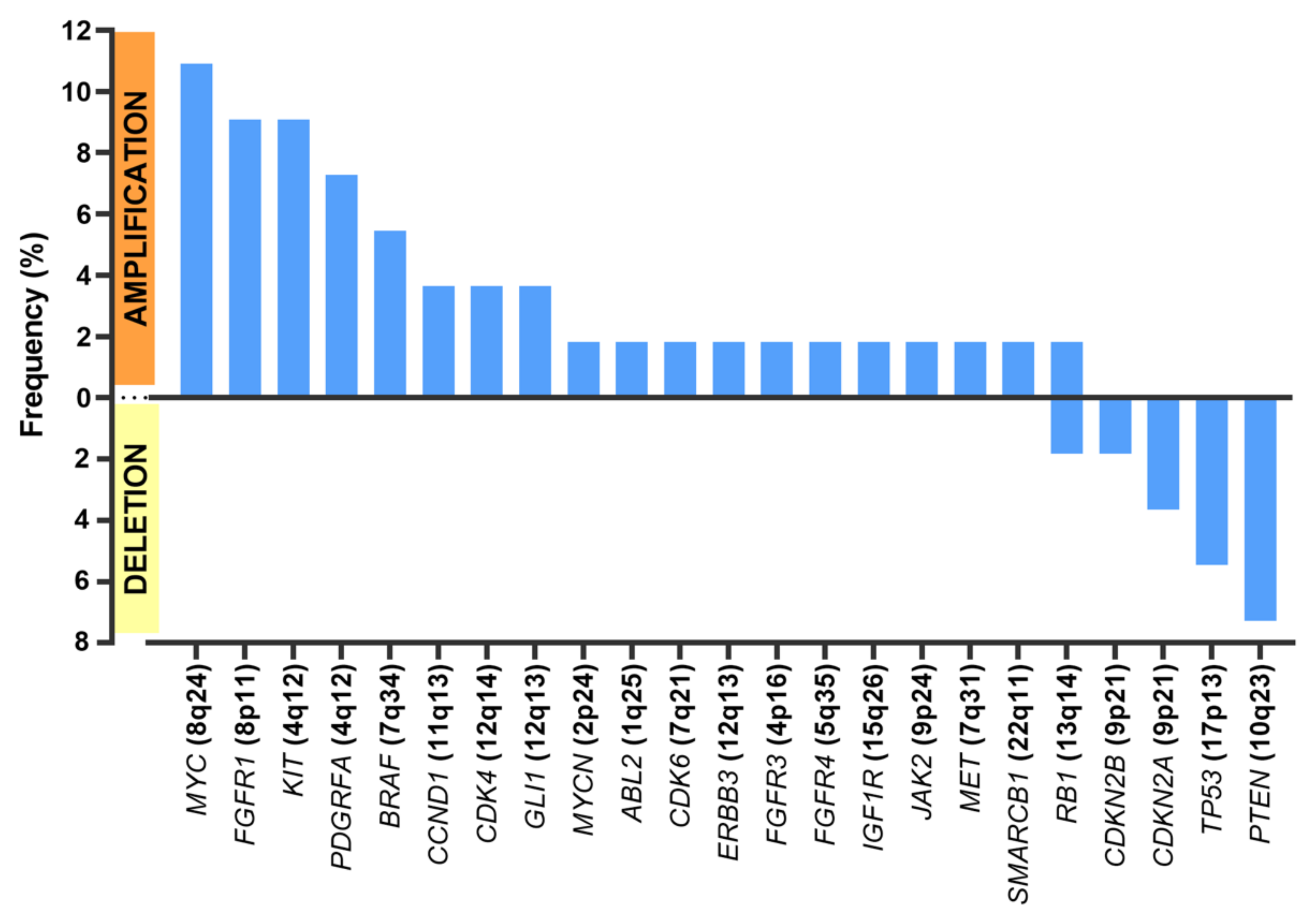
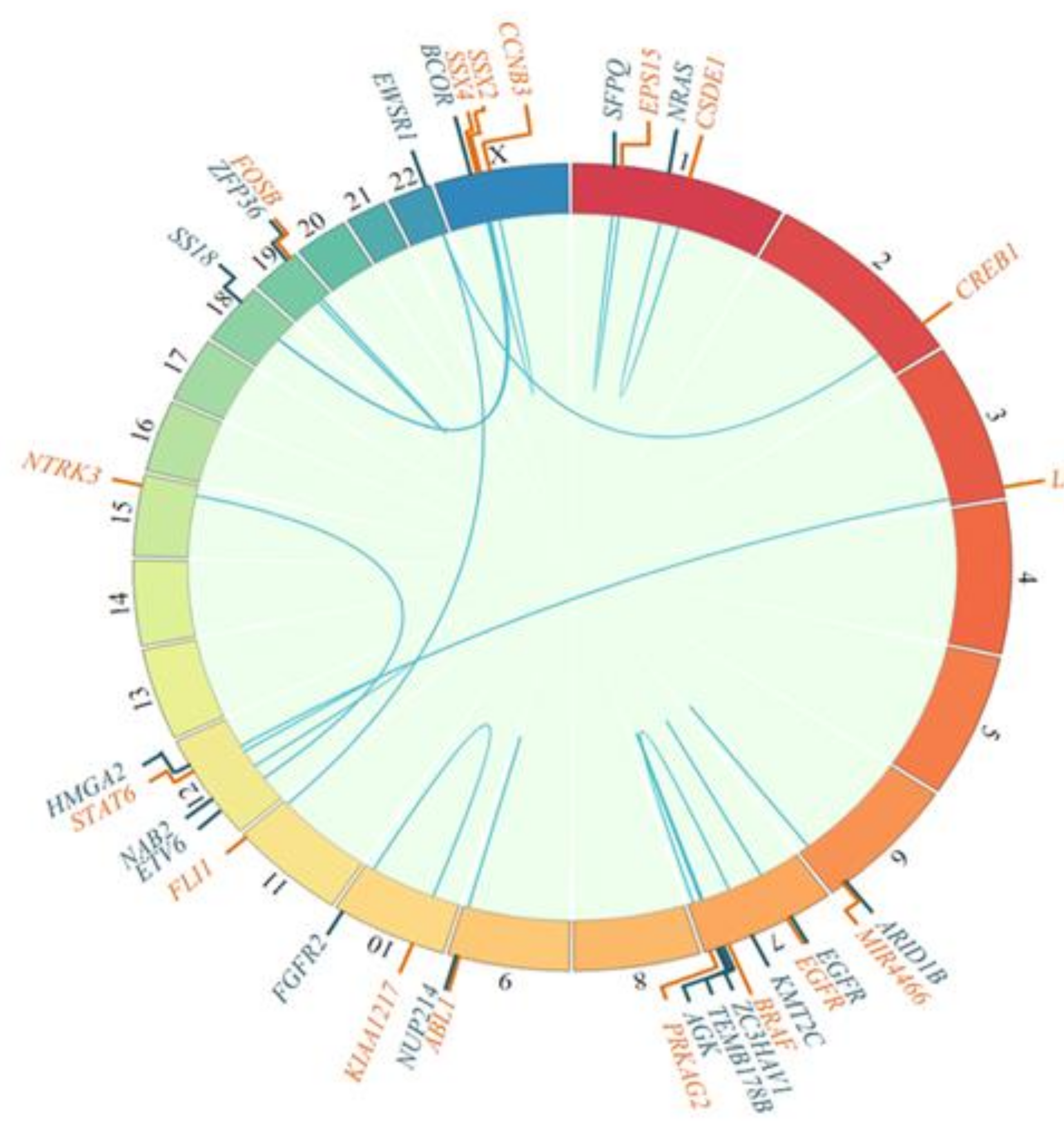
3.2. Genetic Alterations Detected in Sarcomas
3.2.1. Osteosarcoma
3.2.2. Ewing’s Sarcoma
3.2.3. Other Rare Sarcomas
3.3. Clinical Relevance of the Genomic Alterations Detected in Pediatric Sarcomas and Clinical Actions Based on the Results of the Genomic Analysis
- Case #2. Patient initially diagnosed with Ewing-like sarcoma (negative for EWS-FLI1 and EWS-ERG by FISH analysis). The gene fusion FUS-DDIT3, which is the diagnostic hallmark of myxoid liposarcoma, was detected in the genomic analysis. The standard treatment for Ewing’s sarcoma (polychemotherapy, radiotherapy, and autologous hematopoietic progenitor transplant) would have been much more aggressive and potentially toxic than that for myxoid liposarcoma (surgery and local radiotherapy).
