
Epithelial-to-Mesenchymal Transition-Derived Heterogeneity in Head and Neck Squamous Cell Carcinomas


{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
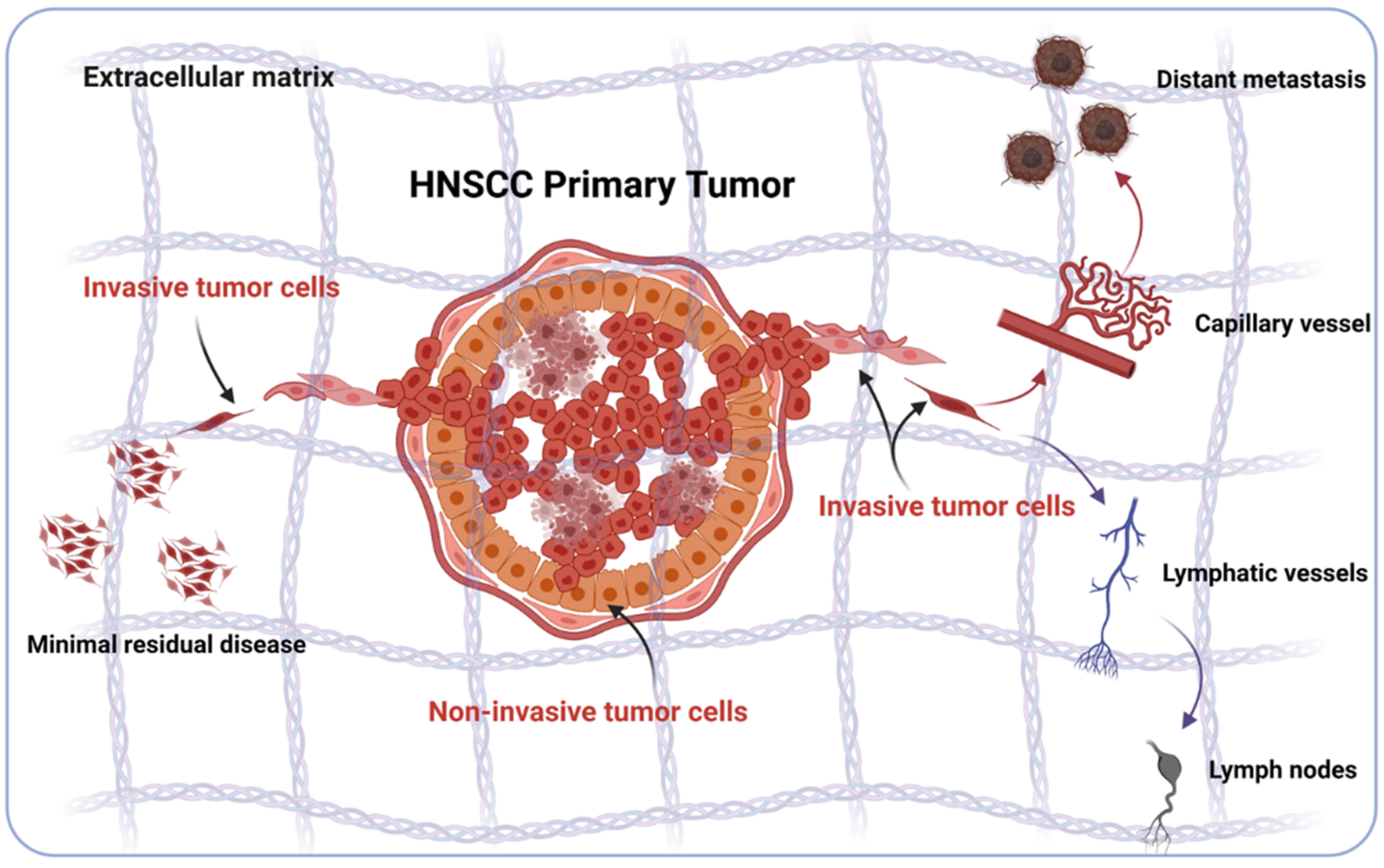
2. Sources of Phenotypic Diversity in HNSCC
3. EMT in HNSCC
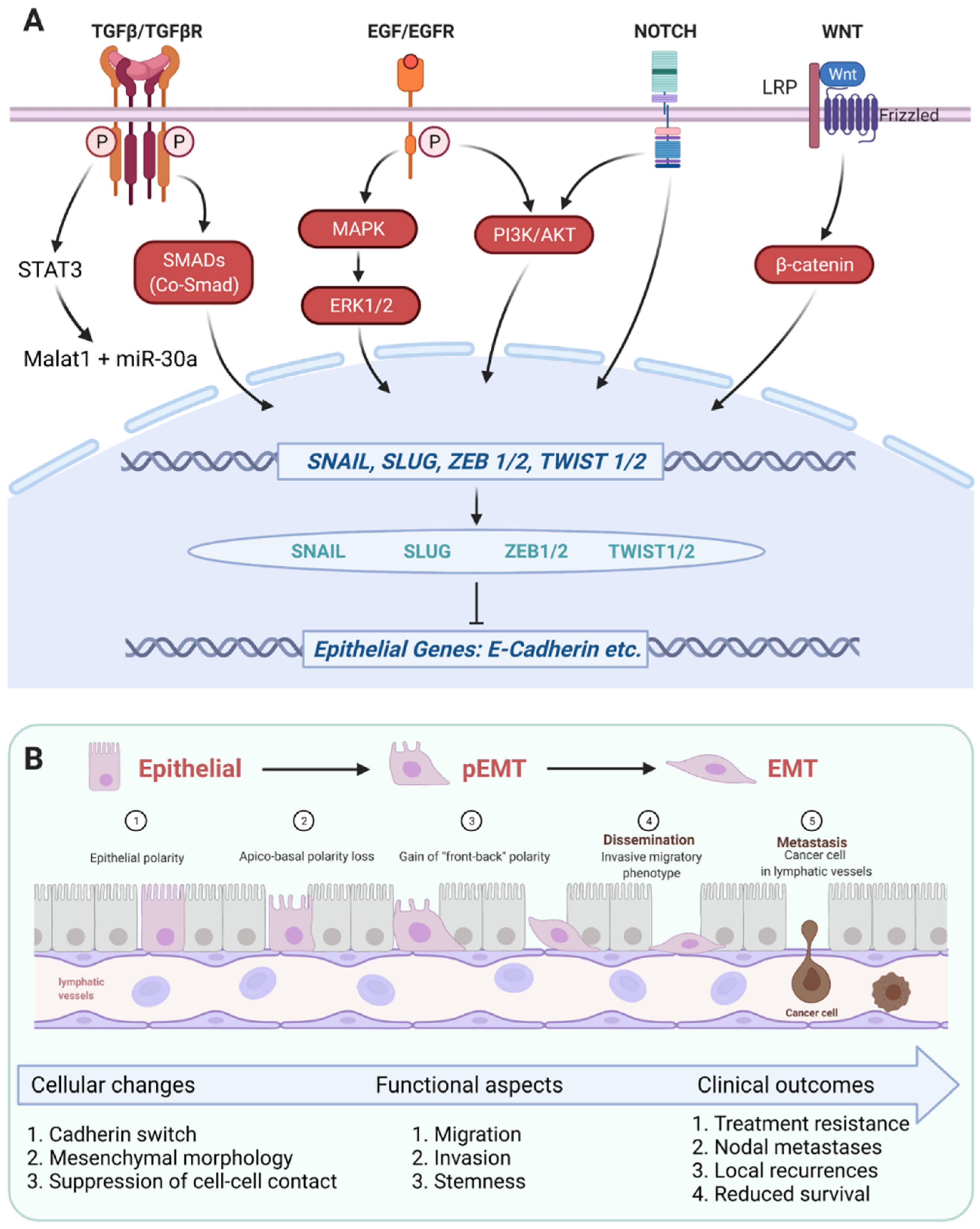
3.1. Regulation of EMT in HNSCC
3.1.1. TGF-β1-Dependent EMT Regulation
3.1.2. EGFR-Dependent EMT Regulation
3.1.3. EMT Transcription Factors in HNSCC
3.2. EMT Gene Signatures
3.3. EMT, Stem-Like Properties, and Treatment Resistance
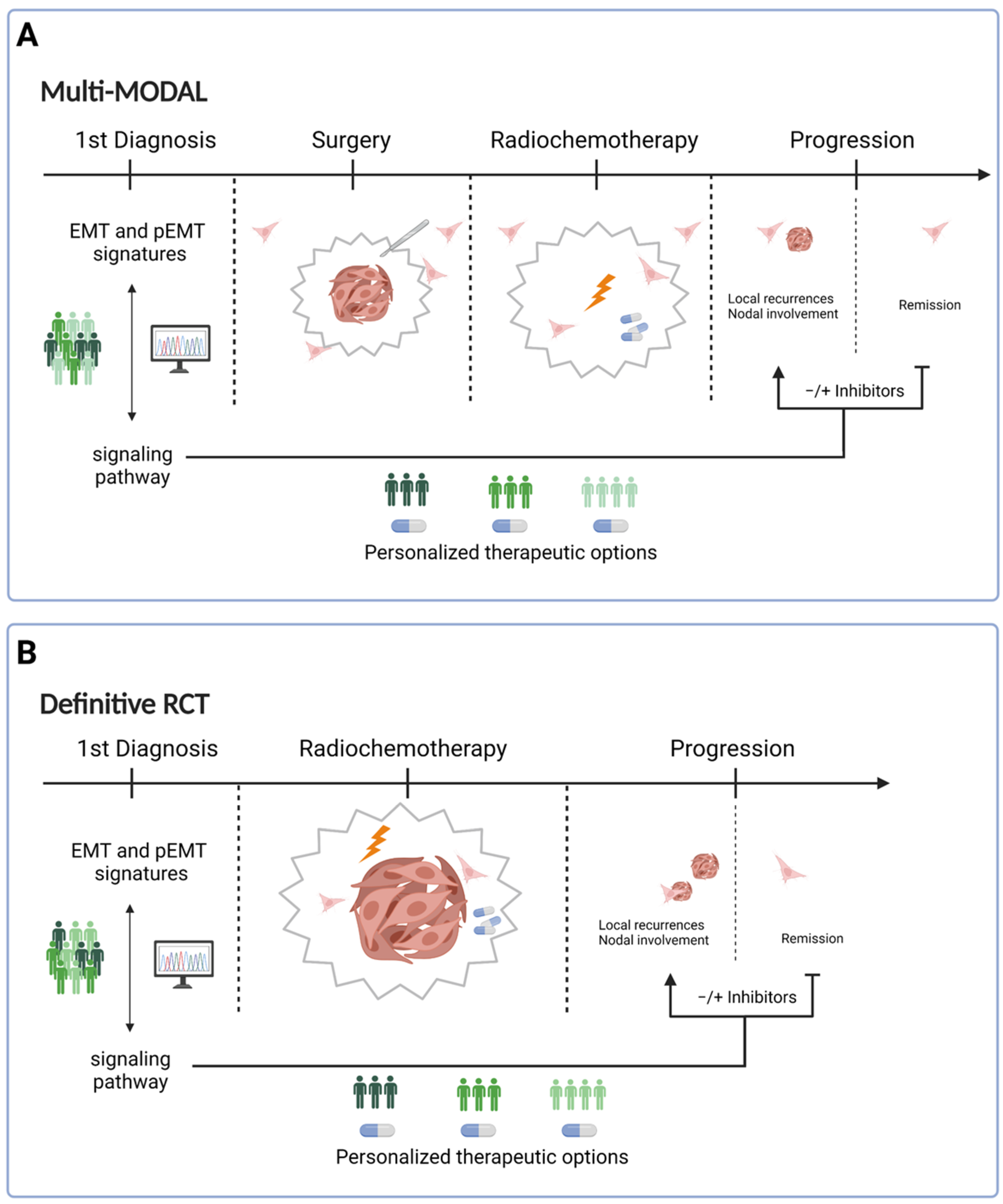
4. Consequences for Treatment
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CAF | Cancer-associate fibroblast |
| CCND1 | Cyclin D1 |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A |
| CXCL1 | C-X-C motif chemokine ligand 1 |
| DFS | Disease-free survival |
| DNA | Deoxyribonucleic acid |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| EMA | European medicines agency |
| EMT-TF | EMT transcription factor |
| EMT | Epithelial-to-mesenchymal transition |
| EpCAM | Epithelial cell adhesion molecule |
| EPIC | Estimating the proportion of immune and cancer cells |
| EZH2 | Enhancer of zeste 2 |
| FDA | Food and drug administration |
| FHCC | Fred Hutchinson cancer center |
| GLI1 | Glioma-associated oncogene 1 |
| GSEA | Gene set enrichment analysis |
| HIF1α | Hypoxia-induced factor 1 alpha |
| HNSCC | Head and neck squamous cell carcinoma |
| HPV | Human papillomavirus |
| IL | Interleukin |
| ITH | Intra- and intertumoral heterogeneity |
| Malat1 | Metastasis-associated lung adenocarcinoma transcript 1 |
| MATH | Mutant allele tumor heterogeneity |
| MDACC | MD Anderson cancer center |
| miRNA | MicroRNA |
| MMP | Matrix metalloproteinase |
| MRD | Minimal residual disease |
| MTD | Maximal tolerable dose |
| MYC | Myc oncogene (Myelocytomatosis oncogene homolog) |
| NPC | Nasopharyngeal carcinoma |
| OS | Overall survival |
| OSCC | Oral squamous cell carcinoma |
| P53 | Tumor protein P53 |
| PD-L1 | Programmed death ligand 1 |
| PD1 | Programmed cell death protein 1 |
| pEMT | Partial EMT |
| PI3KCA | Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha |
| PRRX1 | Paired-related homeobox 1 |
| R(C)T | Radio(chemo)therapy |
| RNA | Ribonucleic acid |
| SCC | Squamous cell carcinoma |
| scRNAseq | Single cell RNA sequencing |
| SMAD | Sma and Mad protein homologs |
| TCGA | The cancer genome atlas |
| TGFβ | Transforming growth factor beta |
| TGFβR | Transforming growth factor beta receptor |
| WNT | Wingless integrated |
| ZEB | Zinc finger E-box-binding homeobox protein |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mroz, E.A.; Tward, A.D.; Pickering, C.R.; Myers, J.N.; Ferris, R.L.; Rocco, J.W. High intratumor genetic heterogeneity is related to worse outcome in patients with head and neck squamous cell carcinoma. Cancer 2013, 119, 3034–3042. [Google Scholar] [CrossRef]
- Raynaud, F.; Mina, M.; Tavernari, D.; Ciriello, G. Pan-cancer inference of intra-tumor heterogeneity reveals associations with different forms of genomic instability. PLoS Genet. 2018, 14, e1007669. [Google Scholar] [CrossRef] [Green Version]
- Morris, L.G.; Riaz, N.; Desrichard, A.; Senbabaoglu, Y.; Hakimi, A.A.; Makarov, V.; Reis-Filho, J.S.; Chan, T.A. Pan-cancer analysis of intratumor heterogeneity as a prognostic determinant of survival. Oncotarget 2016, 7, 10051–10063. [Google Scholar] [CrossRef] [Green Version]
- Zeki, S.S.; McDonald, S.A.; Graham, T.A. Field cancerization in Barrett’s esophagus. Discov. Med. 2011, 12, 371–379. [Google Scholar]
- Slaughter, D.P.; Southwick, H.W.; Smejkal, W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer 1953, 6, 963–968. [Google Scholar] [CrossRef]
- Curtius, K.; Wright, N.A.; Graham, T.A. An evolutionary perspective on field cancerization. Nat. Rev. Cancer 2018, 18, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Braakhuis, B.J.; Tabor, M.P.; Kummer, J.A.; Leemans, C.R.; Brakenhoff, R.H. A genetic explanation of Slaughter’s concept of field cancerization: Evidence and clinical implications. Cancer Res. 2003, 63, 1727–1730. [Google Scholar]
- Leemans, C.R.; Braakhuis, B.J.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef]
