
Network-Based Analysis to Identify Drivers of Metastatic Prostate Cancer Using GoNetic

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
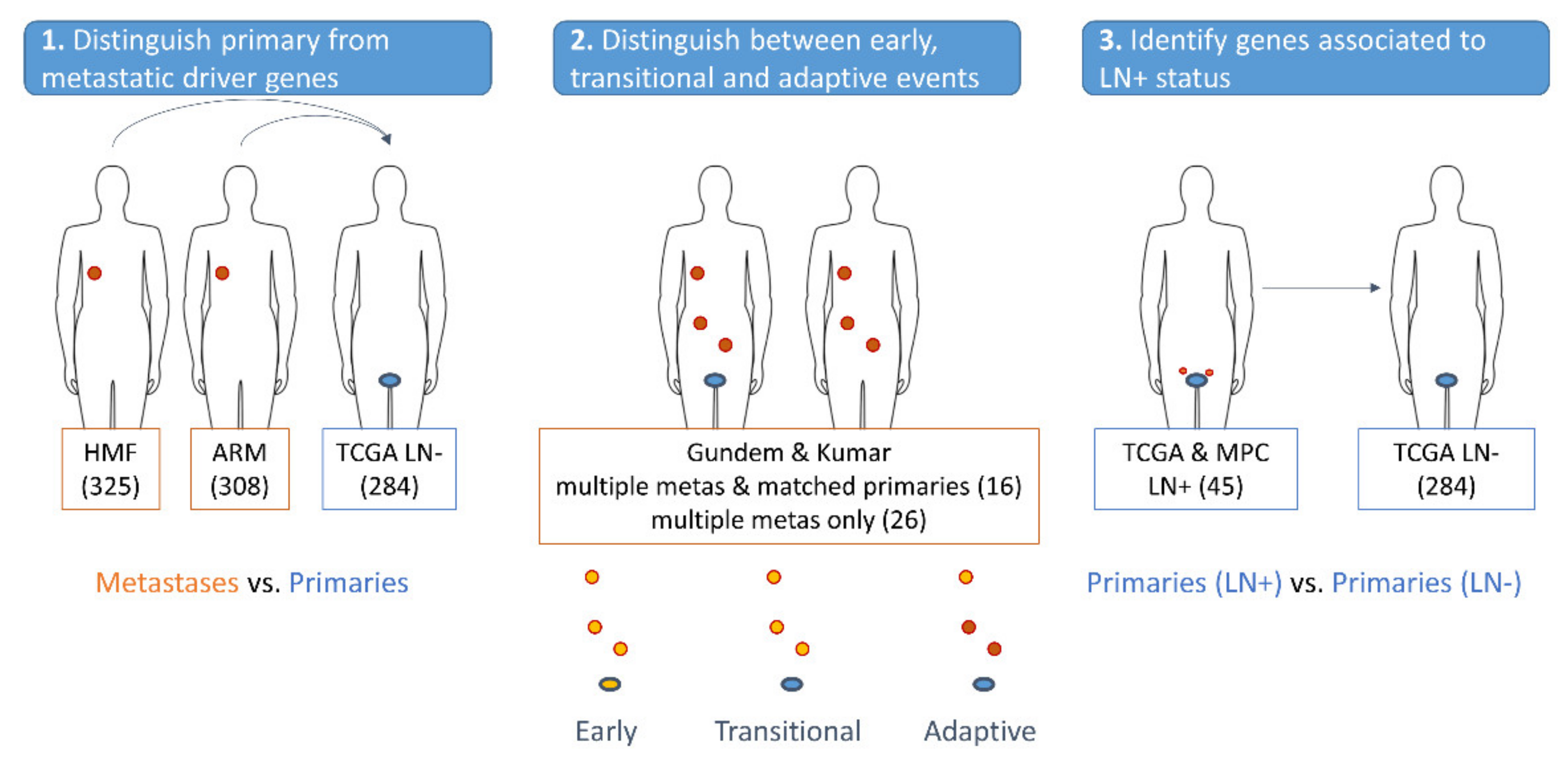
2.1. Discovery and Validation Cohorts
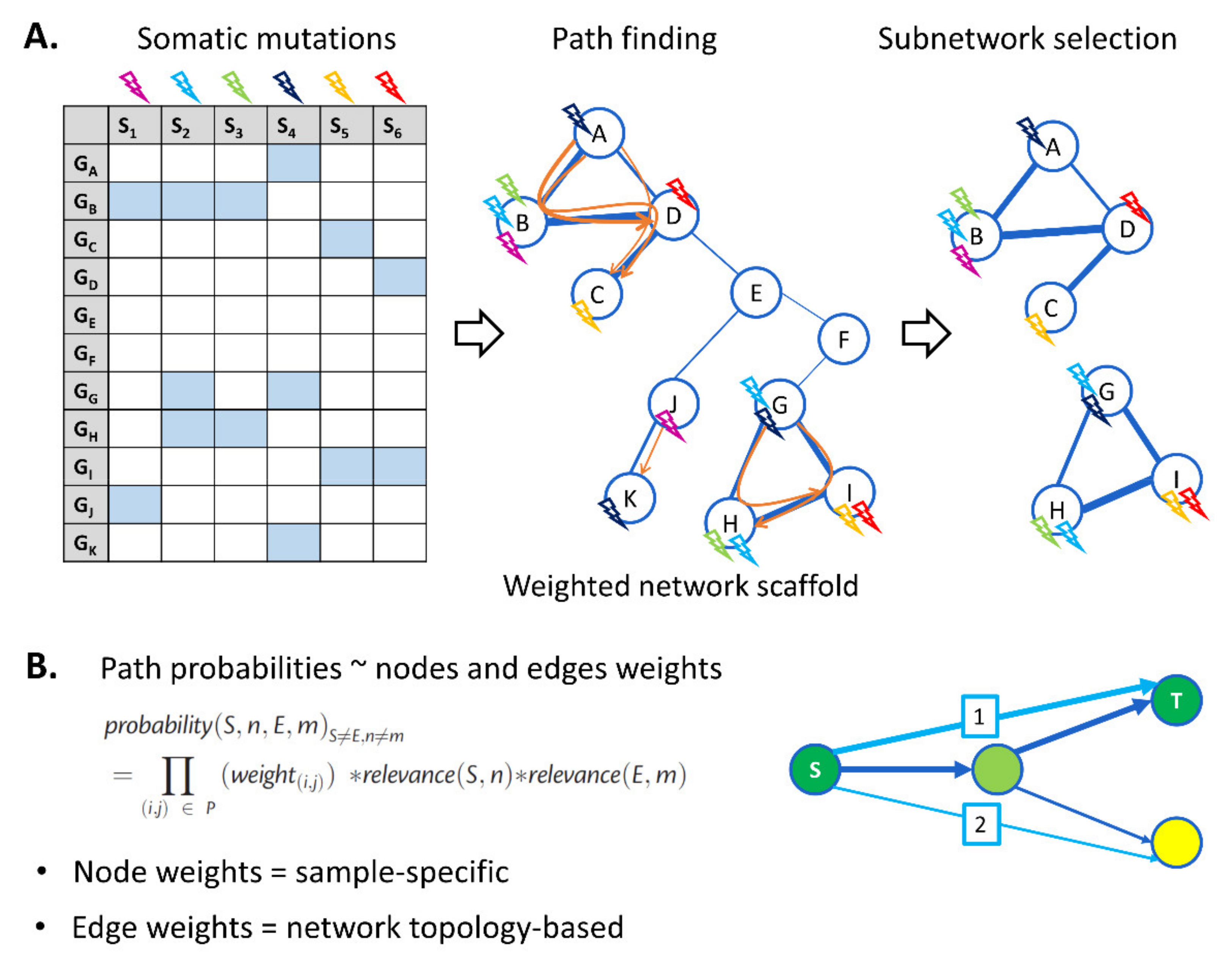
2.2. Novel Network-Based Driver Identification Method: GoNetic
2.3. GoNetic Benchmarking
2.4. OnCompare: Procedure to Compare the Mutation Rate of Driver Genes between Sample Groups
2.5. Analysis of the Discovery Cohort
2.6. SomInaClust Analysis on the HMF Data Set
2.7. Gene Ontology Enrichment Analysis
2.8. Validation of the Other Cohorts
3. Results
3.1. GoNetic: A Flexible Network-Based Driver Identification Method
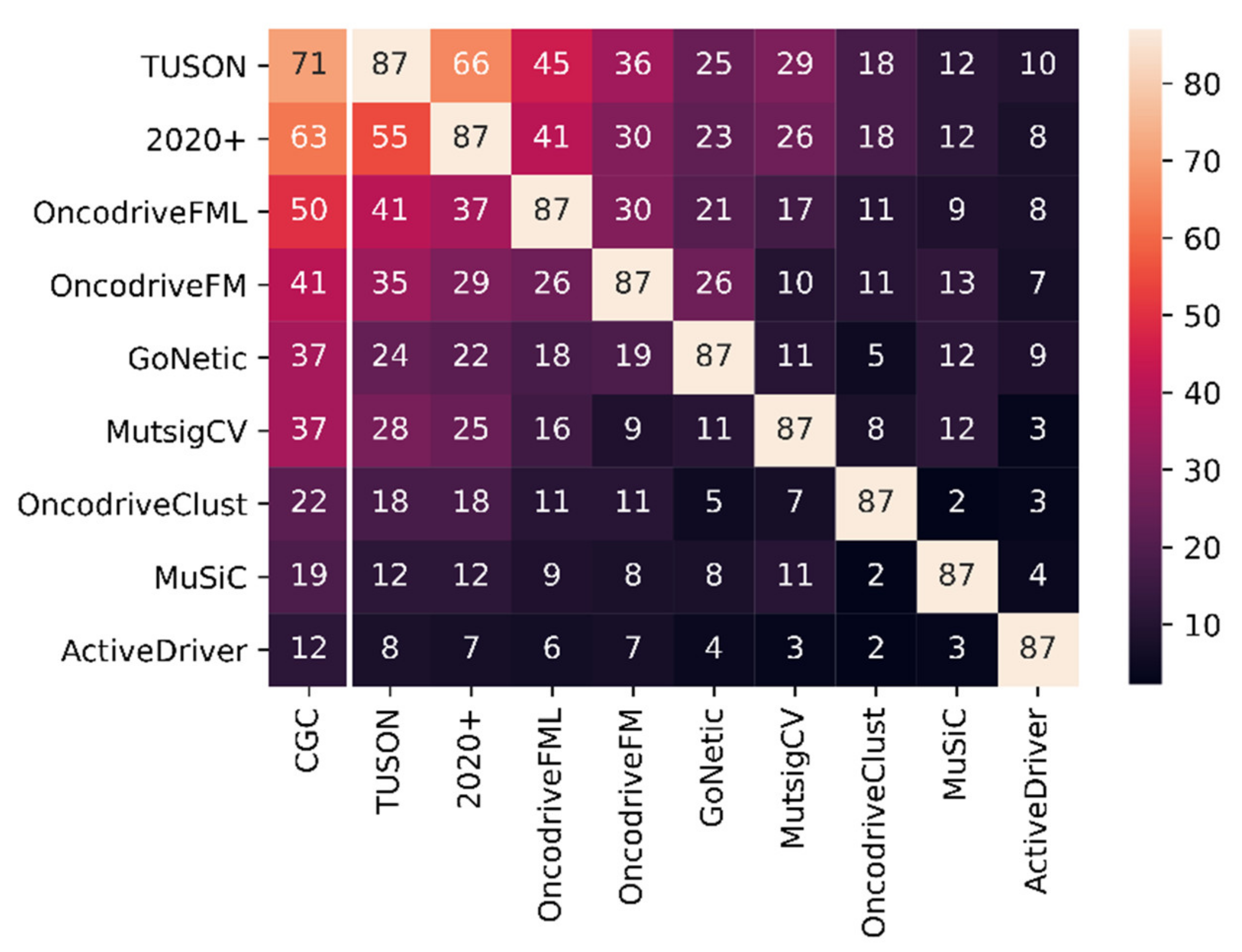
3.2. GoNetic Benchmark with the State-of-the-Art
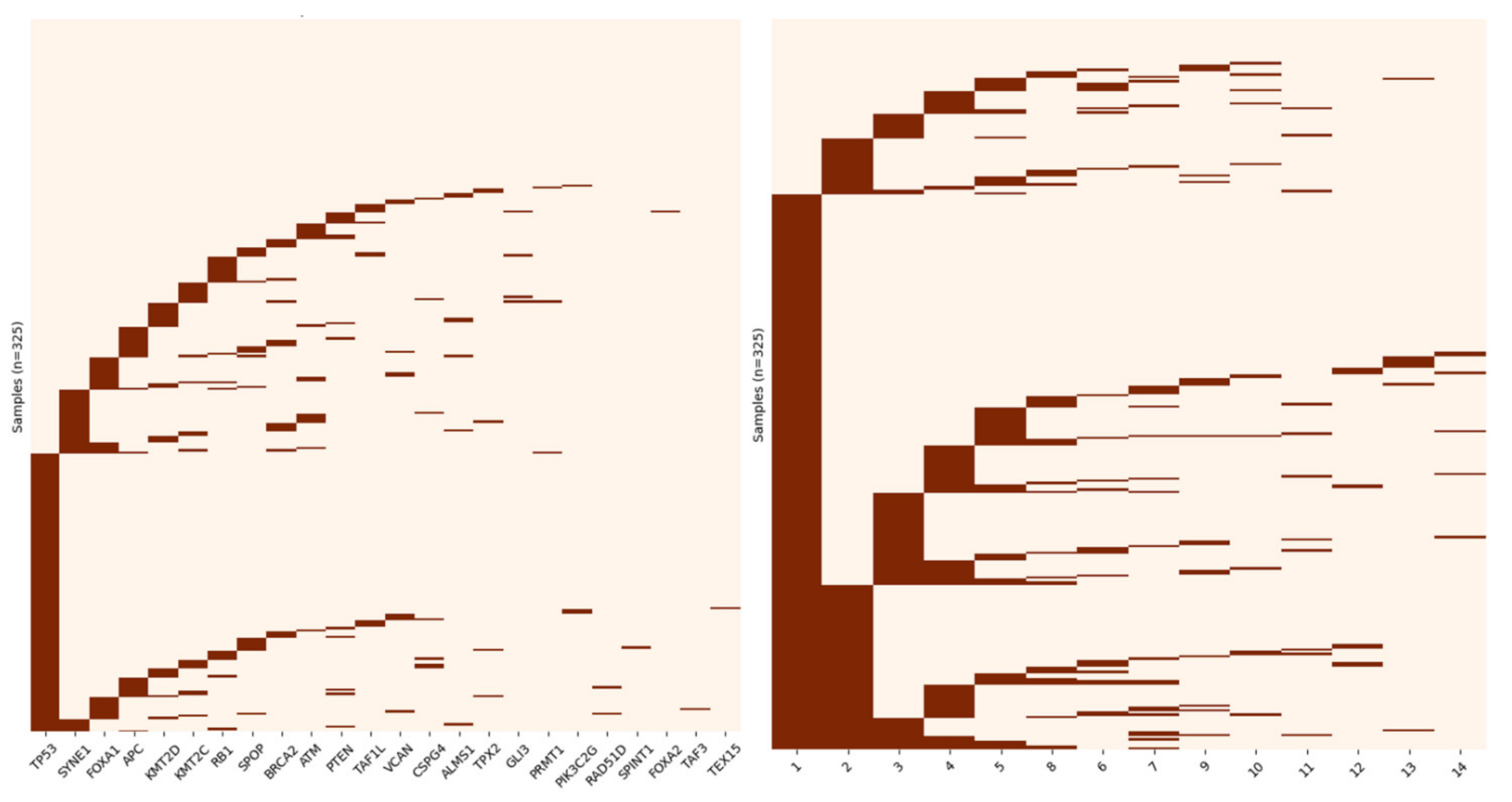
3.3. Application of GoNetic to the HMF Data Set
3.4. Distinguishing Primary from Metastatic Drivers Using Mutational Enrichment Analysis
3.5. Association of the Identified Drivers with Tumor Evolution
3.6. Identifying Drivers Prognostic of Disease Staging
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. GO Enrichment Analysis of the Subnetworks Prioritized by GoNetic
Appendix A.2. Comparison of the Results of GoNetic with Those Obtained by Previous Studies
Appendix A.3. SomInaClust on GoNetic Candidate Drivers
References
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Reyna, M.A.; Haan, D.; Paczkowska, M.; Verbeke, L.P.C.; Vazquez, M.; Kahraman, A.; Pulido-Tamayo, S.; Barenboim, J.; Wadi, L.; Dhingra, P.; et al. Pathway and network analysis of more than 2500 whole cancer genomes. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Leiserson, M.D.M.; Vandin, F.; Wu, H.-T.; Dobson, J.R.; Eldridge, J.V.; Thomas, J.L.; Papoutsaki, A.; Kim, Y.; Niu, B.; McLellan, M.; et al. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat. Genet. 2015, 47, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Dimitrakopoulos, C.; Hindupur, S.K.; Häfliger, L.; Behr, J.; Montazeri, H.; Hall, M.N.; Beerenwinkel, N. Network-based integration of multi-omics data for prioritizing cancer genes. Bioinformatics 2018, 34, 2441–2448. [Google Scholar] [CrossRef] [PubMed]