- Case #10. Patient initially diagnosed with Ewing’s sarcoma, but was negative for EWS-FLI1 and EWS-ERG by FISH analysis. The genomic analysis confirmed the negative FISH results. The final diagnosis was undifferentiated pleomorphic sarcoma. The most common treatment regimen in Ewing’s sarcoma is polychemotherapy; while in pleomorphic sarcoma, the role of chemotherapy is unclear, and surgical resection is central.
- Case #11. Initial diagnosis was inconclusive: suspected undifferentiated pleomorphic sarcoma or liposarcoma. The definitive diagnosis was solitary fibrous tumor (SFT), identified by the pathognomonic gene fusion: NAB2-STAT6. SFT is a neoplasm of intermediate biological potential, and surgical resection alone is the standard treatment in most cases, although in some cases radio or even chemotherapy is indicated.
- Cases #13 and #21. Initial diagnosis was osteosarcoma. The gene fusion EWS-FLI1, which is pathognomonic of Ewing´s sarcoma, was detected by genomic analyses. Ewing’s sarcoma and osteosarcoma treatment regimens are different, and treatment for Ewing’s sarcoma frequently involves radiotherapy.
- Case #17. Initially diagnosed with a low-grade tumor after ruling out synovial sarcoma (there was not SS18 rearrangement). Genomic analysis detected the gene fusion BCOR-CCNB3, which, in the new WHO classification, is pathognomonic for BCOR sarcoma, a high-grade tumor. Treatment for BCOR sarcoma includes chemotherapy; surgical resection alone is insufficient.
- Case #40. This patient was positive for SS18 by immunochemistry in the pathological study and consequently diagnosed as having synovial sarcoma. In the genomic analysis, however, SS18 rearrangement was not detected, and the sarcoma was reclassified as pleomorphic undifferentiated sarcoma. Patients with synovial sarcoma rearrangements might benefit from first-line combination treatment of ifosfamide with doxorubicin.
- Case #49. Initial diagnosis was Ewing´s sarcoma. There were no EWSR1 rearrangements in the genomic panel, but two truncating mutations were present in SMARCB1. The diagnosis was changed to rhabdoid soft tissue tumor, which requires intensive multiagent chemotherapy, and could benefit from a selective EZH2 inhibitor.
- Case #50. Initial diagnosis was Ewing´s sarcoma, as the patient was positive by FISH for the classical EWSR1 rearrangements. The patient underwent four cycles of treatment according to the EuroEwing 2012 protocol before genomic analysis detected the EWSR1-CREB1 rearrangement, the diagnosis was changed to angiomatoid fibrous histiocytoma, and chemotherapy was suspended. Radiotherapy was continued.
3.4. Detection of Potentially Actionable Genomic Alterations
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peris-Bonet, R.; Salmerón, D.; Martínez-Beneito, M.A.; Galceran, J.; Marcos-Gragera, R.; Felipe, S.; González, V.; Sánchez de Toledo Codina, J. Spanish Childhood Cancer Epidemiology Working Group. Spanish Childhood Cancer Epidemiology Working Group. Childhood cancer incidence and survival in Spain. Ann. Oncol. 2010, 21, iii103–iii110. [Google Scholar] [CrossRef]
- Gatta, G.; Botta, L.; Rossi, S.; Aareleid, T.; Bielska-Lasota, M.; Clavel, J.; Dimitrova, N.; Jakab, Z.; Kaatsch, P.; Lacour, B.; et al. Childhood cancer survival in Europe 1999–2007: Results of EUROCARE-5—A population-based study. Lancet Oncol. 2014, 15, 35–47. [Google Scholar] [CrossRef]
- Groisberg, R.; Roszik, J.; Conley, A.; Patel, S.R.; Subbiah, V. The role of next-generation sequencing in sarcomas: Evolution from light microscope to molecular microscope. Curr. Oncol. Rep. 2017, 19, 1–8. [Google Scholar] [CrossRef]