- Adjei Boakye, E.; Buchanan, P.; Hinyard, L.; Osazuwa-Peters, N.; Schootman, M.; Piccirillo, J.F. Incidence and Risk of Second Primary Malignant Neoplasm After a First Head and Neck Squamous Cell Carcinoma. JAMA Otolaryngol. Head Neck Surg. 2018, 144, 727–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Williams, M.J.; Werner, B.; Barnes, C.P.; Graham, T.A.; Sottoriva, A. Identification of neutral tumor evolution across cancer types. Nat. Genet. 2016, 48, 238–244. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Chen, L.; Savage, S.R.; Eguez, R.V.; Dou, Y.; Li, Y.; da Veiga Leprevost, F.; Jaehnig, E.J.; Lei, J.T.; Wen, B.; et al. Proteogenomic insights into the biology and treatment of HPV-negative head and neck squamous cell carcinoma. Cancer Cell 2021, 39, 361–379.e16. [Google Scholar] [CrossRef]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castilho, R.M.; Squarize, C.H.; Almeida, L.O. Epigenetic Modifications and Head and Neck Cancer: Implications for Tumor Progression and Resistance to Therapy. Int. J. Mol. Sci. 2017, 18, 1506. [Google Scholar] [CrossRef] [PubMed]
- Merlo, L.M.; Pepper, J.W.; Reid, B.J.; Maley, C.C. Cancer as an evolutionary and ecological process. Nat. Rev. Cancer 2006, 6, 924–935. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef]
- Jurmeister, P.; Bockmayr, M.; Seegerer, P.; Bockmayr, T.; Treue, D.; Montavon, G.; Vollbrecht, C.; Arnold, A.; Teichmann, D.; Bressem, K.; et al. Machine learning analysis of DNA methylation profiles distinguishes primary lung squamous cell carcinomas from head and neck metastases. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Worsham, M.J.; Stephen, J.K.; Chen, K.M.; Havard, S.; Shah, V.; Gardner, G.; Schweitzer, V.G. Delineating an epigenetic continuum in head and neck cancer. Cancer Lett. 2014, 342, 178–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Towle, R.; Truong, D.; Hogg, K.; Robinson, W.P.; Poh, C.F.; Garnis, C. Global analysis of DNA methylation changes during progression of oral cancer. Oral Oncol. 2013, 49, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Coombes, M.M.; Briggs, K.L.; Bone, J.R.; Clayman, G.L.; El-Naggar, A.K.; Dent, S.Y. Resetting the histone code at CDKN2A in HNSCC by inhibition of DNA methylation. Oncogene 2003, 22, 8902–8911. [Google Scholar] [CrossRef] [Green Version]
- Masood, Y.; Kqueen, C.Y.; Rajadurai, P. Role of miRNA in head and neck squamous cell carcinoma. Expert Rev. Anticancer Ther. 2015, 15, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Hess, J.; Unger, K.; Maihoefer, C.; Schuttrumpf, L.; Wintergerst, L.; Heider, T.; Weber, P.; Marschner, S.; Braselmann, H.; Samaga, D.; et al. A Five-MicroRNA Signature Predicts Survival and Disease Control of Patients with Head and Neck Cancer Negative for HPV Infection. Clin. Cancer Res. 2019, 25, 1505–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiello, N.M.; Brabletz, T.; Kang, Y.; Nieto, M.A.; Weinberg, R.A.; Stanger, B.Z. Upholding a role for EMT in pancreatic cancer metastasis. Nature 2017, 547, E7–E8. [Google Scholar] [CrossRef]
- Ye, X.; Brabletz, T.; Kang, Y.; Longmore, G.D.; Nieto, M.A.; Stanger, B.Z.; Yang, J.; Weinberg, R.A. Upholding a role for EMT in breast cancer metastasis. Nature 2017, 547, E1–E3. [Google Scholar] [CrossRef]
- Sproll, C.; Fluegen, G.; Stoecklein, N.H. Minimal Residual Disease in Head and Neck Cancer and Esophageal Cancer. Adv. Exp. Med. Biol. 2018, 1100, 55–82. [Google Scholar] [CrossRef]
- Lang, S.; Wollenberg, B.; Dellian, M.; Steuer-Vogt, M.K.; Schwenzer, K.; Sautier, W.; Chucholowski, M.; Eckel, R.; Faas, I.; Wilmes, E.; et al. Clinical and epidemiological data of patients with malignomas of the head and neck. Laryngorhinootologie 2002, 81, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Weinberg, R.A. Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef] [Green Version]
- Daniel, Y.; Lelou, E.; Aninat, C.; Corlu, A.; Cabillic, F. Interplay between Metabolism Reprogramming and Epithelial-to-Mesenchymal Transition in Cancer Stem Cells. Cancers 2021, 13, 1973. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, N.; Bhattacharya, S.; Steele, R.; Phillips, N.; Ray, R.B. Involvement of c-Fos in the Promotion of Cancer Stem-like Cell Properties in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2017, 23, 3120–3128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Lim, C.T. Tumor dissemination: An EMT affair. Cancer Cell 2013, 23, 272–273. [Google Scholar] [CrossRef] [Green Version]
- Pang, X.; Tang, Y.L.; Liang, X.H. Transforming growth factor-beta signaling in head and neck squamous cell carcinoma: Insights into cellular responses. Oncol. Lett. 2018, 16, 4799–4806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massague, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Massague, J. TGF-beta signaling in development and disease. FEBS Lett. 2012, 586, 1833. [Google Scholar] [CrossRef] [Green Version]
