1. Introduction
Gastric cancer (GC) is a complex and heterogeneous disease from morphological, molecular, and cellular standpoints [
1]. Such tumor heterogeneity has been demonstrated in numerous histological and molecular classifications. The Lauren classification separates gastric adenocarcinomas into intestinal, diffuse, and mixed subtypes, which were found to be associated with varying stomach cancer risks [
2,
3]. The Asian Cancer Research Group (ACRG) classified GC into four molecular subtypes that, based on gene expression data, were associated with distinct molecular alterations, disease progression, and survival outcomes [
4]. These subtypes are epithelial-to-mesenchymal transition (EMT), MSS/TP53-, MSS/TP53+, and microsatellite instability (MSI). More recently, The Cancer Genome Atlas (TCGA) research network characterized GC into four genomic subtypes by integrating data from six molecular platforms: array-based somatic copy number analysis, whole-exome sequencing, array-based DNA methylation profiling, messenger RNA sequencing, microRNA (miRNA) sequencing, and reverse-phase protein array, as well as Microsatellite instability (MSI) testing [
5]. These genomic subtypes are EBV-positivity (EBV), MSI-high status (MSI), genomically stable (GS), and those exhibiting chromosomal instability (CIN). Each subtype displays distinct molecular and genomic patterns.
Tumor heterogeneity, including the results from the tumor microenvironment (TME), continues to confound researchers’ understanding of tumor growth and the development of an effective therapy [
6,
7]. Tumors are complex ecosystems that are affected by numerous stromal and immune factors, which dampen or enhance the effects of genetic epithelial alterations [
8,
9,
10,
11]. The TME is comprised of tumor cells, tumor stromal cells, endothelial cells, immune cells, and the non-cellular components of extracellular matrix proteins [
12,
13]. Some essential components of the TME, including cancer-associated fibroblasts (CAF) [
14,
15], tumor-infiltrating lymphocytes (TIL) [
16,
17], tumor-associated macrophages (TAM) [
18], and other cellular components [
19,
20,
21], have been evaluated to help researchers better understand the role of the TME in gastric cancer risk. Most of these studies have focused on the subsets of cellular components of TME: typically, the stromal and immune cell populations. However, the prognostic values of diverse gastric epithelial cell types in GC risk were still unclear. To our knowledge, a systematic analysis of the prognostic value of diverse epithelial cell types, including cancer cells, MSC, PMC and PC, emerged in early gastric cancer for predicting survival has not been described.
Digital cytometry enables the examination of heterogeneous bulk tissue transcriptomes at the cellular level in addition to using computational methods to quantify cell type composition. In recent years, single-cell RNA-sequencing (scRNA-seq) techniques have offered new insights into tissue samples at the resolution of single cells. The availability of single cell transcriptomic profiles, which are used to build cell-type-specific signature matrices, promote the development of statistical deconvolution methods for estimating cell type compositions in heterogeneous mixture samples [
22,
23].