- Reimand, J.; Bader, G.D. Systematic analysis of somatic mutations in phosphorylation signaling predicts novel cancer drivers. Mol. Syst. Biol. 2013, 9, 637. [Google Scholar] [CrossRef] [PubMed]
- Reyna, M.A.; Leiserson, M.D.M.; Raphael, B.J. Hierarchical HotNet: Identifying hierarchies of altered subnetworks. Bioinformatics 2018, 34, i972–i980. [Google Scholar] [CrossRef] [PubMed]
- Hofree, M.; Shen, J.P.; Carter, H.; Gross, A.; Ideker, T. Network-based stratification of tumor mutations. Nat. Methods 2013, 10, 1108–1115. [Google Scholar] [CrossRef]
- Verbeke, L.P.C.; Van den Eynden, J.; Fierro, A.C.; Demeester, P.; Fostier, J.; Marchal, K. Pathway Relevance Ranking for Tumor Samples through Network-Based Data Integration. PLoS ONE 2015, 10, e0133503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Shihab, H.A.; Rogers, M.F.; Gough, J.; Mort, M.; Cooper, D.N.; Day, I.N.M.; Gaunt, T.R.; Campbell, C. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 2015, 31, 1536–1543. [Google Scholar] [CrossRef] [Green Version]
- Helgstrand, J.T.; Røder, M.A.; Klemann, N.; Toft, B.G.; Lichtensztajn, D.Y.; Brooks, J.D.; Brasso, K.; Vainer, B.; Iversen, P. Trends in incidence and 5-year mortality in men with newly diagnosed, metastatic prostate cancer-A population-based analysis of 2 national cohorts. Cancer 2018, 124, 2931–2938. [Google Scholar] [CrossRef] [Green Version]
- Van Dessel, L.F.; van Riet, J.; Smits, M.; Zhu, Y.; Hamberg, P.; van der Heijden, M.S.; Bergman, A.M.; van Oort, I.M.; de Wit, R.; Voest, E.E.; et al. The genomic landscape of metastatic castration-resistant prostate cancers reveals multiple distinct genotypes with potential clinical impact. Nat. Commun. 2019, 10, 5251. [Google Scholar] [CrossRef] [Green Version]
- Gundem, G.; Van Loo, P.; Kremeyer, B.; Alexandrov, L.B.; Tubio, J.M.C.; Papaemmanuil, E.; Brewer, D.S.; Kallio, H.M.L.; Högnäs, G.; Annala, M.; et al. The evolutionary history of lethal metastatic prostate cancer. Nature 2015, 520, 353–357. [Google Scholar] [CrossRef]