- Massague, J.; Blain, S.W.; Lo, R.S. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 2000, 103, 295–309. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Liu, Y.; Huang, D.; Dai, Y.; Cai, G.; Sun, J.; Xu, T.; Tian, Y.; Zhang, X. TGF-beta1 mediates epithelial to mesenchymal transition via the TGF-beta/Smad pathway in squamous cell carcinoma of the head and neck. Oncol. Rep. 2011, 25, 1581–1587. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.; Ryu, H.; Kim, S.; Joo, M.; Jeon, H.J.; Lee, M.W.; Song, I.C.; Kim, M.N.; Kim, J.M.; Lee, H.J. CXCR7 promotes migration and invasion in head and neck squamous cell carcinoma by upregulating TGF-beta1/Smad2/3 signaling. Sci. Rep. 2019, 9, 18100. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wu, C.; Zhang, C.; Li, Z.; Zhu, T.; Chen, J.; Ren, Y.; Wang, X.; Zhang, L.; Zhou, X. TGF-beta-induced STAT3 overexpression promotes human head and neck squamous cell carcinoma invasion and metastasis through malat1/miR-30a interactions. Cancer Lett. 2018, 436, 52–62. [Google Scholar] [CrossRef]
- Zhang, W.; Li, J.; Wu, Y.; Ge, H.; Song, Y.; Wang, D.; Yuan, H.; Jiang, H.; Wang, Y.; Cheng, J. TEAD4 overexpression promotes epithelial-mesenchymal transition and associates with aggressiveness and adverse prognosis in head neck squamous cell carcinoma. Cancer Cell Int. 2018, 18, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, D.H.; Dabelsteen, E.; Specht, L.; Fiehn, A.M.; Therkildsen, M.H.; Jonson, L.; Vikesaa, J.; Nielsen, F.C.; von Buchwald, C. Molecular profiling of tumour budding implicates TGFbeta-mediated epithelial-mesenchymal transition as a therapeutic target in oral squamous cell carcinoma. J. Pathol. 2015, 236, 505–516. [Google Scholar] [CrossRef]
- Chen, L.; Sun, D.Z.; Fu, Y.G.; Yang, P.Z.; Lv, H.Q.; Gao, Y.; Zhang, X.Y. Upregulation of microRNA-141 suppresses epithelial-mesenchymal transition and lymph node metastasis in laryngeal cancer through HOXC6-dependent TGF-beta signaling pathway. Cell Signal. 2020, 66, 109444. [Google Scholar] [CrossRef] [PubMed]
- Gluck, C.; Glathar, A.; Tsompana, M.; Nowak, N.; Garrett-Sinha, L.A.; Buck, M.J.; Sinha, S. Molecular dissection of the oncogenic role of ETS1 in the mesenchymal subtypes of head and neck squamous cell carcinoma. PLoS Genet. 2019, 15, e1008250. [Google Scholar] [CrossRef] [Green Version]
- Theodoraki, M.N.; Yerneni, S.S.; Brunner, C.; Theodorakis, J.; Hoffmann, T.K.; Whiteside, T.L. Plasma-derived Exosomes Reverse Epithelial-to-Mesenchymal Transition after Photodynamic Therapy of Patients with Head and Neck Cancer. Oncoscience 2018, 5, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Fanelli, M.F.; Oliveira, T.B.; Braun, A.C.; Corassa, M.; Abdallah, E.A.; Nicolau, U.R.; da Silva Alves, V.; Garcia, D.; Calsavara, V.F.; Kowalski, L.P.; et al. Evaluation of incidence, significance, and prognostic role of circulating tumor microemboli and transforming growth factor-beta receptor I in head and neck cancer. Head Neck 2017, 39, 2283–2292. [Google Scholar] [CrossRef]
- Ohnuki, H.; Jiang, K.; Wang, D.; Salvucci, O.; Kwak, H.; Sanchez-Martin, D.; Maric, D.; Tosato, G. Tumor-infiltrating myeloid cells activate Dll4/Notch/TGF-beta signaling to drive malignant progression. Cancer Res. 2014, 74, 2038–2049. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Zheng, M.; Zhang, M.; Yang, X.; Li, L.; Wang, S.S.; Wu, J.S.; Yu, X.H.; Wu, J.B.; Pang, X.; et al. PRRX1 Regulates Cellular Phenotype Plasticity and Dormancy of Head and Neck Squamous Cell Carcinoma through miR-642b-3p. Neoplasia 2019, 21, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, P.; Hollmann, A.; Kitz, J.; Afthonidou, A.; Simon, F.; Shakhtour, J.; Mack, B.; Kranz, G.; Libl, D.; Leu, M.; et al. High Expression of EpCAM and Sox2 is a Positive Prognosticator of Clinical Outcome for Head and Neck Carcinoma. Sci. Rep. 2018, 8, 14582. [Google Scholar] [CrossRef] [Green Version]
- Schinke, H.; Pan, M.; Akyol, M.; Zhou, J.; Shi, E.; Kranz, G.; Libl, D.; Quadt, T.; Simon, F.; Canis, M.; et al. SLUG-related partial epithelial-to-mesenchymal transition is a transcriptomic prognosticator of head and neck cancer survival. Mol. Oncol. 2021. [Google Scholar] [CrossRef]
- Kumai, T.; Oikawa, K.; Aoki, N.; Kimura, S.; Harabuchi, Y.; Celis, E.; Kobayashi, H. Tumor-derived TGF-beta and prostaglandin E2 attenuate anti-tumor immune responses in head and neck squamous cell carcinoma treated with EGFR inhibitor. J. Transl. Med. 2014, 12, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wang, W.; Chen, S.; Cai, J.; Ban, Y.; Peng, Q.; Zhou, Y.; Zeng, Z.; Li, X.; Xiong, W.; et al. FOXA1 reprograms the TGF-beta-stimulated transcriptional program from a metastasis promoter to a tumor suppressor in nasopharyngeal carcinoma. Cancer Lett. 2019, 442, 1–14. [Google Scholar] [CrossRef]
- Tao, Y.; Sturgis, E.M.; Huang, Z.; Sun, Y.; Dahlstrom, K.R.; Wei, Q.; Li, G. A TGF-beta1 genetic variant at the miRNA187 binding site significantly modifies risk of HPV16-associated oropharyngeal cancer. Int. J. Cancer 2018, 143, 1327–1334. [Google Scholar] [CrossRef] [Green Version]