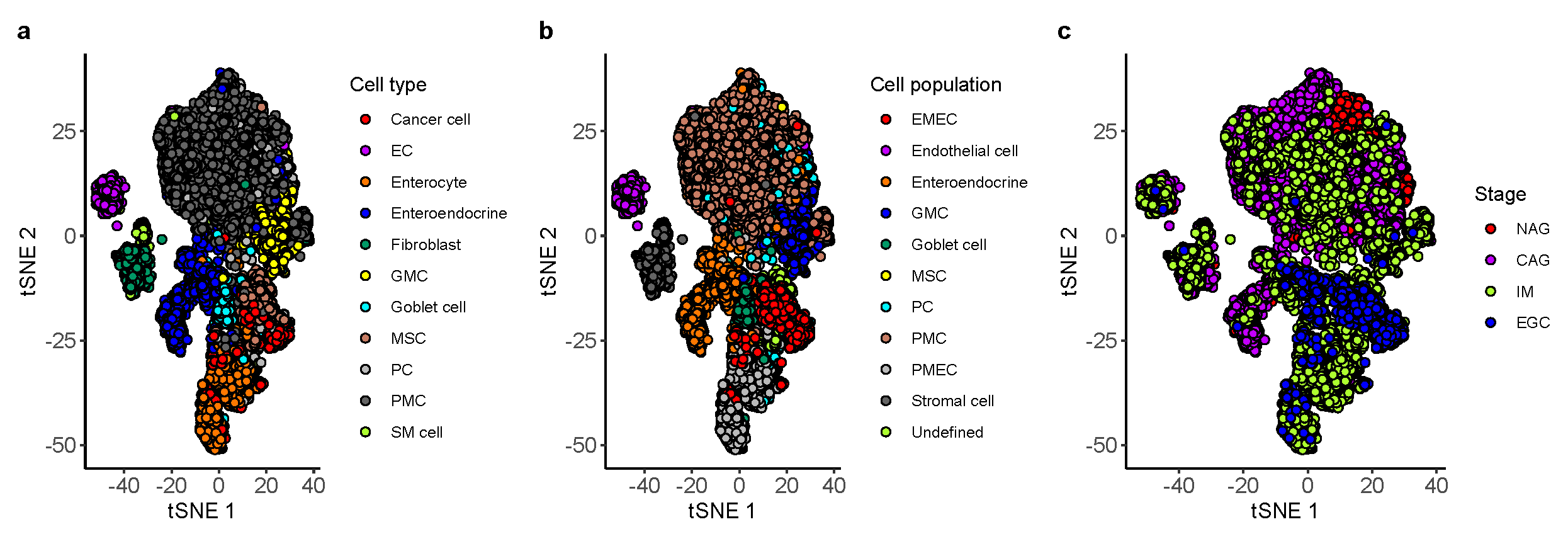
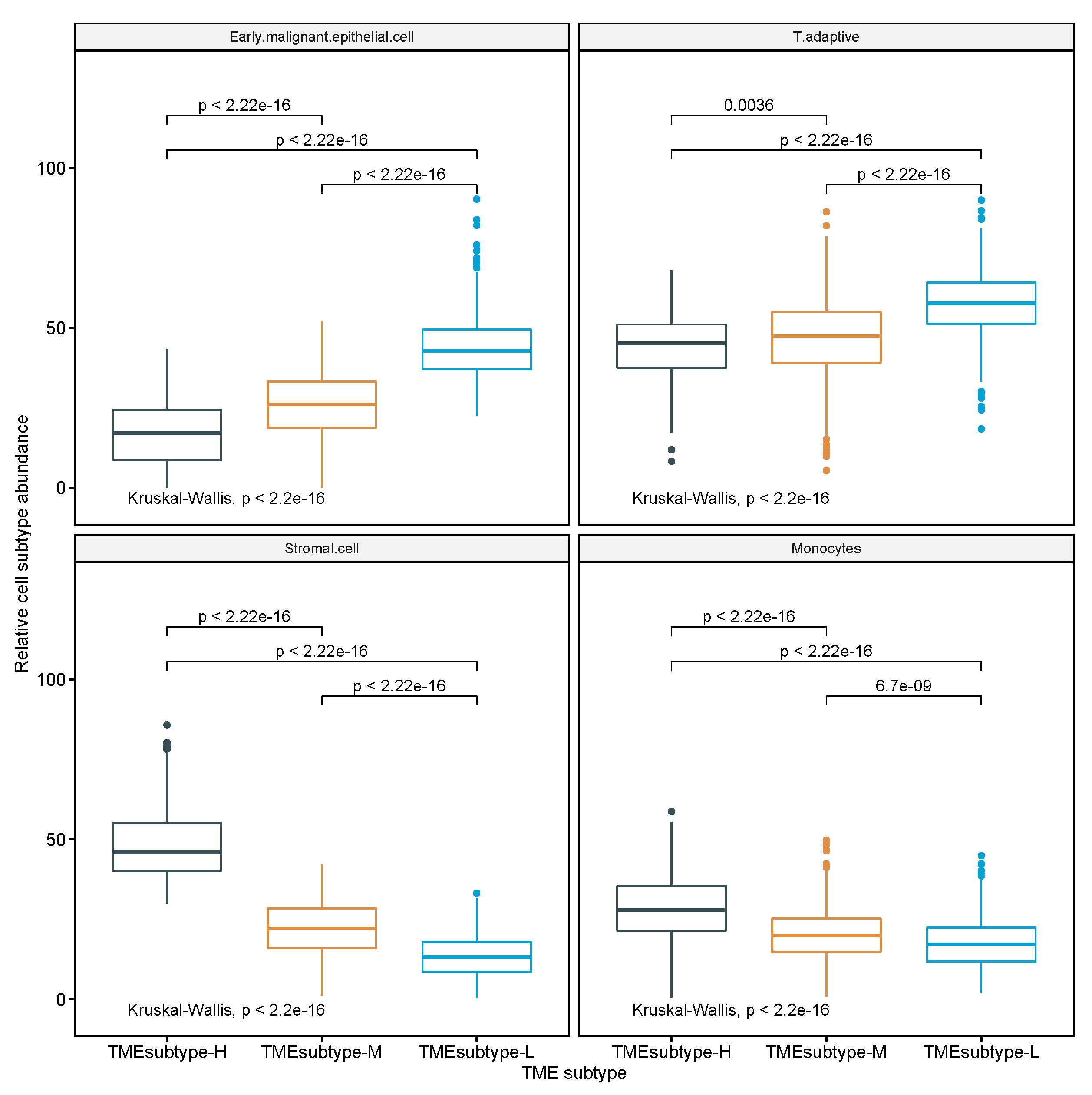
This study attempted to investigate the TME of GC with comprehensive epithelial, stromal, and immune cell profiling by combining single-cell and bulk expression profiles. We undertook a comprehensive analysis on 10 non-immune and 7 immune cell populations of the TME of GC, evaluated the prognostic role of a novel TME signature score called STEM score in four independent GC cohorts, and stratified the GC patients into three TME subtypes based on the abundance of four STEM populations (i.e., Stromal cell, adaptive T cell, EMEC, and Monocyte).
4. Discussion
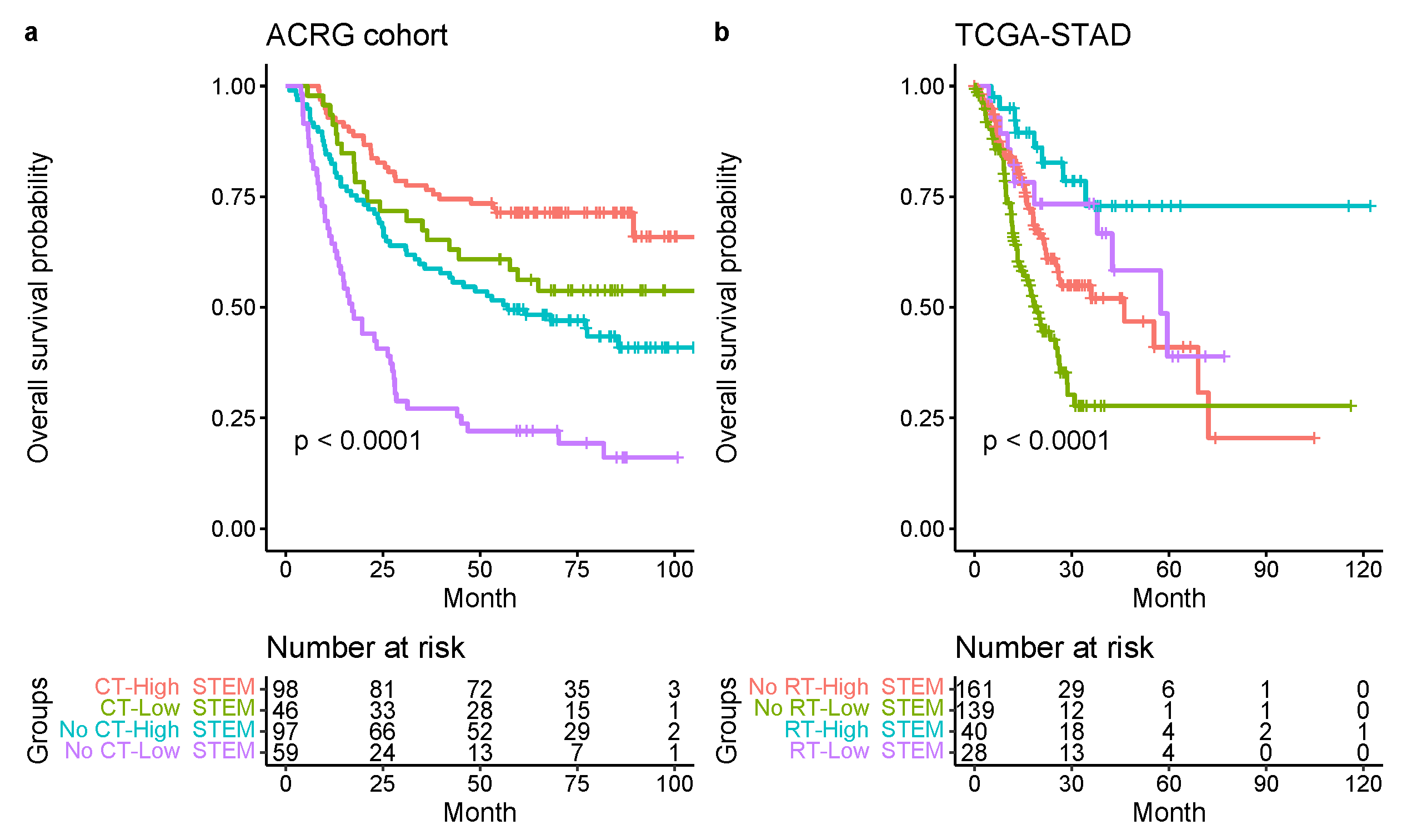
In the present study, a comprehensive and systematic analysis of diverse epithelial, stromal, and immune cell types within the TME and their associations with GC risk was developed. We examined several large cohorts of GC patients at the cellular level and found a new and strong independent prognostic factor (STEM score) for