- Kumar, A.; Coleman, I.; Morrissey, C.; Zhang, X.; True, L.D.; Gulati, R.; Etzioni, R.; Bolouri, H.; Montgomery, B.; White, T.; et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 2016, 22, 369–378. [Google Scholar] [CrossRef]
- Crowdis, J.; Balch, S.; Sterlin, L.; Thomas, B.S.; Camp, S.Y.; Dunphy, M.; Anastasio, E.; Shah, S.; Damon, A.L.; Ramos, R.; et al. A patient-driven clinicogenomic partnership through the Metastatic Prostate Cancer Project. bioRxiv 2021. [Google Scholar] [CrossRef]
- De Maeyer, D.; Renkens, J.; Cloots, L.; De Raedt, L.; Marchal, K. PheNetic: Network-based interpretation of unstructured gene lists in E. coli. Mol. Biosyst. 2013, 9, 1594–1603. [Google Scholar] [CrossRef]
- De Maeyer, D.; Weytjens, B.; Renkens, J.; De Raedt, L.; Marchal, K. PheNetic: Network-based interpretation of molecular profiling data. Nucleic Acids Res. 2015, 43, W244–W250. [Google Scholar] [CrossRef] [Green Version]
- Perez-Romero, C.A.; Weytjens, B.; Decap, D.; Swings, T.; Michiels, J.; De Maeyer, D.; Marchal, K. IAMBEE: A web-service for the identification of adaptive pathways from parallel evolved clonal populations. Nucleic Acids Res. 2019, 47, W151–W157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gitter, A.; Klein-Seetharaman, J.; Gupta, A.; Bar-Joseph, Z. Discovering pathways by orienting edges in protein interaction networks. Nucleic Acids Res. 2011, 39, e22. [Google Scholar] [CrossRef] [PubMed]
- Navlakha, S.; Gitter, A.; Bar-Joseph, Z. A Network-based Approach for Predicting Missing Pathway Interactions. PLOS Comput. Biol. 2012, 8, 1–13. [Google Scholar] [CrossRef]
- Darwiche, A. New advances in compiling CNF to decomposable negation normal form. In Proceedings of the 16th European Conference on Artificial Intelligence (ECAI 2004), Valencia, Spain, 22–27 August 2004; pp. 328–332. [Google Scholar]
