
CSF1R Inhibition Combined with GM-CSF Reprograms Macrophages and Disrupts Protumoral Interplays with AML Cells


Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
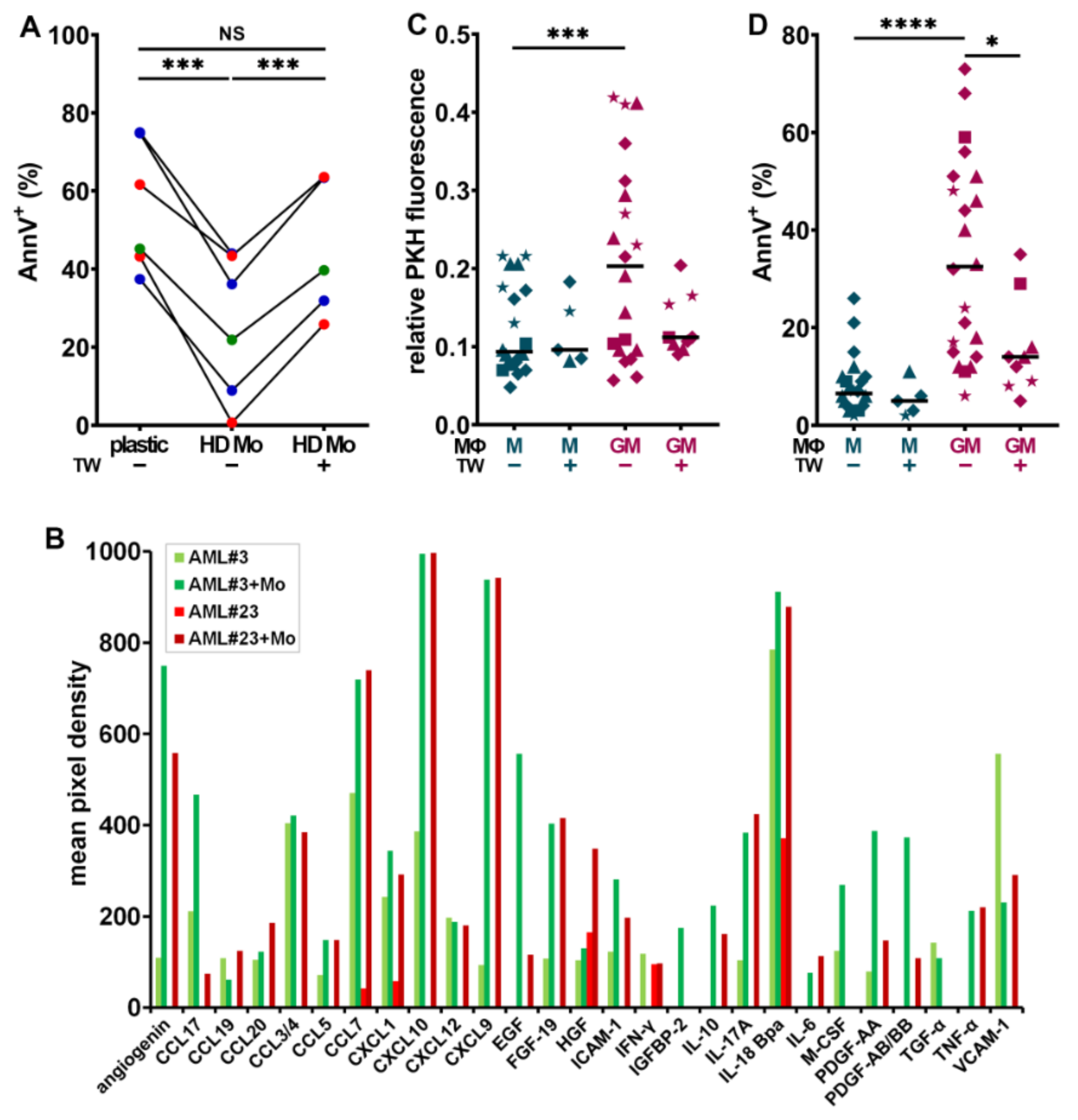{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. Immunophenotypic Analyzes by Flow Cytometry
2.3. Healthy Donor-Derived Macrophages
2.4. Cell Lines and Cell Culture
2.5. PKH Labeling and Apoptosis Detection
2.6. Cleaved Caspase-3 Detection
2.7. Cytokine Arrays
2.8. Myeloblast Resistance to Venetoclax and Midostaurin
2.9. Statistical Analysis
3. Results
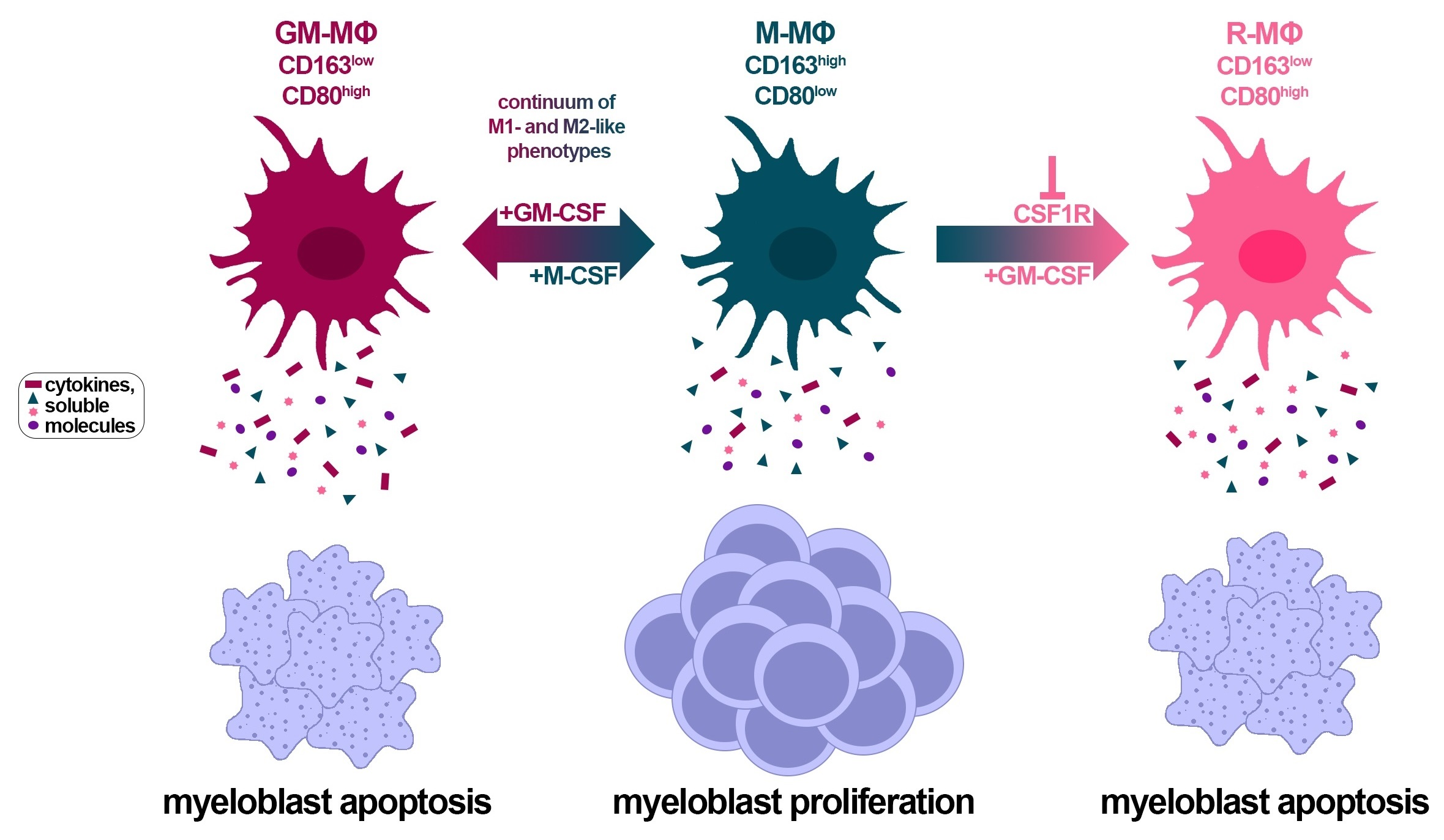
3.1. Presence of CD163-Expressing Macrophages in BM from AML Patients
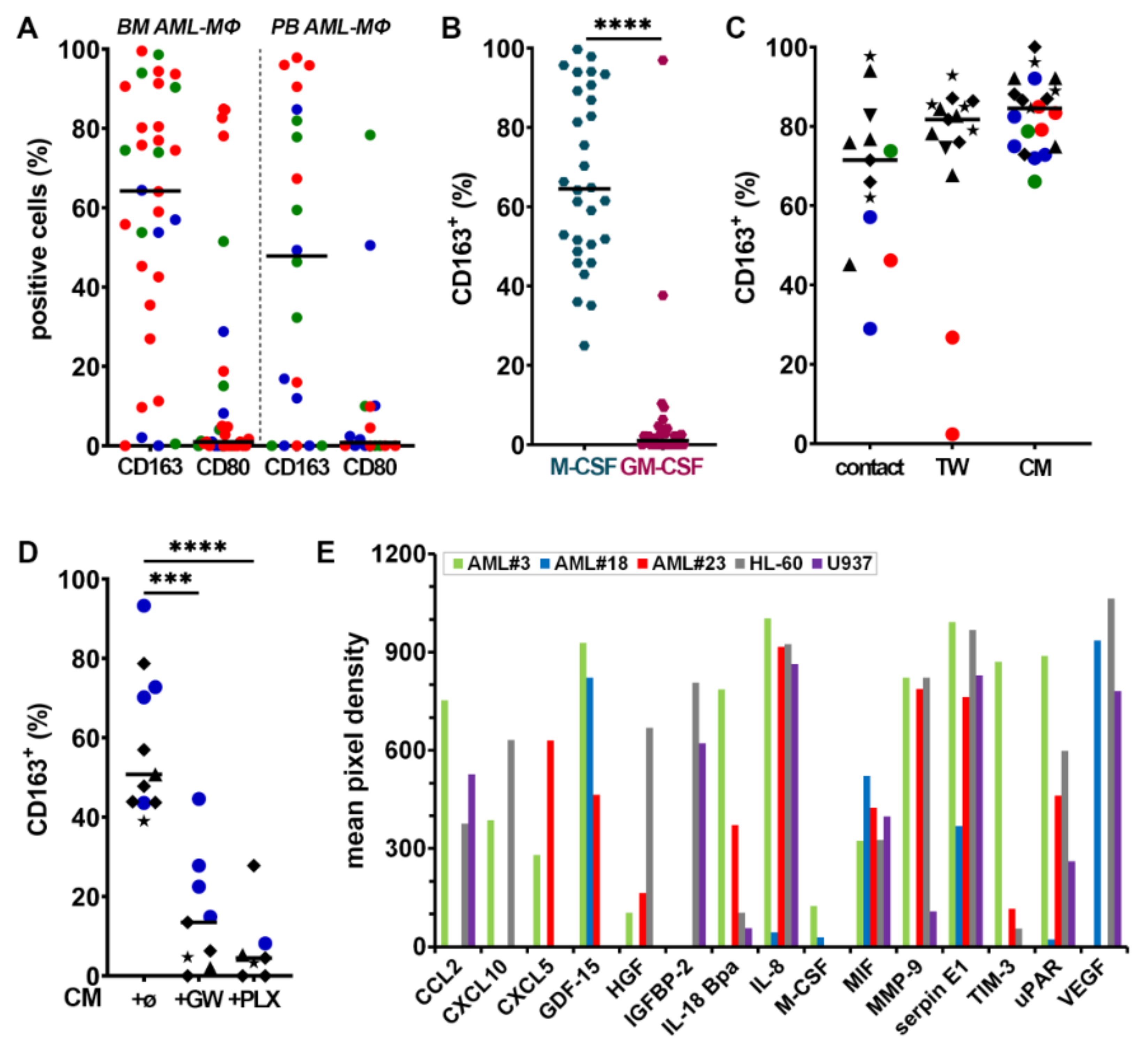
3.2. Myeloblasts Polarize HD Monocytes to Macrophages
3.3. HD Macrophages Promote Primary Myeloblast Survival
3.4. CD163+ MΦs Activated with M-CSF Support Myeloblast Survival and Proliferation
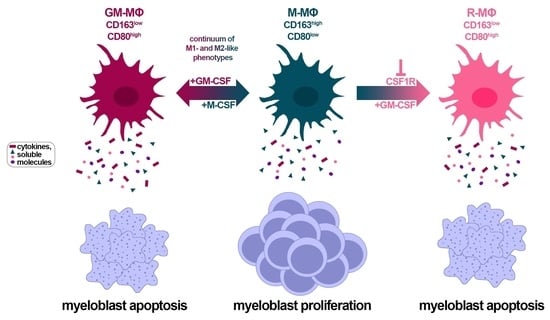
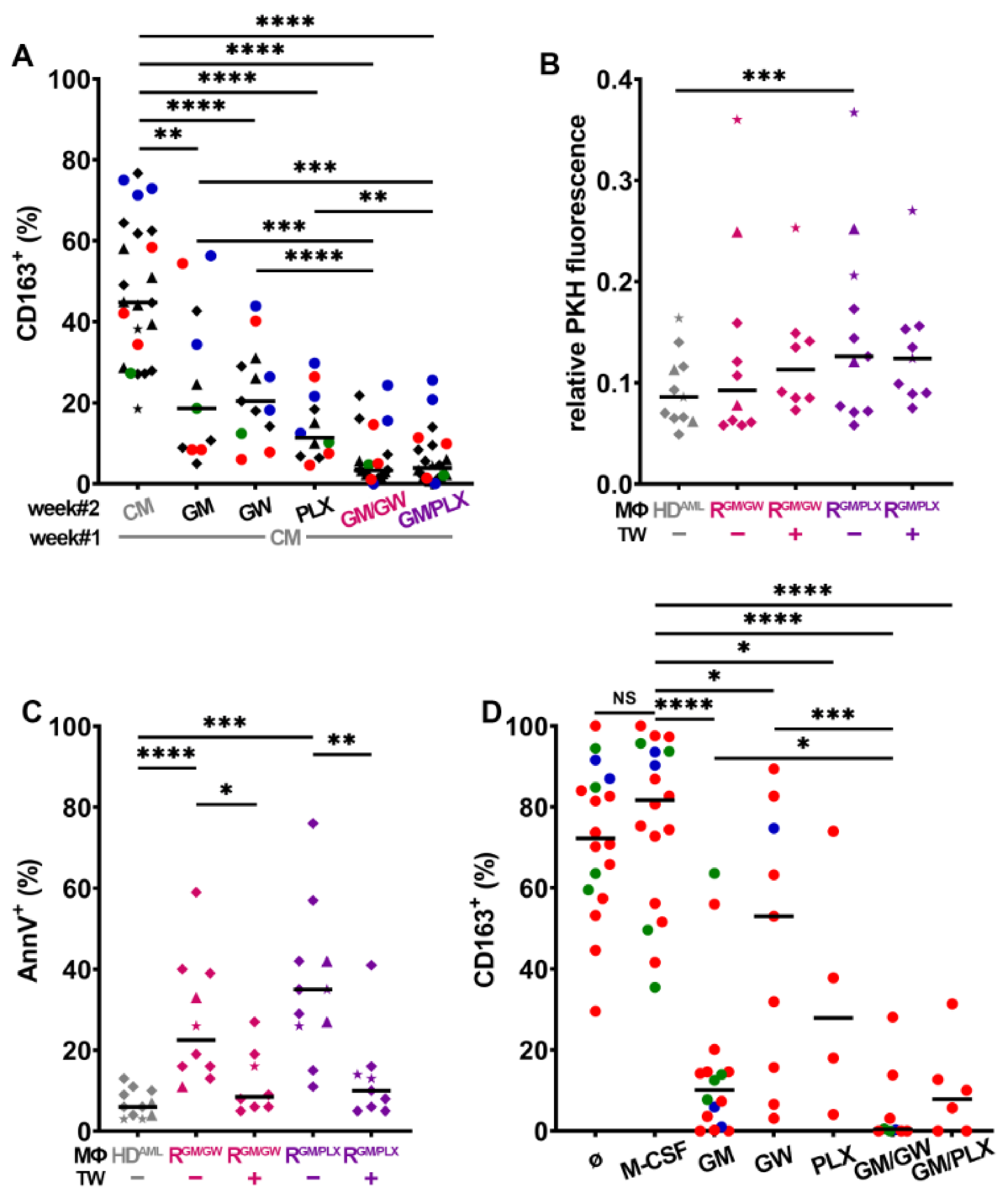
3.5. CSF1R Inhibition and Exposure to GM-CSF Reprograms CD163+ M- and HDAML-MΦs
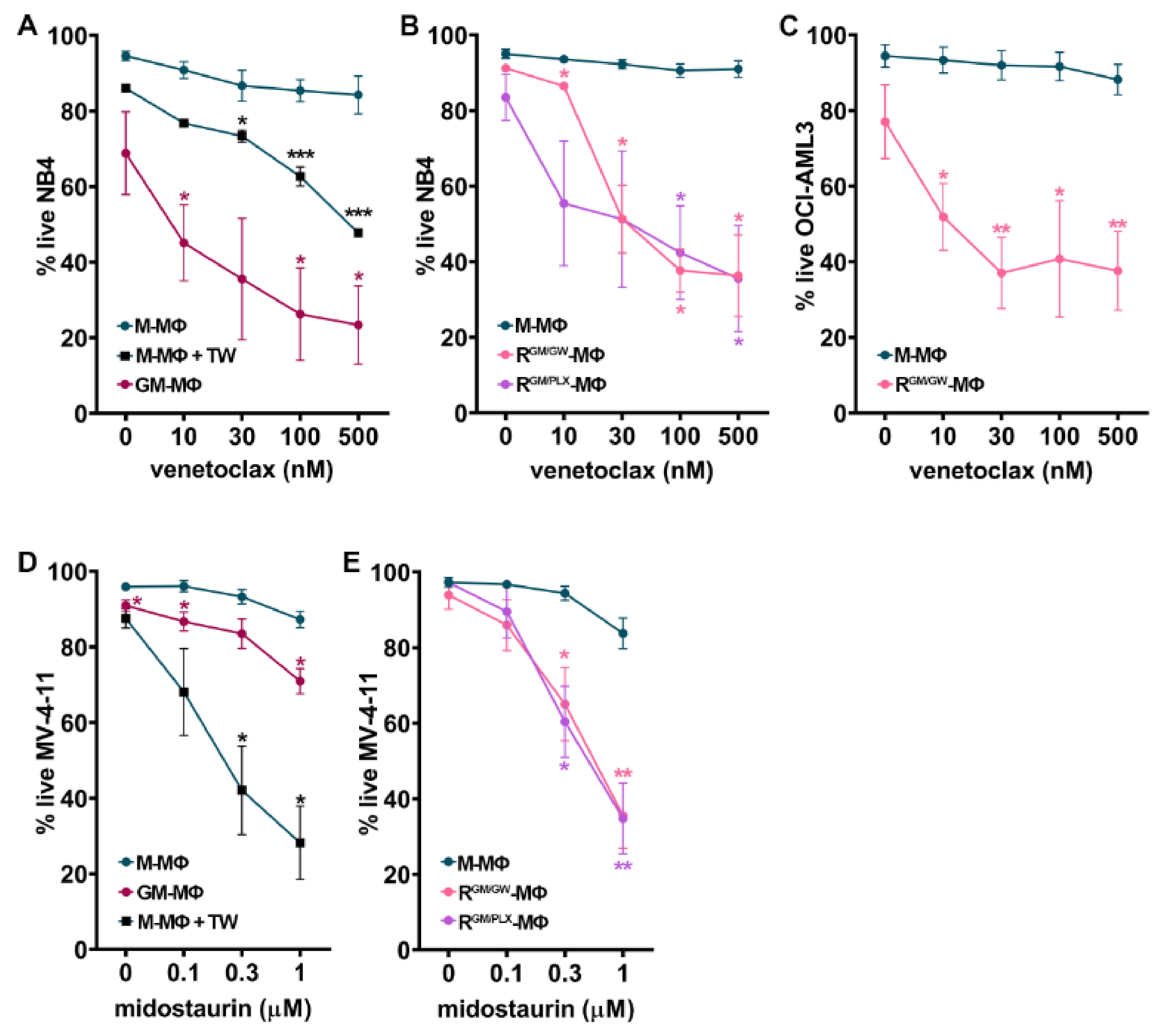
3.6. Macrophage Reprogramming Reverses Drug Resistance
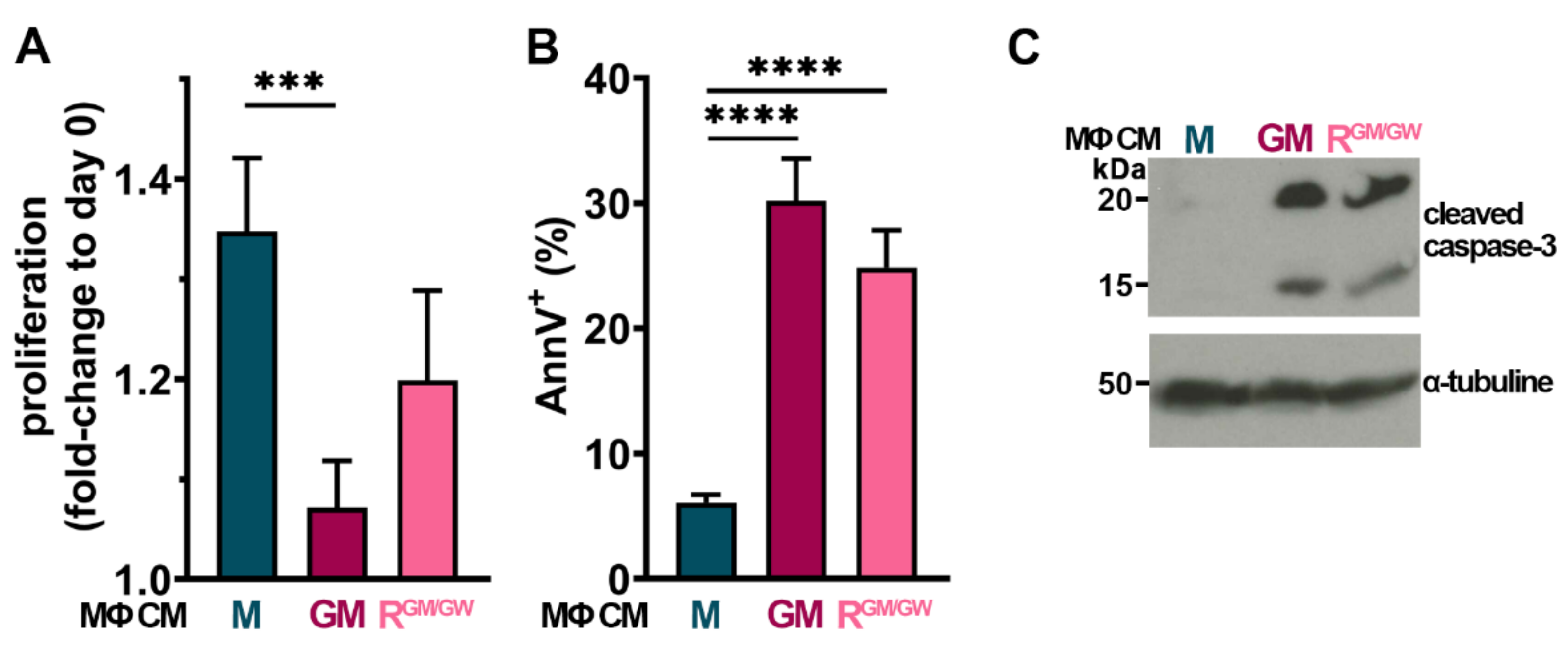
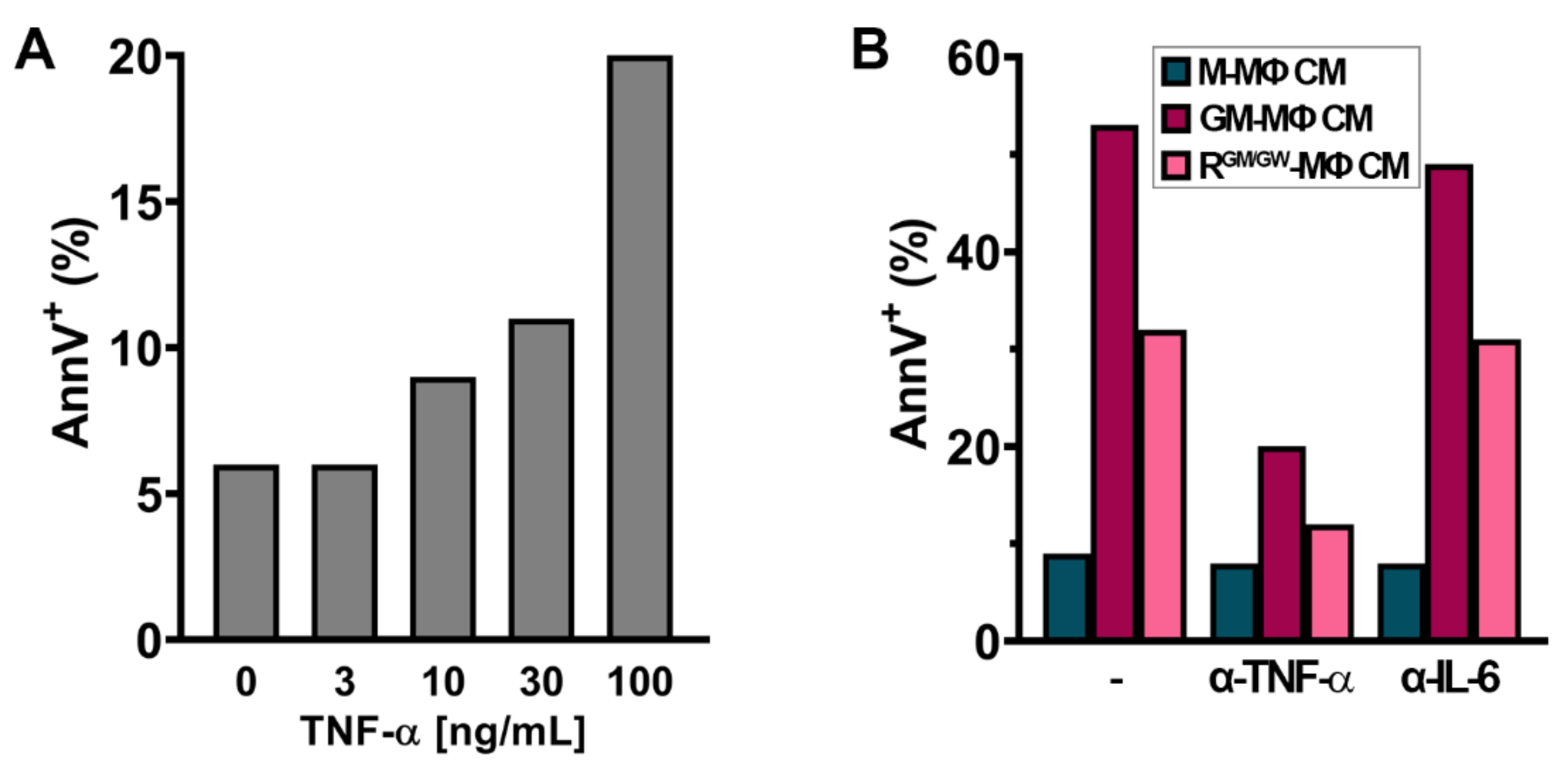
3.7. Factors Secreted by GM- and R-MΦs Play a Role in Myeloblast Apoptosis
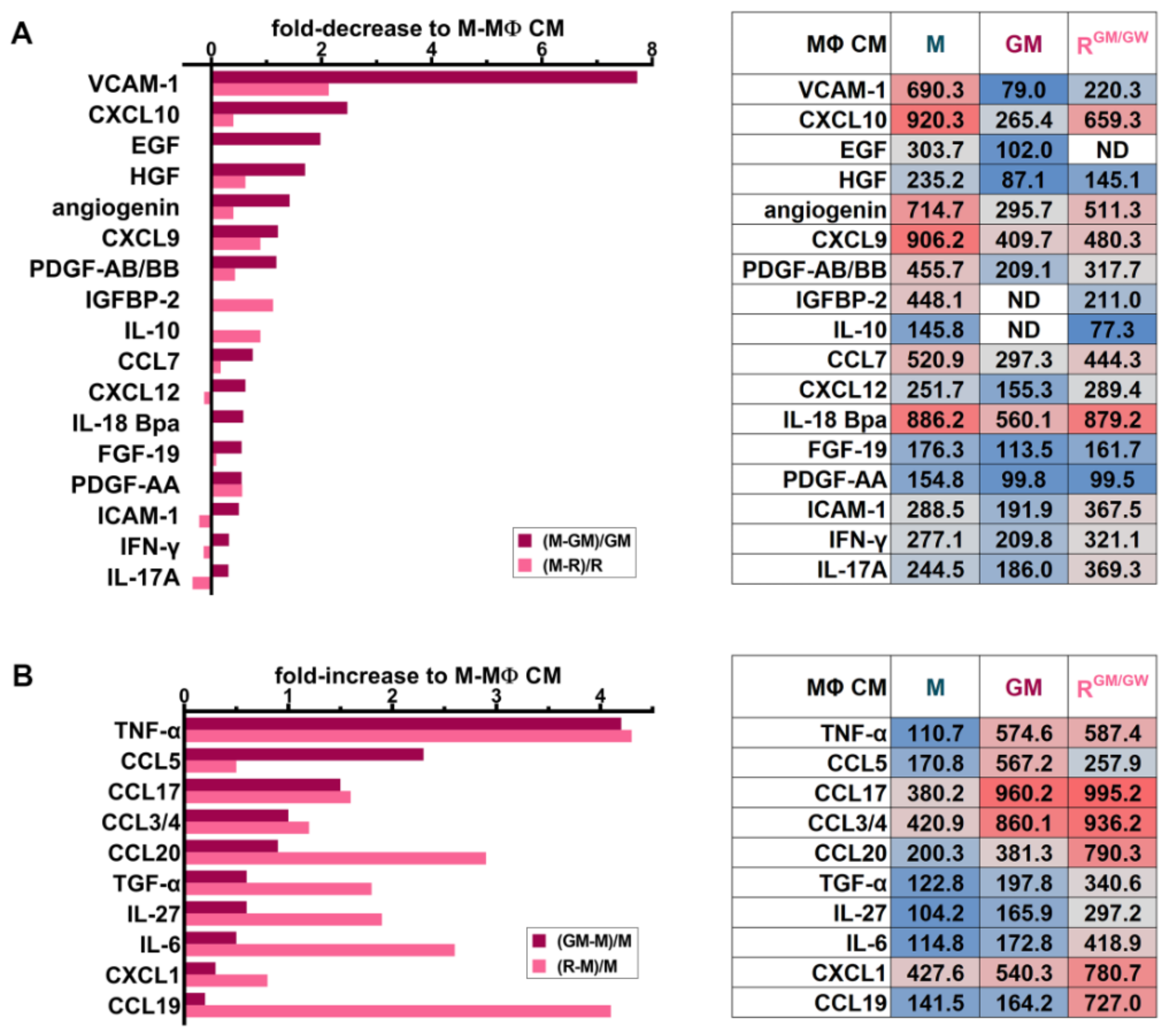
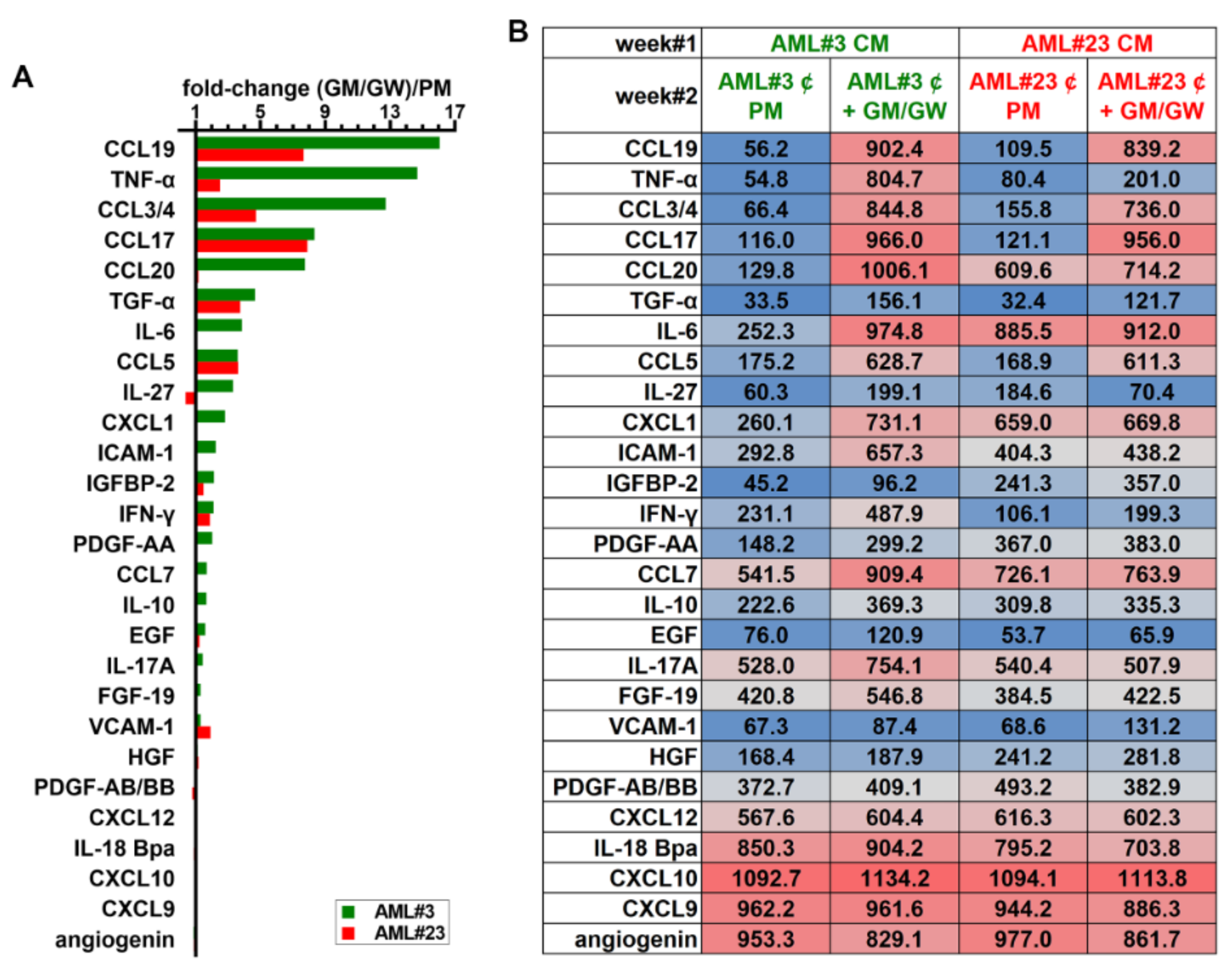
3.8. Identification of Soluble Molecules Secreted by MΦs
3.9. TNF-α Induces Apoptosis in U937 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burnett, A.K.; Hills, R.K.; Russell, N. Twenty five years of UK trials in acute myeloid leukaemia: What have we learned? Br. J. Haematol. 2020, 188, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, K.; Wang, F.; Jahn, K.; Hu, T.; Tanaka, T.; Sasaki, Y.; Kuipers, J.; Loghavi, S.; Wang, S.A.; Yan, Y.; et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat. Commun. 2020, 11, 5327. [Google Scholar] [CrossRef] [PubMed]
- Son, B.; Lee, S.; Youn, H.; Kim, E.; Kim, W.; Youn, B. The role of tumor microenvironment in therapeutic resistance. Oncotarget 2017, 8, 3933–3945. [Google Scholar] [CrossRef] [Green Version]
- Gurnari, C.; Pagliuca, S.; Visconte, V. Deciphering the Therapeutic Resistance in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2020, 21, 8505. [Google Scholar] [CrossRef]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Etzerodt, A.; Tsalkitzi, K.; Maniecki, M.; Damsky, W.; Delfini, M.; Baudoin, E.; Moulin, M.; Bosenberg, M.; Graversen, J.H.; Auphan-Anezin, N.; et al. Specific targeting of CD163(+) TAMs mobilizes inflammatory monocytes and promotes T cell-mediated tumor regression. J. Exp. Med. 2019, 216, 2394–2411. [Google Scholar] [CrossRef]