- Bragado, P.; Estrada, Y.; Parikh, F.; Krause, S.; Capobianco, C.; Farina, H.G.; Schewe, D.M.; Aguirre-Ghiso, J.A. TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nat. Cell Biol. 2013, 15, 1351–1361. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Tang, Q.; Chen, X.; Fan, W.; Zhou, Z.; Huang, W.; Liang, F. TGF-beta1 and IL-17A comediate the protumor phenotype of neutrophils to regulate the epithelial-mesenchymal transition in oral squamous cell carcinoma. J. Oral Pathol. Med. 2021, 50, 353–361. [Google Scholar] [CrossRef]
- Taniguchi, S.; Elhance, A.; Van Duzer, A.; Kumar, S.; Leitenberger, J.J.; Oshimori, N. Tumor-initiating cells establish an IL-33-TGF-beta niche signaling loop to promote cancer progression. Science 2020, 369, eaay1813. [Google Scholar] [CrossRef] [PubMed]
- Jung, A.R.; Jung, C.H.; Noh, J.K.; Lee, Y.C.; Eun, Y.G. Epithelial-mesenchymal transition gene signature is associated with prognosis and tumor microenvironment in head and neck squamous cell carcinoma. Sci. Rep. 2020, 10, 3652. [Google Scholar] [CrossRef]
- Grandis, J.R.; Tweardy, D.J. TGF-alpha and EGFR in head and neck cancer. J. Cell Biochem. Suppl. 1993, 17F, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Grandis, J.R.; Tweardy, D.J. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res. 1993, 53, 3579–3584. [Google Scholar]
- Ford, A.C.; Grandis, J.R. Targeting epidermal growth factor receptor in head and neck cancer. Head Neck 2003, 25, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.S.; Pathak, S.; Frankenthaler, R.; Gallick, G.E.; Sacks, P.G. Effect of epidermal growth factor (EGF) on a newly established head and neck squamous carcinoma cell line. Otolaryngol. Head Neck Surg. 1988, 99, 567–573. [Google Scholar] [CrossRef]
- Grandis, J.R.; Tweardy, D.J. The role of peptide growth factors in head and neck carcinoma. Otolaryngol. Clin. N. Am. 1992, 25, 1105–1115. [Google Scholar] [CrossRef]
- Sturgis, E.M.; Sacks, P.G.; Masui, H.; Mendelsohn, J.; Schantz, S.P. Effects of antiepidermal growth factor receptor antibody 528 on the proliferation and differentiation of head and neck cancer. Otolaryngol. Head Neck Surg. 1994, 111, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Aboud-Pirak, E.; Hurwitz, E.; Pirak, M.E.; Bellot, F.; Schlessinger, J.; Sela, M. Efficacy of antibodies to epidermal growth factor receptor against KB carcinoma in vitro and in nude mice. J. Natl. Cancer Inst. 1988, 80, 1605–1611. [Google Scholar] [CrossRef]
- Graham, J.; Muhsin, M.; Kirkpatrick, P. Cetuximab. Nat. Rev. Drug Discov. 2004, 3, 549–550. [Google Scholar] [CrossRef]
- Goldberg, R.M. Cetuximab. Nat. Rev. Drug Discov. 2005, 1, S10–S11. [Google Scholar] [CrossRef]
- Taberna, M.; Oliva, M.; Mesia, R. Cetuximab-Containing Combinations in Locally Advanced and Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2019, 9, 383. [Google Scholar] [CrossRef]
- Patel, A.N.; Mehnert, J.M.; Kim, S. Treatment of recurrent metastatic head and neck cancer: Focus on cetuximab. Clin. Med. Insights Ear Nose Throat 2012, 5, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Jiang, G.; Blume-Jensen, P.; Hunter, T. Epidermal growth factor-induced tumor cell invasion and metastasis initiated by dephosphorylation and downregulation of focal adhesion kinase. Mol. Cell Biol. 2001, 21, 4016–4031. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Ghosh, S.; Wang, Z.; Hunter, T. Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of beta-catenin, and enhanced tumor cell invasion. Cancer Cell 2003, 4, 499–515. [Google Scholar] [CrossRef] [Green Version]
- Pan, M.; Schinke, H.; Luxenburger, E.; Kranz, G.; Shakhtour, J.; Libl, D.; Huang, Y.; Gaber, A.; Pavsic, M.; Lenarcic, B.; et al. EpCAM ectodomain EpEX is a ligand of EGFR that counteracts EGF-mediated epithelial-mesenchymal transition through modulation of phospho-ERK1/2 in head and neck cancers. PLoS Biol. 2018, 16, e2006624. [Google Scholar] [CrossRef]
- Zuo, J.H.; Zhu, W.; Li, M.Y.; Li, X.H.; Yi, H.; Zeng, G.Q.; Wan, X.X.; He, Q.Y.; Li, J.H.; Qu, J.Q.; et al. Activation of EGFR promotes squamous carcinoma SCC10A cell migration and invasion via inducing EMT-like phenotype change and MMP-9-mediated degradation of E-cadherin. J. Cell Biochem. 2011, 112, 2508–2517. [Google Scholar] [CrossRef]