- Tokheim, C.J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; Karchin, R. Evaluating the evaluation of cancer driver genes. Proc. Natl. Acad. Sci. USA 2016, 113, 14330–14335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davoli, T.; Xu, A.W.; Mengwasser, K.E.; Sack, L.M.; Yoon, J.C.; Park, P.J.; Elledge, S.J. XCumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 2013, 155, 948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mularoni, L.; Sabarinathan, R.; Deu-Pons, J.; Gonzalez-Perez, A.; López-Bigas, N. OncodriveFML: A general framework to identify coding and non-coding regions with cancer driver mutations. Genome Biol. 2016, 17, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Tamborero, D.; Gonzalez-Perez, A.; Lopez-Bigas, N. OncodriveCLUST: Exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics 2013, 29, 2238–2244. [Google Scholar] [CrossRef] [PubMed]
- Dees, N.D.; Zhang, Q.; Kandoth, C.; Wendl, M.C.; Schierding, W.; Koboldt, D.C.; Mooney, T.B.; Callaway, M.B.; Dooling, D.; Mardis, E.R.; et al. MuSiC: Identifying mutational significance in cancer genomes. Genome Res. 2012, 22, 1589–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Perez, A.; Lopez-Bigas, N. Functional impact bias reveals cancer drivers. Nucleic Acids Res. 2012, 40, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Haw, R. Functional interaction network construction and analysis for disease discovery. In Protein Bioinformatics; Springer: Berlin/Heidelberg, Germany, 2017; pp. 235–253. [Google Scholar]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Jian, X.; Boerwinkle, E. dbNSFP: A lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 2011, 32, 894–899. [Google Scholar] [CrossRef]
- Ferlaino, M.; Rogers, M.F.; Shihab, H.A.; Mort, M.; Cooper, D.N.; Gaunt, T.R.; Campbell, C. An integrative approach to predicting the functional effects of small indels in non-coding regions of the human genome. BMC Bioinform. 2017, 18, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Van den Eynden, J.; Fierro, A.C.; Verbeke, L.P.C.; Marchal, K. SomInaClust: Detection of cancer genes based on somatic mutation patterns of inactivation and clustering. BMC Bioinform. 