- Mills, C.D. M1 and M2 Macrophages: Oracles of Health and Disease. Crit. Rev. Immunol. 2012, 32, 463–488. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Cassetta, L.; Fragkogianni, S.; Sims, A.H.; Swierczak, A.; Forrester, L.M.; Zhang, H.; Soong, D.Y.H.; Cotechini, T.; Anur, P.; Lin, E.Y.; et al. Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell 2019, 35, 588–602.e10. [Google Scholar] [CrossRef] [Green Version]
- Neubert, N.J.; Schmittnaegel, M.; Bordry, N.; Nassiri, S.; Wald, N.; Martignier, C.; Tille, L.; Homicsko, K.; Damsky, W.; Maby-El Hajjami, H.; et al. T cell-induced CSF1 promotes melanoma resistance to PD1 blockade. Sci. Transl. Med. 2018, 10, eaan3311. [Google Scholar] [CrossRef] [Green Version]
- Komohara, Y.; Jinushi, M.; Takeya, M. Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer Sci. 2014, 105, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Takizawa, H.; Manz, M.G. Macrophage tolerance: CD47-SIRP-α-mediated signals matter. Nat. Immunol. 2007, 8, 1287–1289. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Cassetta, L.; Kitamura, T. Macrophage targeting: Opening new possibilities for cancer immunotherapy. Immunology 2018, 155, 285–293. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Yang, X.; Feng, W.; Wang, R.; Yang, F.; Wang, L.; Chen, S.; Ru, Y.; Cheng, T.; Zheng, G. Repolarizing heterogeneous leukemia-associated macrophages with more M1 characteristics eliminates their pro-leukemic effects. Oncoimmunology 2018, 7, e1412910. [Google Scholar] [CrossRef]
- Chen, C.; Wang, R.; Feng, W.; Yang, F.; Wang, L.; Yang, X.; Ren, L.; Zheng, G. Peritoneal resident macrophages in mice with MLL-AF9-induced acute myeloid leukemia show an M2-like phenotype. Ann. Transl. Med. 2021, 9, 266. [Google Scholar] [CrossRef]
- Al-Matary, Y.S.; Botezatu, L.; Opalka, B.; Hones, J.M.; Lams, R.F.; Thivakaran, A.; Schutte, J.; Koster, R.; Lennartz, K.; Schroeder, T.; et al. Acute myeloid leukemia cells polarize macrophages towards a leukemia supporting state in a Growth factor independence 1 dependent manner. Haematologica 2016, 101, 1216–1227. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Qu, J.; Cao, W.; Liu, Y.; Zheng, G.; Zhang, E.; Cai, Z. Identification of prognostic genes in the acute myeloid leukemia immune microenvironment based on TCGA data analysis. Cancer Immunol. Immunother. 2019, 68, 1971–1978. [Google Scholar] [CrossRef]
- Shiraishi, D.; Fujiwara, Y.; Horlad, H.; Saito, Y.; Iriki, T.; Tsuboki, J.; Cheng, P.; Nakagata, N.; Mizuta, H.; Bekki, H.; et al. CD163 Is Required for Protumoral Activation of Macrophages in Human and Murine Sarcoma. Cancer Res. 2018, 78, 3255–3266. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Yrigoyen, M.; Cassetta, L.; Pollard, J.W. Macrophage targeting in cancer. Ann. N. Y. Acad. Sci. 2020, 1499, 18–41. [Google Scholar] [CrossRef]
- Edwards, D.K.t.; Watanabe-Smith, K.; Rofelty, A.; Damnernsawad, A.; Laderas, T.; Lamble, A.; Lind, E.F.; Kaempf, A.; Mori, M.; Rosenberg, M.; et al. CSF1R inhibitors exhibit antitumor activity in acute myeloid leukemia by blocking paracrine signals from support cells. Blood 2019, 133, 588–599. [Google Scholar] [CrossRef]