- Sbaraglia, M.; Righi, A.; Gambarotti, M.; Dei Tos, A.P. Ewing sarcoma and Ewing-like tumors. Virchows Arch. 2020, 476, 109–119. [Google Scholar] [CrossRef]
- Rau, R.E.; Loh, M.L. Using genomics to define pediatric blood cancers and inform practice. Hematol. Am. Soc. Hematol. Educ. Program Book 2018, 1, 286–300. [Google Scholar] [CrossRef] [Green Version]
- Pincez, T.; Clément, N.; Lapouble, E.; Pierron, G.; Kamal, M.; Bieche, I.; Bernard, V.; Fréneaux, P.; Michon, J.; Orbach, D.; et al. Feasibility and clinical integration of molecular profiling for target identification in pediatric solid tumors. Pediatr. Blood Cancer 2017, 64, e26365. [Google Scholar] [CrossRef]
- Tirtei, E.; Cereda, M.; De Luna, E.; Quarello, P.; Asaftei, S.D.; Fagioli, F. Omic approaches to pediatric bone sarcomas. Pediatr. Blood Cancer 2020, 67, e28072. [Google Scholar] [CrossRef]
- Montella, L.; Altucci, L.; Sarno, F.; Buonerba, C.; De Simone, S.; Facchini, B.A.; Franzese, E.; De Vita, F.; Tafuto, S.; Berretta, M.; et al. Toward a Personalized Therapy in Soft-Tissue Sarcomas: State of the Art and Future Directions. Cancers 2021, 13, 2359. [Google Scholar] [CrossRef]
- Gounder, M.M.; Ali, S.M.; Robinson, V.; Bailey, M.; Ferraro, R.; Patel, N.M.; Krishnan, A.; Millis, S.Z.; Dickson, M.A.; D’Angelo, S.P.; et al. Impact of next-generation sequencing (NGS) on diagnostic and therapeutic options in soft-tissue and bone sarcoma. J. Clin. Oncol. 2017, 35 (Suppl. S15), 11001. [Google Scholar]
- Italiano, A.; Di Mauro, I.; Rapp, J.; Pierron, G.; Auger, N.; Alberti, L.; Chibon, F.; Escande, F.; Voegeli, A.C.; Ghnassia, J.P.; et al. Clinical effect of molecular methods in sarcoma diagnosis (GENSARC): A prospective, multicentre, observational study. Lancet Oncol. 2016, 17, 532–538. [Google Scholar] [CrossRef]
- Lucchesi, C.; Khalifa, E.; Laizet, Y.; Soubeyran, I.; Mathoulin-Pelissier, S.; Chomienne, C.; Italiano, A. Targetable alterations in adult patients with soft-tissue sarcomas: Insights for personalized therapy. JAMA Oncol. 2018, 4, 1398–1404. [Google Scholar] [CrossRef]
- Vyse, S.; Thway, K.; Huang, P.H.; Jones, R.L. Next-generation sequencing for the management of sarcomas with no known driver mutations. Curr. Opin. Oncol. 2021, 33, 315–322. [Google Scholar] [CrossRef]
- Xiao, X.; Garbutt, C.C.; Hornicek, F.; Guo, Z.; Duan, Z. Advances in chromosomal translocations and fusion genes in sarcomas and potential therapeutic applications. Cancer Treat. Rev. 2018, 63, 61–70. [Google Scholar] [CrossRef]
- Dufresne, A.; Brahmi, M.; Karanian, M.; Blay, J.Y. Using biology to guide the treatment of sarcomas and aggressive connective-tissue tumors. Nat. Rev. Clin. Oncol. 2018, 15, 443–458. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [Green Version]
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A precision oncology knowledge base. JCO Precis. Oncol. 2017, 1, 1–16. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- McLeod, C.; Gout, A.M.; Zhou, X.; Thrasher, A.; Rahbarinia, D.; Brady, S.W.; Macias, M.; Birch, K.; Finkelstein, D.; Sunny, J.; et al. St. Jude Cloud: A Pediatric Cancer Genomic Data-Sharing Ecosystem. Cancer Discov. 2021, 11, 1082–1099. [Google Scholar] [CrossRef]
- Hu, X.; Wang, Q.; Tang, M.; Barthel, F.; Amin, S.; Yoshihara, K.; Lang, F.M.; Martinez-Ledesma, E.; Lee, S.H.; Zheng, S.; et al. TumorFusions: An integrative resource for cancer-associated transcript fusions. Nucleic Acids Res. 2018, 46, D1144–D1149. [Google Scholar] [CrossRef] [Green Version]