2015, 16, 125. [Google Scholar] [CrossRef] [Green Version]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.J.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Grasso, C.S.; Wu, Y.-M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2012, 12, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.-P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, M.F.; Lawrence, M.S.; Demichelis, F.; Drier, Y.; Cibulskis, K.; Sivachenko, A.Y.; Sboner, A.; Esgueva, R.; Pflueger, D.; Sougnez, C.; et al. The genomic complexity of primary human prostate cancer. Nature 2011, 470, 214–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Yang, L.; Li, T.; Zhang, Y. Cadherin signaling in cancer: Its functions and role as a therapeutic target. Front. Oncol. 2019, 9, 989. [Google Scholar] [CrossRef] [PubMed]
- Berx, G.; Van Roy, F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a003129. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Ouadid-Ahidouch, H.; Skryma, R.; Shuba, Y. Remodelling of Ca2+ transport in cancer: How it contributes to cancer hallmarks? Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2014, 369, 20130097. [Google Scholar] [CrossRef] [Green Version]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Calcium in tumour metastasis: New roles for known actors. Nat. Rev. Cancer 2011, 11, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Mariot, P.; Prevarskaya, N.; Roudbaraki, M.M.; Le Bourhis, X.; Van Coppenolle, F.; Vanoverberghe, K.; Skryma, R. Evidence of functional ryanodine receptor involved in apoptosis of prostate cancer (LNCaP) cells. Prostate 2000, 43, 205–214. [Google Scholar] [CrossRef]
- Ding, D.; Yao, Y.; Zhang, S.; Su, C.; Zhang, Y. C-type lectins facilitate tumor metastasis. Oncol. Lett. 2017, 13, 13–21. [Google Scholar] [CrossRef]
- Tzeng, S.-F.; Tsai, C.-H.; Chao, T.-K.; Chou, Y.-C.; Yang, Y.-C.; Tsai, M.-H.; Cha, T.-L.; Hsiao, P.-W. O-Glycosylation-mediated signaling circuit drives metastatic castration-resistant prostate cancer. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, fj201800687. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Steeg, P.S. Endocytosis: A pivotal pathway for regulating metastasis. Br. J. Cancer 2021, 124, 66–75. [Google Scholar] [CrossRef]