- Gutierrez-Gonzalez, A.; Martinez-Moreno, M.; Samaniego, R.; Arellano-Sanchez, N.; Salinas-Munoz, L.; Relloso, M.; Valeri, A.; Martinez-Lopez, J.; Corbi, A.L.; Hidalgo, A.; et al. Evaluation of the potential therapeutic benefits of macrophage reprogramming in multiple myeloma. Blood 2016, 128, 2241–2252. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Liu, F.; Liu, L. Prognostic significance of PD-L1 in solid tumor: An updated meta-analysis. Medicine 2017, 96, e6369. [Google Scholar] [CrossRef]
- Papin, A.; Tessoulin, B.; Bellanger, C.; Moreau, A.; Le Bris, Y.; Maisonneuve, H.; Moreau, P.; Touzeau, C.; Amiot, M.; Pellat-Deceunynck, C.; et al. CSF1R and BTK inhibitions as novel strategies to disrupt the dialog between mantle cell lymphoma and macrophages. Leukemia 2019, 33, 2442–2453. [Google Scholar] [CrossRef]
- Valero, J.G.; Matas-Cespedes, A.; Arenas, F.; Rodriguez, V.; Carreras, J.; Serrat, N.; Guerrero-Hernandez, M.; Yahiaoui, A.; Balague, O.; Martin, S.; et al. The receptor of the colony-stimulating factor-1 (CSF-1R) is a novel prognostic factor and therapeutic target in follicular lymphoma. Leukemia 2021, 35, 2635–2649. [Google Scholar] [CrossRef]
- Spertini, C.; Baïsse, B.; Bellone, M.; Gikic, M.; Smirnova, T.; Spertini, O. Acute Myeloid and Lymphoblastic Leukemia Cell Interactions with Endothelial Selectins: Critical Role of PSGL-1, CD44 and CD43. Cancers 2019, 11, 1253. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, S.; Campo, E.; Harris, N.; Jaffe, E.; Pileri, S.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; IARC Press: Lyon, France, 2016. [Google Scholar]
- Bene, M.C.; Nebe, T.; Bettelheim, P.; Buldini, B.; Bumbea, H.; Kern, W.; Lacombe, F.; Lemez, P.; Marinov, I.; Matutes, E.; et al. Immunophenotyping of acute leukemia and lymphoproliferative disorders: A consensus proposal of the European LeukemiaNet Work Package 10. Leukemia 2011, 25, 567–574. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, L.; Smirnova, T.; Kedrin, D.; Wyckoff, J.; Zhu, L.; Stanley, E.R.; Cox, D.; Muller, W.J.; Pollard, J.W.; Van Rooijen, N.; et al. The EGF/CSF-1 paracrine invasion loop can be triggered by heregulin beta1 and CXCL12. Cancer Res. 2009, 69, 3221–3227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.J.; Gu, Y.; Wang, C.Z.; Jin, Y.; Wen, X.M.; Ma, J.C.; Tang, L.J.; Mao, Z.W.; Qian, J.; Lin, J. The M2 macrophage marker CD206: A novel prognostic indicator for acute myeloid leukemia. Oncoimmunology 2020, 9, 1683347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fayard, B.; Bianchi, F.; Dey, J.; Moreno, E.; Djaffer, S.; Hynes, N.E.; Monard, D. The serine protease inhibitor protease nexin-1 controls mammary cancer metastasis through LRP-1-mediated MMP-9 expression. Cancer Res. 2009, 69, 5690–5698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wischhusen, J.; Melero, I.; Fridman, W.H. Growth/Differentiation Factor-15 (GDF-15): From Biomarker to Novel Targetable Immune Checkpoint. Front. Immunol. 