GC patients. The STEM score was defined as the arithmetic sum of the two most significant TME factors: the EMEC-to-stromal ratio and the adaptive-T-cell-to-monocyte ratio. Our results suggest that high-risk patient groups (STEM score ) had significantly shorter OS times than patients in the low-risk group (STEM score ). The STEM score includes four major epithelial, stromal, and immune cell populations and greatly facilitated the quantitative characterization of GC TME in a comprehensive manner.
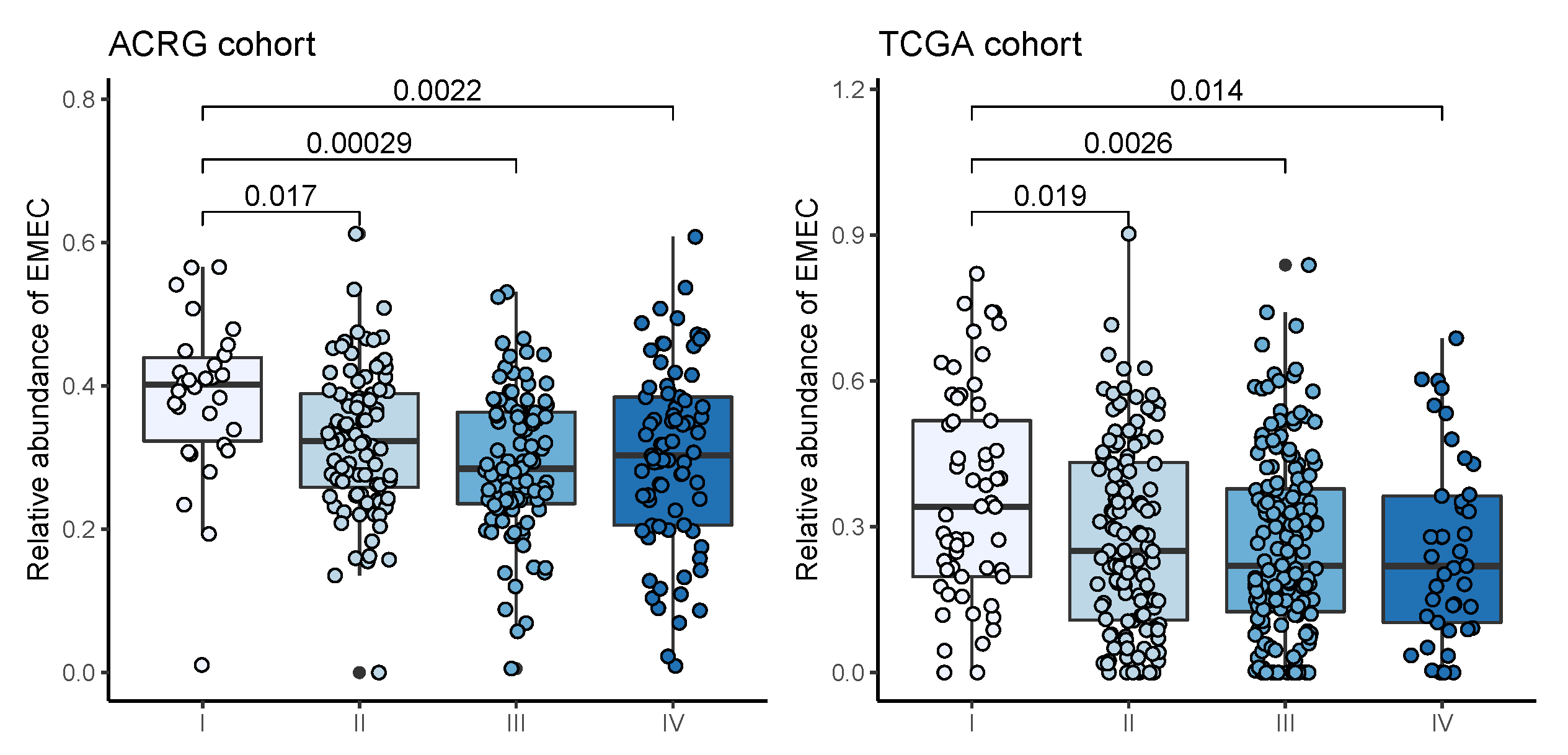
Stromal cells, especially cancer-associated fibroblasts (CAFs), in the TME have been found to promote growth and survival of malignant cells [
36]. Many studies have found that cancer cells release factors promoting fibroblasts to secrete tumor-promoting chemokines [
37]. The interactions of tumors and CAFs can lead to increased malignancy in many cancer types [
38,
39]. Several studies of GC suggest that a low tumor-to-stroma ratio (TSR) is associated with a poor prognosis [