- Nickerson, M.L.; Im, K.M.; Misner, K.J.; Tan, W.; Lou, H.; Gold, B.; Wells, D.W.; Bravo, H.C.; Fredrikson, K.M.; Harkins, T.T.; et al. Somatic alterations contributing to metastasis of a castration-resistant prostate cancer. Hum. Mutat. 2013, 34, 1231–1241. [Google Scholar] [CrossRef] [Green Version]
- Tavassoli, P.; Wafa, L.A.; Cheng, H.; Zoubeidi, A.; Fazli, L.; Gleave, M.; Snoek, R.; Rennie, P.S. TAF1 differentially enhances androgen receptor transcriptional activity via its N-terminal kinase and ubiquitin-activating and -conjugating domains. Mol. Endocrinol. 2010, 24, 696–708. [Google Scholar] [CrossRef] [Green Version]
- Zhong, S.; Yan, H.; Chen, Z.; Li, Y.; Shen, Y.; Wang, Y.; Li, L.; Sheng, S.; Wang, Y. Overexpression of TAF1L promotes cell proliferation, migration and invasion in esophageal squamous cell carcinoma. J. Cancer 2019, 10, 979. [Google Scholar] [CrossRef] [Green Version]
- Ilieva, K.M.; Cheung, A.; Mele, S.; Chiaruttini, G.; Crescioli, S.; Griffin, M.; Nakamura, M.; Spicer, J.F.; Tsoka, S.; Lacy, K.E.; et al. Chondroitin sulfate proteoglycan 4 and its potential as an antibody immunotherapy target across different tumor types. Front. Immunol. 2018, 8, 1911. [Google Scholar] [CrossRef] [Green Version]
- Maistro, S.; dos Santos Xavier, C.; Serio, P.A.M.P.; Katayama, M.L.H.; Roela, R.A.; Koike Folgueira, M.A.A. Cancer driver genes in prostate cancer from young men. J. Clin. Oncol. 2019, 37, e16586. [Google Scholar] [CrossRef]
- Nie, M.; Pan, X.; Tao, H.; Xu, M.; Liu, S.; Sun, W.; Wu, J.; Zou, X. Clinical and prognostic significance of MYH11 in lung cancer. Oncol Lett 2020, 19, 3899–3906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Xu, Y.; Pan, C.; Yan, L.; Wang, Z.; Zhu, X. The emerging role of SPOP protein in tumorigenesis and cancer therapy. Mol. Cancer 2020, 19, 2. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; Chae, D.