2020, 11, 951. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, H.; Sun, M.; Deng, X.; Wu, X.; Ma, Y.; Li, M.; Shuoa, S.M.; You, Q.; Miao, L. CXCL5/CXCR2 axis in tumor microenvironment as potential diagnostic biomarker and therapeutic target. Cancer Commun. 2020, 40, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Kubota, Y.; Takubo, K.; Shimizu, T.; Ohno, H.; Kishi, K.; Shibuya, M.; Saya, H.; Suda, T. M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J. Exp. Med. 2009, 206, 1089–1102. [Google Scholar] [CrossRef]
- Kobori, T.; Hamasaki, S.; Kitaura, A.; Yamazaki, Y.; Nishinaka, T.; Niwa, A.; Nakao, S.; Wake, H.; Mori, S.; Yoshino, T.; et al. Interleukin-18 Amplifies Macrophage Polarization and Morphological Alteration, Leading to Excessive Angiogenesis. Front. Immunol. 2018, 9, 334. [Google Scholar] [CrossRef]
- Récher, C. Clinical Implications of Inflammation in Acute Myeloid Leukemia. Front. Oncol. 2021, 11, 623952. [Google Scholar] [CrossRef]
- Sanchez-Correa, B.; Bergua, J.M.; Campos, C.; Gayoso, I.; Arcos, M.J.; Banas, H.; Morgado, S.; Casado, J.G.; Solana, R.; Tarazona, R. Cytokine profiles in acute myeloid leukemia patients at diagnosis: Survival is inversely correlated with IL-6 and directly correlated with IL-10 levels. Cytokine 2013, 61, 885–891. [Google Scholar] [CrossRef]
- Li, T.; Forbes, M.E.; Fuller, G.N.; Li, J.; Yang, X.; Zhang, W. IGFBP2: Integrative hub of developmental and oncogenic signaling network. Oncogene 2020, 39, 2243–2257. [Google Scholar] [CrossRef]
- Kühnl, A.; Kaiser, M.; Neumann, M.; Fransecky, L.; Heesch, S.; Radmacher, M.; Marcucci, G.; Bloomfield, C.D.; Hofmann, W.K.; Thiel, E.; et al. High expression of IGFBP2 is associated with chemoresistance in adult acute myeloid leukemia. Leuk. Res. 2011, 35, 1585–1590. [Google Scholar] [CrossRef] [Green Version]
- Doepfner, K.T.; Spertini, O.; Arcaro, A. Autocrine insulin-like growth factor-I signaling promotes growth and survival of human acute myeloid leukemia cells via the phosphoinositide 3-kinase/Akt pathway. Leukemia 2007, 21, 1921–1930. [Google Scholar] [CrossRef]
- Girard-Guyonvarc’h, C.; Palomo, J.; Martin, P.; Rodriguez, E.; Troccaz, S.; Palmer, G.; Gabay, C. Unopposed IL-18 signaling leads to severe TLR9-induced macrophage activation syndrome in mice. Blood 2018, 131, 1430–1441. [Google Scholar] [CrossRef]
- Smirnova, T.; Bonapace, L.; MacDonald, G.; Kondo, S.; Wyckoff, J.; Ebersbach, H.; Fayard, B.; Doelemeyer, A.; Coissieux, M.M.; Heideman, M.R.; et al. Serpin E2 promotes breast cancer metastasis by remodeling the tumor matrix and polarizing tumor associated macrophages. Oncotarget 2016, 7, 82289–82304. [Google Scholar] [CrossRef] [Green Version]