- Holz, C.; Niehr, F.; Boyko, M.; Hristozova, T.; Distel, L.; Budach, V.; Tinhofer, I. Epithelial-mesenchymal-transition induced by EGFR activation interferes with cell migration and response to irradiation and cetuximab in head and neck cancer cells. Radiother. Oncol. 2011, 101, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Zhang, Q.; Ishida, Y.; Hajjar, S.; Tang, X.; Shi, H.; Dang, C.V.; Le, A.D. EGF induces epithelial-mesenchymal transition and cancer stem-like cell properties in human oral cancer cells via promoting Warburg effect. Oncotarget 2017, 8, 9557–9571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Dong, Z.; Lauxen, I.S.; Filho, M.S.; Nor, J.E. Endothelial cell-secreted EGF induces epithelial to mesenchymal transition and endows head and neck cancer cells with stem-like phenotype. Cancer Res. 2014, 74, 2869–2881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, W.; Li, Y.; Zhou, Q.; Xu, Z.; Sun, C.; Tan, X.; Lu, L. Cetuximab inhibits oral squamous cell carcinoma invasion and metastasis via degradation of epidermal growth factor receptor. J. Oral Pathol. Med. 2014, 43, 250–257. [Google Scholar] [CrossRef]
- Grybauskas, M.; Daisne, J.F.; Aleknavicius, E.; Burneckis, A. Early prediction of response to cetuximab and radiotherapy by FDG-PET/CT for the treatment of a locoregionally advanced squamous cell carcinoma of the hypopharynx. Medicina 2014, 50, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Eguchi, T.; Sogawa, C.; Ono, K.; Murakami, J.; Ibaragi, S.; Asaumi, J.I.; Calderwood, S.K.; Okamoto, K.; Kozaki, K.I. Carcinogenic epithelial-mesenchymal transition initiated by oral cancer exosomes is inhibited by anti-EGFR antibody cetuximab. Oral Oncol. 2018, 86, 251–257. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, W.; Zhong, W.Q.; Liu, Z.J.; Li, H.M.; Yu, Z.L.; Zhao, Y.F. Tumor associated macrophages induce epithelial to mesenchymal transition via the EGFR/ERK1/2 pathway in head and neck squamous cell carcinoma. Oncol. Rep. 2018, 40, 2558–2572. [Google Scholar] [CrossRef]
- Watermann, C.; Pasternack, H.; Idel, C.; Ribbat-Idel, J.; Bragelmann, J.; Kuppler, P.; Offermann, A.; Jonigk, D.; Kuhnel, M.P.; Schrock, A.; et al. Recurrent HNSCC Harbor an Immunosuppressive Tumor Immune Microenvironment Suggesting Successful Tumor Immune Evasion. Clin. Cancer Res. 2021, 27, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.H.; Tso, H.C.; Hung, S.H.; Kuan, I.I.; Lai, J.K.; Ke, F.Y.; Chuang, Y.T.; Liu, I.J.; Wang, Y.P.; Chen, R.H.; et al. Extracellular domain of EpCAM enhances tumor progression through EGFR signaling in colon cancer cells. Cancer Lett. 2018, 433, 165–175. [Google Scholar] [CrossRef]
- Maetzel, D.; Denzel, S.; Mack, B.; Canis, M.; Went, P.; Benk, M.; Kieu, C.; Papior, P.; Baeuerle, P.A.; Munz, M.; et al. Nuclear signalling by tumour-associated antigen EpCAM. Nat. Cell Biol. 2009, 11, 162–171. [Google Scholar] [CrossRef]
- Chen, H.N.; Liang, K.H.; Lai, J.K.; Lan, C.H.; Liao, M.Y.; Hung, S.H.; Chuang, Y.T.; Chen, K.C.; Tsuei, W.W.; Wu, H.C. EpCAM Signaling Promotes Tumor Progression and Protein Stability of PD-L1 through the EGFR Pathway. Cancer Res. 2020, 80, 5035–5050. [Google Scholar] [CrossRef]
- Kuan, I.I.; Lee, C.C.; Chen, C.H.; Lu, J.; Kuo, Y.S.; Wu, H.C. The extracellular domain of epithelial cell adhesion molecule (EpCAM) enhances multipotency of mesenchymal stem cells through EGFR-LIN28-LET7 signaling. J. Biol. Chem. 2019, 294, 7769–7786. [Google Scholar] [CrossRef]
- Puram, S.V.; Parikh, A.S.; Tirosh, I. Single cell RNA-seq highlights a role for a partial EMT in head and neck cancer. Mol. Cell Oncol. 2018, 5, e1448244. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Wang, Z.; Xiong, X.; Zhong, Y.; Zhang, W.; Dong, Y.; Li, J.; Zhu, Z.; Zhang, W.; Wu, H.; et al. Membrane-tethered Notch1 exhibits oncogenic property via activation of EGFR-PI3K-AKT pathway in oral squamous cell carcinoma. J. Cell Physiol. 2019, 234, 5940–5952. [Google Scholar] [CrossRef]
- Keysar, S.B.; Le, P.N.; Anderson, R.T.; Morton, J.J.; Bowles, D.W.; Paylor, J.J.; Vogler, B.W.; Thorburn, J.; Fernandez, P.; Glogowska, M.J.; et al. Hedgehog signaling alters reliance on EGF receptor signaling and mediates anti-EGFR therapeutic resistance in head and neck cancer. Cancer Res. 2013, 73, 3381–3392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stemmler, M.P.; Eccles, R.L.; Brabletz, S.; Brabletz, T. Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 2019, 21, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Liu, H.; Zhang, M.; Huang, Z.; Zhou, H.; Zhu, Y.; Tao, Y.; Xie, N.; Liu, X.; Hou, J.; et al. Prognostic value of epithelial-mesenchymal transition-inducing transcription factors in head and neck squamous cell carcinoma: A meta-analysis. Head Neck 2020, 42, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cheng, Q.; Zhou, Y.; Wang, Y.; Chen, X. Slug is a key mediator of hypoxia induced cadherin switch in HNSCC: Correlations with poor prognosis. Oral Oncol. 2013, 49, 1043–1050. [Google Scholar] [CrossRef]