-K.; Lee, S.-H.; Lim, Y.; An, J.; Chae, C.H.; Kim, B.C.; Bhak, J.; Bolser, D.; Cho, D.-H. Efficient mutation screening for cervical cancers from circulating tumor DNA in blood. BMC Cancer 2020, 20, 694. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Zhou, J.; Zhang, W.; Li, P.; Liu, Y.; Shi, H.; Zhang, C.; Wang, Y.; Zhou, C.; Peng, C.; et al. RNF213 gene mutation in circulating tumor DNA detected by targeted next-generation sequencing in the assisted discrimination of early-stage lung cancer from pulmonary nodules. Thorac. Cancer 2021, 12, 181–193. [Google Scholar] [CrossRef]
- Yang, Z.; Yue, Z.; Ma, X.; Xu, Z. Calcium homeostasis: A potential vicious cycle of bone metastasis in breast cancers. Front. Oncol. 2020, 10, 293. [Google Scholar] [CrossRef]
- Bourgot, I.; Primac, I.; Louis, T.; Noël, A.; Maquoi, E. Reciprocal Interplay Between Fibrillar Collagens and Collagen-Binding Integrins: Implications in Cancer Progression and Metastasis. Front. Oncol. 2020, 10, 1488. [Google Scholar] [CrossRef] [PubMed]
- Juan-Rivera, M.C.; Martínez-Ferrer, M. Integrin inhibitors in prostate cancer. Cancers 2018, 10, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quaglia, F.; Krishn, S.R.; Wang, Y.; Goodrich, D.W.; McCue, P.; Kossenkov, A.V.; Mandigo, A.C.; Knudsen, K.E.; Weinreb, P.H.; Corey, E.; et al. Differential expression of αVβ3 and αVβ6 integrins in prostate cancer progression. PLoS ONE 2021, 16, e0244985. [Google Scholar] [CrossRef]
- Wang, H.; Ren, R.; Yang, Z.; Cai, J.; Du, S.; Shen, X. The COL11A1/Akt/CREB signaling axis enables mitochondrial-mediated apoptotic evasion to promote chemoresistance in pancreatic cancer cells through modulating BAX/BCL-2 function. J. Cancer 2021, 12, 1406–1420. [Google Scholar] [CrossRef]
- Lyu, P.; Zhang, S.-D.; Yuen, H.-F.; McCrudden, C.M.; Wen, Q.; Chan, K.-W.; Kwok, H.F. Identification of TWIST-interacting genes in prostate cancer. Sci. China Life Sci. 2017, 60, 386–396. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Panel A | Panel B | |||||
|---|---|---|---|---|---|---|