- Vilgelm, A.E.; Richmond, A. Chemokines Modulate Immune Surveillance in Tumorigenesis, Metastasis, and Response to Immunotherapy. Front. Immunol. 2019, 10, 333. [Google Scholar] [CrossRef] [Green Version]
- Schepers, K.; Campbell, T.B.; Passegue, E. Normal and leukemic stem cell niches: Insights and therapeutic opportunities. Cell Stem Cell 2015, 16, 254–267. [Google Scholar] [CrossRef] [Green Version]
- Binder, S.; Luciano, M.; Horejs-Hoeck, J. The cytokine network in acute myeloid leukemia (AML): A focus on pro- and anti-inflammatory mediators. Cytokine Growth Factor Rev. 2018, 43, 8–15. [Google Scholar] [CrossRef]
- Wyckoff, J.; Wang, W.; Lin, E.Y.; Wang, Y.; Pixley, F.; Stanley, E.R.; Graf, T.; Pollard, J.W.; Segall, J.; Condeelis, J. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004, 64, 7022–7029. [Google Scholar] [CrossRef] [Green Version]
- Boimel, P.J.; Smirnova, T.; Zhou, Z.N.; Wyckoff, J.; Park, H.; Coniglio, S.J.; Qian, B.Z.; Stanley, E.R.; Cox, D.; Pollard, J.W.; et al. Contribution of CXCL12 secretion to invasion of breast cancer cells. Breast Cancer Res. 2012, 14, R23. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Cai, Y.; Liu, L.; Wu, Y.; Xiong, X. Crucial biological functions of CCL7 in cancer. PeerJ 2018, 6, e4928. [Google Scholar] [CrossRef]
- Prabhala, R.H.; Pelluru, D.; Fulciniti, M.; Prabhala, H.K.; Nanjappa, P.; Song, W.; Pai, C.; Amin, S.; Tai, Y.T.; Richardson, P.G.; et al. Elevated IL-17 produced by TH17 cells promotes myeloma cell growth and inhibits immune function in multiple myeloma. Blood 2010, 115, 5385–5392. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Molecular Pathways: Deciphering Mechanisms of Resistance to Macrophage-Targeted Therapies. Clin. Cancer Res. 2017, 23, 876–884. [Google Scholar] [CrossRef] [Green Version]
- Chao, M.P.; Takimoto, C.H.; Feng, D.D.; McKenna, K.; Gip, P.; Liu, J.; Volkmer, J.P.; Weissman, I.L.; Majeti, R. Therapeutic Targeting of the Macrophage Immune Checkpoint CD47 in Myeloid Malignancies. Front. Oncol. 2019, 9, 1380. [Google Scholar] [CrossRef]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; Baty, J.D.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Jacamo, R.; Chen, Y.; Wang, Z.; Ma, W.; Zhang, M.; Spaeth, E.L.; Wang, Y.; Battula, V.L.; Mak, P.Y.; Schallmoser, K.; et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-κB mediates chemoresistance. Blood 2014, 123, 2691–2702. [Google Scholar] [CrossRef]
- Abdul-Aziz, A.M.; Shafat, M.S.; Mehta, T.K.; Di Palma, F.; Lawes, M.J.; Rushworth, S.A.; Bowles, K.M. MIF-Induced Stromal PKCβ/IL8 Is Essential in Human Acute Myeloid Leukemia. Cancer Res. 2017, 77, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Cho, B.S.; Zeng, Z.; Mu, H.; Wang, Z.; Konoplev, S.; McQueen, T.; Protopopova, M.; Cortes, J.; Marszalek, J.R.; Peng, S.B.; et al. Antileukemia activity of the novel peptidic CXCR4 antagonist LY2510924 as monotherapy and in combination with chemotherapy. Blood 2015, 126, 222–232. [Google Scholar] [CrossRef]