19,
40]. Herein, we analyzed a gastric scRNA-seq data set that covered diverse epithelial cell types isolated from patients with NAG, CAG, IM, and EGC, and identified the EMEC population. The EMEC-to-stromal cell ratio was shown to have significant correlation to OS, which agreed with previous studies [
19,
40] on the positive prognostic value of TSR. The EMEC population is comprised of cancer cells, MSCs, PCs, and PMCs that emerged uniformly in the EGC biopsy and that were predominantly present in tumor samples but not typically found in adjacent normal samples. Additionally, significantly higher EMEC populations were detected in patients with stage I cancer than stage II, III, or IV, suggesting the value of the EMEC population in the early detection of gastric cancer.
There is increasing evidence that suggests a strong infiltration of T cells, especially CD8+ T cells, into the TME correlates with a good prognosis in many types of cancer, and this has implications for the success of active cancer immunotherapy [
41,
42]. Studies have shown that CD8+ T cells play a vital role in mediating anti-tumor immunity, and cytotoxic CD8+ memory T cells kill tumor cells by recognizing tumor-associated antigens presented on major histocompatibility complex class I [
43,
44,
45]. High numbers of CD4+ T helper 1 cells in the TME also correlate with a better prognosis [
45]. Tumor-associated macrophages were found to enhance malignant cell migration, invasion, and metastases [
46]. Monocytes can give rise to macrophages, so the abundance of monocytes may lead to increased production of macrophages. In our study of GC cohorts, an increased adaptive T cell to monocyte ratio was significantly associated with increased OS. This is in line with studies by [
47] of haematologic malignancies and [
48] of stage III colon cancer. The studies of haematologic malignancies and stage III colon cancer demonstrate that an elevated lymphocyte-to-monocyte ratio (LMR) yields a better survival outcome.