- Cappellesso, R.; Marioni, G.; Crescenzi, M.; Giacomelli, L.; Guzzardo, V.; Mussato, A.; Staffieri, A.; Martini, A.; Blandamura, S.; Fassina, A. The prognostic role of the epithelial-mesenchymal transition markers E-cadherin and Slug in laryngeal squamous cell carcinoma. Histopathology 2015, 67, 491–500. [Google Scholar] [CrossRef]
- Katafiasz, D.; Smith, L.M.; Wahl, J.K., 3rd. Slug (SNAI2) expression in oral SCC cells results in altered cell-cell adhesion and increased motility. Cell Adh. Migr. 2011, 5, 315–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, J.H.; Lee, S.H.; Lim, Y.C. Wnt/beta-catenin/Slug pathway contributes to tumor invasion and lymph node metastasis in head and neck squamous cell carcinoma. Clin. Exp. Metastasis 2021, 38, 163–174. [Google Scholar] [CrossRef]
- Schinke, H.; Heider, T.; Herkommer, T.; Simon, F.; Blancke Soares, A.; Kranz, G.; Samaga, D.; Dajka, L.; Feuchtinger, A.; Walch, A.; et al. Digital scoring of EpCAM and slug expression as prognostic markers in head and neck squamous cell carcinomas. Mol. Oncol. 2020, 15, 1040–1053. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.H.; Lee, S.H.; Koo, B.S.; Kim, J.M.; Huang, S.; Cho, J.H.; Eun, Y.G.; Shin, H.A.; Lim, Y.C. Slug is a novel molecular target for head and neck squamous cell carcinoma stem-like cells. Oral Oncol. 2020, 111, 104948. [Google Scholar] [CrossRef] [PubMed]
- Riechelmann, H.; Steinbichler, T.B.; Sprung, S.; Santer, M.; Runge, A.; Ganswindt, U.; Gamerith, G.; Dudas, J. The Epithelial-Mesenchymal Transcription Factor Slug Predicts Survival Benefit of Up-Front Surgery in Head and Neck Cancer. Cancers 2021, 13, 772. [Google Scholar] [CrossRef]
- Dennis, M.; Wang, G.; Luo, J.; Lin, Y.; Dohadwala, M.; Abemayor, E.; Elashoff, D.A.; Sharma, S.; Dubinett, S.M.; St John, M.A. Snail controls the mesenchymal phenotype and drives erlotinib resistance in oral epithelial and head and neck squamous cell carcinoma cells. Otolaryngol. Head Neck Surg. 2012, 147, 726–732. [Google Scholar] [CrossRef] [Green Version]
- Masui, T.; Ota, I.; Yook, J.I.; Mikami, S.; Yane, K.; Yamanaka, T.; Hosoi, H. Snail-induced epithelial-mesenchymal transition promotes cancer stem cell-like phenotype in head and neck cancer cells. Int. J. Oncol. 2014, 44, 693–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Mallen-St. Clair, J.; Wang, G.; Luo, J.; Palma-Diaz, F.; Lai, C.; Elashoff, D.A.; Sharma, S.; Dubinett, S.M.; St. John, M. p38 MAPK mediates epithelial-mesenchymal transition by regulating p38IP and Snail in head and neck squamous cell carcinoma. Oral Oncol. 2016, 60, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Kumar, B.; Datta, J.; Teknos, T.N.; Kumar, P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol. Cancer Res. 2011, 9, 1658–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ota, I.; Masui, T.; Kurihara, M.; Yook, J.I.; Mikami, S.; Kimura, T.; Shimada, K.; Konishi, N.; Yane, K.; Yamanaka, T.; et al. Snail-induced EMT promotes cancer stem cell-like properties in head and neck cancer cells. Oncol. Rep. 2016, 35, 261–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.F.; Chen, J.Y.; Ho, Y.H.; Hsu, W.H.; Wu, L.C.; Lan, H.Y.; Hsu, D.S.; Tai, S.K.; Chang, Y.C.; Yang, M.H. Snail-induced claudin-11 prompts collective migration for tumour progression. Nat. Cell Biol. 2019, 21, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Way, T.D.; Huang, J.T.; Chou, C.H.; Huang, C.H.; Yang, M.H.; Ho, C.T. Emodin represses TWIST1-induced epithelial-mesenchymal transitions in head and neck squamous cell carcinoma cells by inhibiting the beta-catenin and Akt pathways. Eur. J. Cancer 2014, 50, 366–378. [Google Scholar] [CrossRef] [PubMed]
- da Silva, S.D.; Alaoui-Jamali, M.A.; Soares, F.A.; Carraro, D.M.; Brentani, H.P.; Hier, M.; Rogatto, S.R.; Kowalski, L.P. TWIST1 is a molecular marker for a poor prognosis in oral cancer and represents a potential therapeutic target. Cancer 2014, 120, 352–362. [Google Scholar] [CrossRef] [Green Version]
- Gasparotto, D.; Polesel, J.; Marzotto, A.; Colladel, R.; Piccinin, S.; Modena, P.; Grizzo, A.; Sulfaro, S.; Serraino, D.; Barzan, L.; et al. Overexpression of TWIST2 correlates with poor prognosis in head and neck squamous cell carcinomas. Oncotarget 2011, 2, 1165–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.H.; Parker, J.S.; Ely, K.; Carter, J.; Yi, Y.; Murphy, B.A.; Ang, K.K.; El-Naggar, A.K.; Zanation, A.M.; Cmelak, A.J.; et al. Gene expression profiles identify epithelial-to-mesenchymal transition and activation of nuclear factor-kappaB signaling as characteristics of a high-risk head and neck squamous cell carcinoma. Cancer Res. 2006, 66, 8210–8218. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.H.; Chang, S.Y.; Chiou, S.H.; Liu, C.J.; Chi, C.W.; Chen, P.M.; Teng, S.C.; Wu, K.J. Overexpression of NBS1 induces epithelial-mesenchymal transition and co-expression of NBS1 and Snail predicts metastasis of head and neck cancer. Oncogene 2007, 26, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Domnich, M.; Riedesel, J.; Pylaeva, E.; Kurten, C.H.L.; Buer, J.; Lang, S.; Jablonska, J. Oral Neutrophils: Underestimated Players in Oral Cancer. Front. Immunol. 2020, 11, 565683. [Google Scholar] [CrossRef] [PubMed]