- Sanchez-Martin, L.; Estecha, A.; Samaniego, R.; Sanchez-Ramon, S.; Vega, M.A.; Sanchez-Mateos, P. The chemokine CXCL12 regulates monocyte-macrophage differentiation and RUNX3 expression. Blood 2011, 117, 88–97. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Cho, Y.B. CCL7 Signaling in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1231, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Urra, S.; Fischer, M.C.; Martinez, J.R.; Veliz, L.; Orellana, P.; Solar, A.; Bohmwald, K.; Kalergis, A.; Riedel, C.; Corvalan, A.H.; et al. Differential expression profile of CXCR3 splicing variants is associated with thyroid neoplasia. Potential role in papillary thyroid carcinoma oncogenesis? Oncotarget 2018, 9, 2445–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Q.; Lu, P.; Xia, Y.; Ding, S.; Fan, Y.; Li, X.; Han, P.; Liu, J.; Tian, D.; Liu, M. CXCL9: Evidence and contradictions for its role in tumor progression. Cancer Med. 2016, 5, 3246–3259. [Google Scholar] [CrossRef] [PubMed]
- Marques, R.E.; Guabiraba, R.; Russo, R.C.; Teixeira, M.M. Targeting CCL5 in inflammation. Expert Opin. Ther. Targets 2013, 17, 1439–1460. [Google Scholar] [CrossRef] [PubMed]
- Achuthan, A.; Cook, A.D.; Lee, M.C.; Saleh, R.; Khiew, H.W.; Chang, M.W.; Louis, C.; Fleetwood, A.J.; Lacey, D.C.; Christensen, A.D.; et al. Granulocyte macrophage colony-stimulating factor induces CCL17 production via IRF4 to mediate inflammation. J. Clin. Investig. 2016, 126, 3453–3466. [Google Scholar] [CrossRef] [Green Version]
- Ruytinx, P.; Proost, P.; Van Damme, J.; Struyf, S. Chemokine-Induced Macrophage Polarization in Inflammatory Conditions. Front. Immunol. 2018, 9, 1930. [Google Scholar] [CrossRef] [Green Version]
- Zlotnik, A.; Yoshie, O. Chemokines: A new classification system and their role in immunity. Immunity 2000, 12, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Chiba, Y.; Mizoguchi, I.; Furusawa, J.; Hasegawa, H.; Ohashi, M.; Xu, M.; Owaki, T.; Yoshimoto, T. Interleukin-27 Exerts Its Antitumor Effects by Promoting Differentiation of Hematopoietic Stem Cells to M1 Macrophages. Cancer Res. 2018, 78, 182–194. [Google Scholar] [CrossRef] [Green Version]
- Larrick, J.W.; Wright, S.C. Cytotoxic mechanism of tumor necrosis factor-alpha. FASEB J. 1990, 4, 3215–3223. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Dander, E.; Fallati, A.; Gulic, T.; Pagni, F.; Gaspari, S.; Silvestri, D.; Cricri, G.; Bedini, G.; Portale, F.; Buracchi, C.; et al. Monocyte-macrophage polarization and recruitment pathways in the tumour microenvironment of B-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2021, 193, 1157–1171. [Google Scholar] [CrossRef]