- Piñeiro-Yáñez, E.; Reboiro-Jato, M.; Gómez-López, G.; Perales-Patón, J.; Troulé, K.; Rodríguez, J.M.; Tejero, H.; Shimamura, T.; López-Casas, P.P.; Carretero, J.; et al. PanDrugs: A novel method to prioritize anticancer drug treatments according to individual genomic data. Genome Med. 2018, 10, 1–11. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Gross, A.M.; Wolters, P.L.; Dombi, E.; Baldwin, A.; Whitcomb, P.; Fisher, M.J.; Weiss, B.; Kim, A.; Bornhorst, M.; Shah, A.C.; et al. Selumetinib in children with inoperable plexiform neurofibromas. N. Engl. J. Med. 2020, 382, 1430–1442. [Google Scholar] [CrossRef]
- Dombi, E.; Baldwin, A.; Marcus, L.J.; Fisher, M.J.; Weiss, B.; Kim, A.; Whitcomb, P.; Martin, S.; Aschbacher-Smith, L.E.; Rizvi, T.A.; et al. Activity of selumetinib in neurofibromatosis type 1–related plexiform neurofibromas. N. Engl. J. Med. 2016, 375, 2550–2560. [Google Scholar] [CrossRef]
- Ku, B.M.; Bae, Y.H.; Lee, K.Y.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; Park, K.; Ahn, M.J. Entrectinib resistance mechanisms in ROS1-rearranged non-small cell lung cancer. Investig. New Drugs 2020, 38, 360–368. [Google Scholar] [CrossRef] [Green Version]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumors: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Doebele, R.C.; Davis, L.E.; Vaishnavi, A.; Le, A.T.; Estrada-Bernal, A.; Keysar, E.; Jimeno, A.; Varella-Garcia, M.; Aisner, D.L.; Li, Y.; et al. An oncogenic NTRK fusion in a patient with soft-tissue sarcoma with response to the tropomyosin-related kinase inhibitor LOXO-101. Cancer Discov. 2015, 5, 1049–1057. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of larotrectinib in TRK fusion–positive cancers in adults and children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Anderson, W.J.; Doyle, L.A. Updates from the 2020 World Health Organization Classification of Soft Tissue and Bone Tumors. Histopathology 2021, 78, 644–657. [Google Scholar] [CrossRef]
- Dancsok, A.R.; Asleh-Aburaya, K.; Nielsen, T.O. Advances in sarcoma diagnostics and treatment. Oncotarget 2017, 8, 7068. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Li, M.M.; Schubert, J.; Wu, J.; Lin, F.; Wertheim, G.B.; Surrey, L.; Luo, M.; Zhong, Y.; Wu, C.; et al. Clinical significance of serial tumor next generation sequencing (NGS) in 155 pediatric cancer patients. J. Clin. Oncol. 2020, 38, e13666. [Google Scholar] [CrossRef]
- Mody, R.J.; Wu, Y.M.; Lonigro, R.J.; Cao, X.; Roychowdhury, S.; Vats, P.; Frank, K.M.; Prensner, J.R.; Asangani, I.; Palanisamy, N.; et al. Integrative clinical sequencing in the management of refractory or relapsed cancer in youth. JAMA 2015, 314, 913–925. [Google Scholar] [CrossRef]
- Harris, M.H.; DuBois, S.G.; Bender, J.L.G.; Kim, A.; Crompton, B.D.; Parker, E.; Dumont, I.P.; Hong, A.L.; Guo, D.; Church, A.; et al. Multicenter feasibility study of tumor molecular profiling to inform therapeutic decisions in advanced pediatric solid tumors: The individualized cancer therapy (iCat) study. JAMA Oncol. 2016, 2, 608–615. [Google Scholar] [CrossRef]
- Parsons, D.W.; Roy, A.; Yang, Y.; Wang, T.; Scollon, S.; Bergstrom, K.; Kerstein, R.A.; Gutierrez, S.; Petersen, A.K.; Bavle, A.; et al. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol. 2016, 2, 616–624. [Google Scholar] [CrossRef]
- Chang, W.; Brohl, A.S.; Patidar, R.; Sindiri, S.; Shern, J.F.; Wei, J.S.; Song, Y.K.; Yohe, M.E.; Gryder, B.; Zhang, S.; et al. MultiDimensional ClinOmics for Precision Therapy of Children and Adolescent Young Adults with Relapsed and Refractory Cancer: A Report from the Center for Cancer Research. Clin. Cancer Res. 2016, 22, 3810–3820. [Google Scholar] [CrossRef] [Green Version]