- Ong, H.S.; Gokavarapu, S.; Tian, Z.; Li, J.; Xu, Q.; Zhang, C.P.; Cao, W. PDGFRA mRNA overexpression is associated with regional metastasis and reduced survival in oral squamous cell carcinoma. J. Oral Pathol. Med. 2018, 47, 652–659. [Google Scholar] [CrossRef]
- Wang, J.; Cui, R.; Clement, C.G.; Nawgiri, R.; Powell, D.W.; Pinchuk, I.V.; Watts, T.L. Activation PDGFR-alpha/AKT Mediated Signaling Pathways in Oral Squamous Cell Carcinoma by Mesenchymal Stem/Stromal Cells Promotes Anti-apoptosis and Decreased Sensitivity to Cisplatin. Front. Oncol. 2020, 10, 552. [Google Scholar] [CrossRef]
- Parikh, A.S.; Puram, S.V.; Faquin, W.C.; Richmon, J.D.; Emerick, K.S.; Deschler, D.G.; Varvares, M.A.; Tirosh, I.; Bernstein, B.E.; Lin, D.T. Immunohistochemical quantification of partial-EMT in oral cavity squamous cell carcinoma primary tumors is associated with nodal metastasis. Oral Oncol. 2019, 99, 104458. [Google Scholar] [CrossRef] [PubMed]
- da Silva, S.D.; Morand, G.B.; Alobaid, F.A.; Hier, M.P.; Mlynarek, A.M.; Alaoui-Jamali, M.A.; Kowalski, L.P. Epithelial-mesenchymal transition (EMT) markers have prognostic impact in multiple primary oral squamous cell carcinoma. Clin. Exp. Metastasis 2015, 32, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Kisoda, S.; Shao, W.; Fujiwara, N.; Mouri, Y.; Tsunematsu, T.; Jin, S.; Arakaki, R.; Ishimaru, N.; Kudo, Y. Prognostic value of partial EMT-related genes in head and neck squamous cell carcinoma by a bioinformatic analysis. Oral Dis. 2020, 26, 1149–1156. [Google Scholar] [CrossRef]
- Pectasides, E.; Rampias, T.; Sasaki, C.; Perisanidis, C.; Kouloulias, V.; Burtness, B.; Zaramboukas, T.; Rimm, D.; Fountzilas, G.; Psyrri, A. Markers of epithelial to mesenchymal transition in association with survival in head and neck squamous cell carcinoma (HNSCC). PLoS ONE 2014, 9, e94273. [Google Scholar] [CrossRef] [PubMed]
- Racle, J.; de Jonge, K.; Baumgaertner, P.; Speiser, D.E.; Gfeller, D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife 2017, 6, e26476. [Google Scholar] [CrossRef]
- Foroutan, M.; Bhuva, D.D.; Lyu, R.; Horan, K.; Cursons, J.; Davis, M.J. Single sample scoring of molecular phenotypes. BMC Bioinform. 2018, 19, 404. [Google Scholar] [CrossRef] [Green Version]
- Tyler, M.; Tirosh, I. Decoupling epithelial-mesenchymal transitions from stromal profiles by integrative expression analysis. Nat. Commun. 2021, 12, 2592. [Google Scholar] [CrossRef]
- Chen, C.; Wei, Y.; Hummel, M.; Hoffmann, T.K.; Gross, M.; Kaufmann, A.M.; Albers, A.E. Evidence for epithelial-mesenchymal transition in cancer stem cells of head and neck squamous cell carcinoma. PLoS ONE 2011, 6, e16466. [Google Scholar] [CrossRef]
- Biddle, A.; Gammon, L.; Liang, X.; Costea, D.E.; Mackenzie, I.C. Phenotypic Plasticity Determines Cancer Stem Cell Therapeutic Resistance in Oral Squamous Cell Carcinoma. EBioMedicine 2016, 4, 138–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biddle, A.; Liang, X.; Gammon, L.; Fazil, B.; Harper, L.J.; Emich, H.; Costea, D.E.; Mackenzie, I.C. Cancer stem cells in squamous cell carcinoma switch between two distinct phenotypes that are preferentially migratory or proliferative. Cancer Res. 2011, 71, 5317–5326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigeishi, H.; Biddle, A.; Gammon, L.; Emich, H.; Rodini, C.O.; Gemenetzidis, E.; Fazil, B.; Sugiyama, M.; Kamata, N.; Mackenzie, I.C. Maintenance of stem cell self-renewal in head and neck cancers requires actions of GSK3beta influenced by CD44 and RHAMM. Stem Cells 2013, 31, 2073–2083. [Google Scholar] [CrossRef]
- Chen, L.; Li, Y.C.; Wu, L.; Yu, G.T.; Zhang, W.F.; Huang, C.F.; Sun, Z.J. TRAF6 regulates tumour metastasis through EMT and CSC phenotypes in head and neck squamous cell carcinoma. J. Cell Mol. Med. 2018, 22, 1337–1349. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Ren, Z.; Yang, X.; Yang, R.; Chen, Y.; Liu, Z.; Dai, Z.; Zhang, Y.; He, Y.; Zhang, C.; et al. Nerve growth factor (NGF)-TrkA axis in head and neck squamous cell carcinoma triggers EMT and confers resistance to the EGFR inhibitor erlotinib. Cancer Lett. 2020, 472, 81–96. [Google Scholar] [CrossRef]