- Skytthe, M.K.; Graversen, J.H.; Moestrup, S.K. Targeting of CD163(+) Macrophages in Inflammatory and Malignant Diseases. Int. J. Mol. Sci. 2020, 21, 5497. [Google Scholar] [CrossRef]
- Etzerodt, A.; Moestrup, S.K. CD163 and inflammation: Biological, diagnostic, and therapeutic aspects. Antioxid. Redox Signal. 2013, 18, 2352–2363. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.M.C.; Achuthan, A.A.; Hamilton, J.A. GM-CSF: A Promising Target in Inflammation and Autoimmunity. Immunotargets Ther. 2020, 9, 225–240. [Google Scholar] [CrossRef]
- Kim, I.K.; Koh, C.H.; Jeon, I.; Shin, K.S.; Kang, T.S.; Bae, E.A.; Seo, H.; Ko, H.J.; Kim, B.S.; Chung, Y.; et al. GM-CSF Promotes Antitumor Immunity by Inducing Th9 Cell Responses. Cancer Immunol. Res. 2019, 7, 498–509. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Gupta, S.C.; Kim, J.H. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood 2012, 119, 651–665. [Google Scholar] [CrossRef] [Green Version]
- Galletti, G.; Scielzo, C.; Barbaglio, F.; Rodriguez, T.V.; Riba, M.; Lazarevic, D.; Cittaro, D.; Simonetti, G.; Ranghetti, P.; Scarfo, L.; et al. Targeting Macrophages Sensitizes Chronic Lymphocytic Leukemia to Apoptosis and Inhibits Disease Progression. Cell Rep. 2016, 14, 1748–1760. [Google Scholar] [CrossRef] [Green Version]
- Kratochvill, F.; Neale, G.; Haverkamp, J.M.; Van de Velde, L.A.; Smith, A.M.; Kawauchi, D.; McEvoy, J.; Roussel, M.F.; Dyer, M.A.; Qualls, J.E.; et al. TNF Counterbalances the Emergence of M2 Tumor Macrophages. Cell Rep. 2015, 12, 1902–1914. [Google Scholar] [CrossRef] [Green Version]
- Cook, A.D.; Lee, M.C.; Saleh, R.; Khiew, H.W.; Christensen, A.D.; Achuthan, A.; Fleetwood, A.J.; Lacey, D.C.; Smith, J.E.; Forster, I.; et al. TNF and granulocyte macrophage-colony stimulating factor interdependence mediates inflammation via CCL17. JCI Insight 2018, 3, e99249. [Google Scholar] [CrossRef] [Green Version]
- Adachi, K.; Kano, Y.; Nagai, T.; Okuyama, N.; Sakoda, Y.; Tamada, K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018, 36, 346–351. [Google Scholar] [CrossRef]
- Duan, D.; Wang, K.; Wei, C.; Feng, D.; Liu, Y.; He, Q.; Xu, X.; Wang, C.; Zhao, S.; Lv, L.; et al. The BCMA-Targeted Fourth-Generation CAR-T Cells Secreting IL-7 and CCL19 for Therapy of Refractory/Recurrent Multiple Myeloma. Front. Immunol. 2021, 12, 609421. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Jones, L.L.; Geiger, T.L. IL-6 Promotes T Cell Proliferation and Expansion under Inflammatory Conditions in Association with Low-Level RORγt Expression. J. Immunol. 2018, 201, 2934–2946. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, T.; Murata, K.; Akira, S.; Tonozuka, Y.; Minoshima, Y.; Feng, S.; Kumagai, H.; Tsuruga, H.; Ikeda, Y.; Asano, S.; et al. STAT5 induces macrophage differentiation of M1 leukemia cells through activation of IL-6 production mediated by NF-kappaB p65. J. Immunol. 