| #CGC | # Significant | % CGC | # CGC | # Significant | % CGC | |
| TUSON | 115 | 243 | 0.4733 | 71 | 87 | 0.8161 |
| 2020+ | 104 | 208 | 0.5000 | 63 | 87 | 0.7241 |
| OncodriveFML | 107 | 679 | 0.1576 | 50 | 87 | 0.5747 |
| OncodriveFM | 143 | 2600 | 0.0550 | 41 | 87 | 0.4713 |
| GoNetic | 81 | 331 | 0.2447 | 37 | 87 | 0.4253 |
| MutSigCV | 71 | 158 | 0.4494 | 37 | 87 | 0.4253 |
| OncodriveClust | 59 | 586 | 0.1007 | 22 | 87 | 0.2529 |
| MuSiC | 173 | 1975 | 0.0876 | 19 | 87 | 0.2184 |
| ActiveDriver | 34 | 417 | 0.0815 | 12 | 87 | 0.1379 |
| Hierarchical HotNet | 41 | 455 | 0.0901 | |||
| Drivers with Mutational Overrepresentation in Metastatic Samples | Primary Drivers or Genes Potentiating Metastasis | ||||
|---|---|---|---|---|---|
| Strong | Intermediate | Weak | No Sign. Enrichment with TMB Correction | No Sign. Enrichment | |
| Primary/early metastatic drivers | TP53 ** | DCHS2 * | KMT2D | FOXA1 | |
| RB1 | MUC2 * | KMT2C | SPOP | ||
| CTNNB1 | ATM PTEN | ||||
| Transitional metastatic drivers | SYNE1 * MUC16 | ||||
| TAF1L ** VCAN | |||||
| Late metastatic drivers | AR | FAT4 | LAMA2 | ||
| CACNA1H | |||||
| Not present in Gundem nor in Kumar | APC CSPG4 * | BRCA2 ** IGF2R * MUC4 | MYH11 ** LRRC7 AMPH | ||
| SHANK1 * | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Schaetzen van Brienen, L.; Miclotte, G.; Larmuseau, M.; Van den Eynden, J.; Marchal, K. Network-Based Analysis to Identify Drivers of Metastatic Prostate Cancer Using GoNetic. Cancers 2021, 13, 5291. https://doi.org/10.3390/cancers13215291
de Schaetzen van Brienen L, Miclotte G, Larmuseau M, Van den Eynden J, Marchal K. Network-Based Analysis to Identify Drivers of Metastatic Prostate Cancer Using GoNetic. Cancers. 2021; 13(21):5291. https://doi.org/10.3390/cancers13215291
Chicago/Turabian Stylede Schaetzen van Brienen, Louise, Giles Miclotte, Maarten Larmuseau, Jimmy Van den Eynden, and Kathleen Marchal. 2021. "Network-Based Analysis to Identify Drivers of Metastatic Prostate Cancer Using GoNetic" Cancers 13, no. 21: 5291. https://doi.org/10.3390/cancers13215291