Molecular signatures associated with distinct clinical outcomes have been studied in many types of cancer [
5,
49,
50]. We identified several gene signatures of the EMEC and stromal cell populations to be independent prognostic factors of OS in multivariate analysis. Several prognostic gene signatures of stromal cells have been previously reported to play oncogenic roles in cancer cell proliferation, migration, or invasion. FERMT2 (also known as Kindlin-2), a focal adhesion protein, has been found to regulate cancer cell proliferation, apoptosis, and chromosomal abnormalities in breast cancer that are associated with tumor stromal invasion, lymph node metastasis, and patient outcome in gastric cancer. Over-expression of FERMT2 promotes tumor formation in breast cancer and was linked with poorer patient outcomes [
51,
52]. TAGLN is expressed in fibroblasts and smooth muscle, and the overexpression of TAGLN has been found in the tumor-induced reactive myofibroblastic stromal tissue in lung adenocarcinoma, as well in carcinomas of the stomach, liver, and oesophagus [
53]. Silencing of TAGLN2, a homologue of TAGLN, has been reported to significantly inhibit cell proliferation and increase apoptosis in bladder cancer [
54]. MYL9 was previously found to be over-expressed in stages III and IV of non-small-cell lung cancer [
55]. Over-expression of MYL9 in tumor cells was associated with poorer OS and recurrence-free survival in esophageal squamous cell carcinoma [
56]. TPM2, a marker of fibroblasts, was previously reported to be associated with poor prognosis in colorectal cancer (CRC) [
57]. The TAGLN, MYL9, and TPM2 were found to be over-expressed in fibroblasts from primary tumors compared to adjacent normal tissues and were associated with a poorer prognosis in the TCGA cohort of colorectal cancer [
57]. PPP1R14A, also known as CPI-17, has been investigated as a prognostic biomarker of gastric cancer [
58]. Previous studies have revealed that PPP1R14A can drive Ras activity and promote tumorigenic transformation [
59,