2001, 167, 3652–3660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, T.; Yakoub, M.A.; Chandler, A.; Christ, A.B.; Yang, G.; Ouerfelli, O.; Rajasekhar, V.K.; Yoshida, A.; Kondo, H.; Hata, T.; et al. CSF1/CSF1R Signaling Inhibitor Pexidartinib (PLX3397) Reprograms Tumor-Associated Macrophages and Stimulates T-cell Infiltration in the Sarcoma Microenvironment. Mol. Cancer Ther. 2021, 20, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef] [Green Version]
- Maiti, A.; Rausch, C.R.; Cortes, J.E.; Pemmaraju, N.; Daver, N.G.; Ravandi, F.; Garcia-Manero, G.; Borthakur, G.; Naqvi, K.; Ohanian, M.; et al. Outcomes of relapsed or refractory acute myeloid leukemia after frontline hypomethylating agent and venetoclax regimens. Haematologica 2021, 106, 894–898. [Google Scholar] [CrossRef]
- Saliba, A.N.; John, A.J.; Kaufmann, S.H. Resistance to venetoclax and hypomethylating agents in acute myeloid leukemia. Cancer Drug Resist. 2021, 4, 125–142. [Google Scholar] [CrossRef]
- Yang, X.; Sexauer, A.; Levis, M. Bone marrow stroma-mediated resistance to FLT3 inhibitors in FLT3-ITD AML is mediated by persistent activation of extracellular regulated kinase. Br. J. Haematol. 2014, 164, 61–72. [Google Scholar] [CrossRef]
- Sletta, K.Y.; Castells, O.; Gjertsen, B.T. Colony Stimulating Factor 1 Receptor in Acute Myeloid Leukemia. Front. Oncol. 2021, 11, 654817. [Google Scholar] [CrossRef]
- Smith, C.C.; Levis, M.J.; Frankfurt, O.; Pagel, J.M.; Roboz, G.J.; Stone, R.M.; Wang, E.S.; Severson, P.L.; West, B.L.; Le, M.H.; et al. A phase 1/2 study of the oral FLT3 inhibitor pexidartinib in relapsed/refractory FLT3-ITD-mutant acute myeloid leukemia. Blood Adv. 2020, 4, 1711–1721. [Google Scholar] [CrossRef]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Zhang, Y.; Wang, J.; Gu, J. Defects in Macrophage Reprogramming in Cancer Therapy: The Negative Impact of PD-L1/PD-1. Front. Immunol. 2021, 12, 690869. [Google Scholar] [CrossRef]
- Ries, C.H.; Hoves, S.; Cannarile, M.A.; Ruttinger, D. CSF-1/CSF-1R targeting agents in clinical development for cancer therapy. Curr. Opin. Pharmacol. 2015, 23, 45–51. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smirnova, T.; Spertini, C.; Spertini, O. CSF1R Inhibition Combined with GM-CSF Reprograms Macrophages and Disrupts Protumoral Interplays with AML Cells. Cancers 2021, 13, 5289. https://doi.org/10.3390/cancers13215289
Smirnova T, Spertini C, Spertini O. CSF1R Inhibition Combined with GM-CSF Reprograms Macrophages and Disrupts Protumoral Interplays with AML Cells. Cancers. 2021; 13(21):5289. https://doi.org/10.3390/cancers13215289
Chicago/Turabian StyleSmirnova, Tatiana, Caroline Spertini, and Olivier Spertini. 2021. "CSF1R Inhibition Combined with GM-CSF Reprograms Macrophages and Disrupts Protumoral Interplays with AML Cells" Cancers 13, no. 21: 5289. https://doi.org/10.3390/cancers13215289