- Steinbichler, T.B.; Alshaimaa, A.; Maria, M.V.; Daniel, D.; Herbert, R.; Jozsef, D.; Ira-Ida, S. Epithelial-mesenchymal crosstalk induces radioresistance in HNSCC cells. Oncotarget 2018, 9, 3641–3652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.W.; Gwak, S.Y.; Shim, G.A.; Liu, L.; Lim, Y.C.; Kim, J.M.; Jung, M.G.; Koo, B.S. EZH2 is associated with poor prognosis in head-and-neck squamous cell carcinoma via regulating the epithelial-to-mesenchymal transition and chemosensitivity. Oral Oncol. 2016, 52, 66–74. [Google Scholar] [CrossRef]
- Cheng, H.; Fertig, E.J.; Ozawa, H.; Hatakeyama, H.; Howard, J.D.; Perez, J.; Considine, M.; Thakar, M.; Ranaweera, R.; Krigsfeld, G.; et al. Decreased SMAD4 expression is associated with induction of epithelial-to-mesenchymal transition and cetuximab resistance in head and neck squamous cell carcinoma. Cancer Biol. Ther. 2015, 16, 1252–1258. [Google Scholar] [CrossRef] [Green Version]
- Maseki, S.; Ijichi, K.; Tanaka, H.; Fujii, M.; Hasegawa, Y.; Ogawa, T.; Murakami, S.; Kondo, E.; Nakanishi, H. Acquisition of EMT phenotype in the gefitinib-resistant cells of a head and neck squamous cell carcinoma cell line through Akt/GSK-3beta/snail signalling pathway. Br. J. Cancer 2012, 106, 1196–1204. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wang, Y.; Zhu, Y.; Yuan, C.; Wang, D.; Zhang, W.; Qi, B.; Qiu, J.; Song, X.; Ye, J.; et al. The Hippo transducer TAZ promotes epithelial to mesenchymal transition and cancer stem cell maintenance in oral cancer. Mol. Oncol. 2015, 9, 1091–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrader, C.H.; Kolb, M.; Zaoui, K.; Flechtenmacher, C.; Grabe, N.; Weber, K.J.; Hielscher, T.; Plinkert, P.K.; Hess, J. Kallikrein-related peptidase 6 regulates epithelial-to-mesenchymal transition and serves as prognostic biomarker for head and neck squamous cell carcinoma patients. Mol. Cancer 2015, 14, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukusumi, T.; Guo, T.W.; Sakai, A.; Ando, M.; Ren, S.; Haft, S.; Liu, C.; Amornphimoltham, P.; Gutkind, J.S.; Califano, J.A. The NOTCH4-HEY1 Pathway Induces Epithelial-Mesenchymal Transition in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2018, 24, 619–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatenby, R.A.; Brown, J.S. Integrating evolutionary dynamics into cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 675–686. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhan, H. Communication between EMT and PD-L1 signaling: New insights into tumor immune evasion. Cancer Lett. 2020, 468, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Zhang, M.; Zhou, C.; Wang, W.; Yang, H.; Ye, W. The role of epithelial-mesenchymal transition in regulating radioresistance. Crit. Rev. Oncol. Hematol. 2020, 150, 102961. [Google Scholar] [CrossRef]
- Du, B.; Shim, J.S. Targeting Epithelial-Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules 2016, 21, 965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonckheere, S.; Adams, J.; De Groote, D.; Campbell, K.; Berx, G.; Goossens, S. Epithelial-Mesenchymal Transition (EMT) as a Therapeutic Target. Cells Tissues Organs 2021, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Voon, D.C.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. The EMT spectrum and therapeutic opportunities. Mol. Oncol. 2017, 11, 878–891. [Google Scholar] [CrossRef] [Green Version]
- Tolcher, A.W.; Berlin, J.D.; Cosaert, J.; Kauh, J.; Chan, E.; Piha-Paul, S.A.; Amaya, A.; Tang, S.; Driscoll, K.; Kimbung, R.; et al. A phase 1 study of anti-TGFbeta receptor type-II monoclonal antibody LY3022859 in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2017, 79, 673–680. [Google Scholar] [CrossRef] [Green Version]
- Ramesh, V.; Brabletz, T.; Ceppi, P. Targeting EMT in Cancer with Repurposed Metabolic Inhibitors. Trends Cancer 2020, 6, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Caponigro, F.; Di Gennaro, E.; Ionna, F.; Longo, F.; Aversa, C.; Pavone, E.; Maglione, M.G.; Di Marzo, M.; Muto, P.; Cavalcanti, E.; et al. Phase II clinical study of valproic acid plus cisplatin and cetuximab in recurrent and/or metastatic squamous cell carcinoma of Head and Neck-V-CHANCE trial. BMC Cancer 2016, 16, 918. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baumeister, P.; Zhou, J.; Canis, M.; Gires, O. Epithelial-to-Mesenchymal Transition-Derived Heterogeneity in Head and Neck Squamous Cell Carcinomas. Cancers 2021, 13, 5355. https://doi.org/10.3390/cancers13215355
Baumeister P, Zhou J, Canis M, Gires O. Epithelial-to-Mesenchymal Transition-Derived Heterogeneity in Head and Neck Squamous Cell Carcinomas. Cancers. 2021; 13(21):5355. https://doi.org/10.3390/cancers13215355
Chicago/Turabian StyleBaumeister, Philipp, Jiefu Zhou, Martin Canis, and Olivier Gires. 2021. "Epithelial-to-Mesenchymal Transition-Derived Heterogeneity in Head and Neck Squamous Cell Carcinomas" Cancers 13, no. 21: 5355. https://doi.org/10.3390/cancers13215355