60]. LAMC1 was found to be a target of miR-29s. Silencing of LAMC1 significantly inhibited cell migration and invasion in prostate cancer cells [
61]. AKAP12 has been investigated as a tumor suppressant in some human primary cancers, including GC [
62,
63]; however, in the present study, we found it significantly over-expressed in the TMEsubtype-H high-risk group, suggesting an increased risk of OS with higher expression levels.
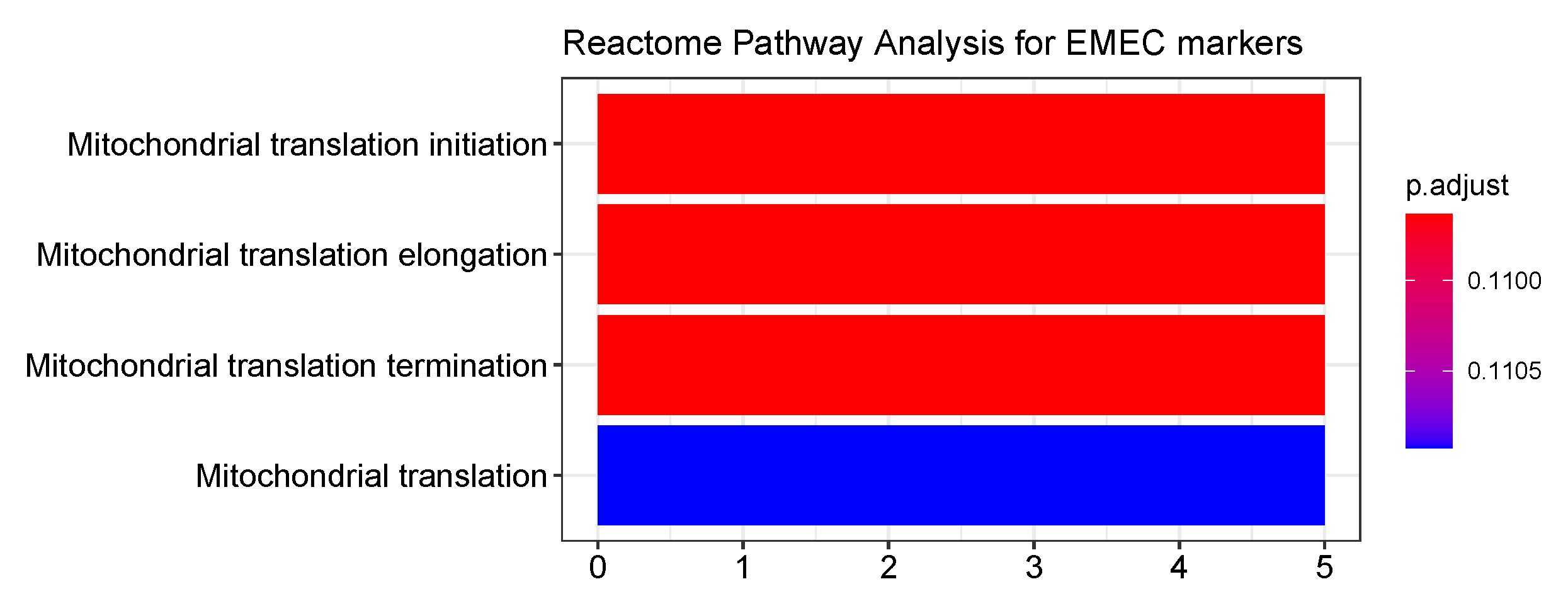
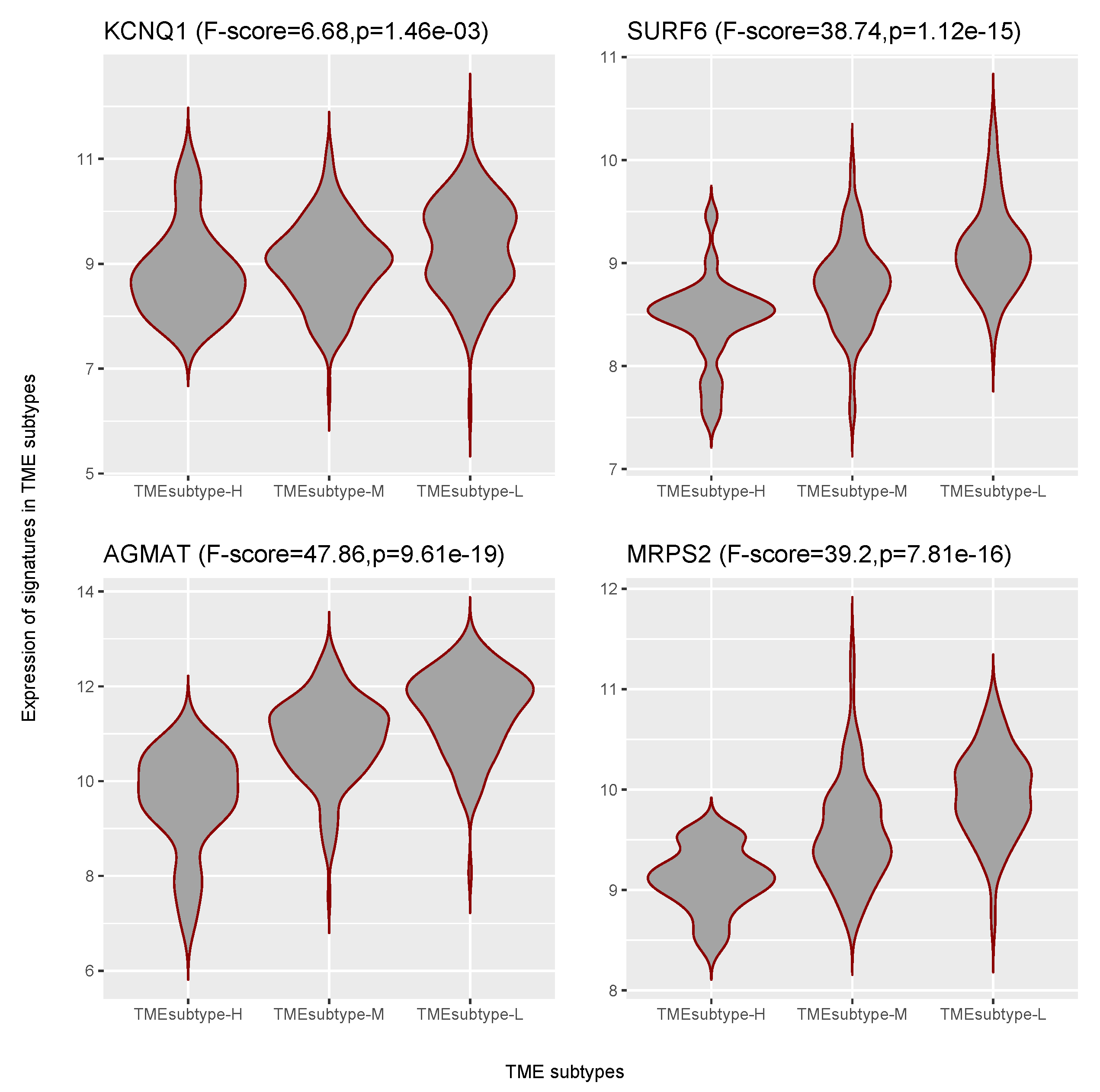
Herein, we identified four prognostic gene signatures of the EMEC population to be positively associated with OS. AGMAT were found to be positively associated with OS in kidney renal clear cell carcinoma [
64]. MRPS2 encoding the mitochondrial ribosomal protein S2 was important for mitoribosome formation and stability and mitochondrial translation. It was reported to predict poor OS in ovarian cancer patients [
65]. However, we found that it was associated with better clinical outcomes in GC patients. KCNQ1 has been shown to distribute widely and be functionally relevant in a variety of epithelial tissues [
66]. There is preliminary evidence to suggest that KCNQ1 is a tumor suppressor in the stomach and colon [
67,
68]. Low or loss of expression of KCNQ1 was previously found to associate with poor disease-free survival in stage II and III colon cancer patients [
69]. Moreover, over-expression of KCNQ1 in the colorectal cancer cell line was found to have trapped
-catenin at the plasma membrane, induced a patent lumen in CRC spheroids, and slowed CRC cell invasion [
68].
In addition, many studies have revealed some gene signatures with potential therapeutic value in cancers. LAMC1 has recently been found to be overexpressed in endometrial cancer and is reported to be a potent biomarker for identifying endometrial cancer patients needing aggressive adjuvant therapy [
70]. Several studies have demonstrated that TAGLN2 expression in cancer cells is associated with increased drug resistance, and selectively suppressed TAGLN2 expression may prevent multidrug resistance in cancer chemotherapy [
71,
72,
73,