- Suehara, Y.; Alex, D.; Bowman, A.; Middha, S.; Zehir, A.; Chakravarty, D.; Wang, L.; Jour, G.; Nafa, K.; Hayashi, T.; et al. Clinical genomic sequencing of pediatric and adult osteosarcoma reveals distinct molecular subsets with potentially targetable alterations. Clin. Cancer Res. 2019, 25, 6346–6356. [Google Scholar] [CrossRef] [Green Version]
- Rubio-San-Simón, A.; Hladun Alvaro, R.; Juan Ribelles, A.; Castañeda Heredia, A.; Guerra-García, P.; Verdú-Amorós, J.; Andrés, M.; Cañete, A.; Rives, S.; Pérez-Martínez, A.; et al. The paediatric cancer clinical research landscape in Spain: A 13-year multicentre experience of the new agents group of the Spanish Society of Paediatric Haematology and Oncology (SEHOP). Clin. Transl. Oncol 2021. online ahead of print. [Google Scholar] [CrossRef]
- Kallen, M.E.; Hornick, J.L. The 2020 WHO Classification: What’s new in soft tissue tumor pathology? Am. J. Surg. Pathol. 2021, 45, e1–e23. [Google Scholar] [CrossRef]
- Cidre-Aranaz, F.; Alonso, J. EWS/FLI1 target genes and therapeutic opportunities in Ewing sarcoma. Front. Oncol. 2015, 5, 162. [Google Scholar] [CrossRef] [Green Version]
- Groisberg, R.; Hong, D.S.; Holla, V.; Janku, F.; Piha-Paul, S.; Ravi, V.; Benjamin, R.; Patel, S.K.; Somaiah, N.; Conley, A.; et al. Clinical genomic profiling to identify actionable alterations for investigational therapies in patients with diverse sarcomas. Oncotarget 2017, 8, 39254–39267. [Google Scholar] [CrossRef]




| Gene | Type of Alteration | N Cases | OncoKB Level | Drugs |
|---|---|---|---|---|
| NF1 | Truncating mutation | 1 | Level 1 | Selumetinib |
| ETV6-NTRK3 | Fusion | 1 | Level 1 | Larotrectinib |
| CDK4 | Amplification | 2 | Level 2B | Palbociclib, abemaciclib |
| KIT | Amplification | 5 | Level 3B | Imatinib, sunitinib, regorafenib, ripetrinib |
| PDGFRA | Amplification | 4 | Level 3B | Imatinib, sunitinib |
| BRAF | Fusion | 3 | Level 3B | Cobimetinib, trametinib |
| IDH1 | Mutation missense | 1 | Level 3B | Ivosidenib |
| MET | Amplification | 1 | Level 3B | Cabozantinib, crizotinib |
| FLI1 | Fusion | 14 | Level 4 | TK216 |
| PTEN | Deletion/Truncating mutation | 5 | Level 4 | AZD8186, GSK2636771 |
| CDKN2A | Deletion/Truncating mutation | 4 | Level 4 | Abemaciclib, ribociclib, palbociclib |
| FGFR1/FGFR3 | Amplification | 4 | Level 4 | AZD4547, erdafitinib, BGJ398, Debio1347 |
| SMARCB1 | Fusion/Truncating mutation | 2 | Level 4 | Tazemetostat |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutiérrez-Jimeno, M.; Alba-Pavón, P.; Astigarraga, I.; Imízcoz, T.; Panizo-Morgado, E.; García-Obregón, S.; Catalán-Lambán, A.; San-Julián, M.; Lamo-Espinosa, J.M.; Echebarria-Barona, A.; et al. Clinical Value of NGS Genomic Studies for Clinical Management of Pediatric and Young Adult Bone Sarcomas. Cancers 2021, 13, 5436. https://doi.org/10.3390/cancers13215436
Gutiérrez-Jimeno M, Alba-Pavón P, Astigarraga I, Imízcoz T, Panizo-Morgado E, García-Obregón S, Catalán-Lambán A, San-Julián M, Lamo-Espinosa JM, Echebarria-Barona A, et al. Clinical Value of NGS Genomic Studies for Clinical Management of Pediatric and Young Adult Bone Sarcomas. Cancers. 2021; 13(21):5436. https://doi.org/10.3390/cancers13215436
Chicago/Turabian StyleGutiérrez-Jimeno, Miriam, Piedad Alba-Pavón, Itziar Astigarraga, Teresa Imízcoz, Elena Panizo-Morgado, Susana García-Obregón, Ana Catalán-Lambán, Mikel San-Julián, José M. Lamo-Espinosa, Aizpea Echebarria-Barona, and et al. 2021. "Clinical Value of NGS Genomic Studies for Clinical Management of Pediatric and Young Adult Bone Sarcomas" Cancers 13, no. 21: 5436. https://doi.org/10.3390/cancers13215436