74]. Over-expression of FERMT2 was previously found to promote melanoma growth and migration, which was attributed to stimulate the downstream MAPK pathway, and was reported to be a potential therapeutic target for treating melanoma [
75].
Strengths of our study include the use of multiple independent validation cohorts, and the use of three transcriptome profiling technologies, including microarray, bulk RNA-seq, and scRNA-seq, which improve the reliability and generalizability of the results. We created and validated the GC10 signature matrix for deconvolution of 10 epithelial and stromal subsets in gastric samples, which enabled the investigation of the heterogeneous tumor microenvironment in GC. Nevertheless, our study had several limitations. The main limitation of this study was the retrospective nature, which might have been subject to selection bias. Moreover, we characterized the cell populations using only gene expression measurements; high-resolution mass-spectrometry-based proteomics profiling may reduce the potential bias of transcriptome profiling technologies. The retrospective nature of this study calls for further validation using a prospective investigation.
In conclusion, we identified significant prognostic factors and gene signatures among diverse epithelial, stromal, and immune cell populations in the GC TME. Our findings indicated that assessment of the early malignant epithelial, stromal, adaptive T cells, and monocyte abundance via the STEM score provided a potent predictor of survival in patients with GC. This study demonstrated the STEM score as an independent prognostic factor for GC. It showed that a lower STEM score was significantly associated with a shorter survival time. The entire GC cohort was stratified into three risk groups (high-, moderate-, and low-risk) based on the four STEM populations, which yielded incremental survival times. The risk stratification model may aid stratification of patients with stage III gastric cancer for radiation therapy and may be informative for refinement of molecular subtypes of GC.



























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}