
Discovery of a Potent and Highly Isoform-Selective Inhibitor of the Neglected Ribosomal Protein S6 Kinase Beta 2 (S6K2)

 , ,
, ,  , , and
, , and
Abstract
:Simple Summary
Abstract
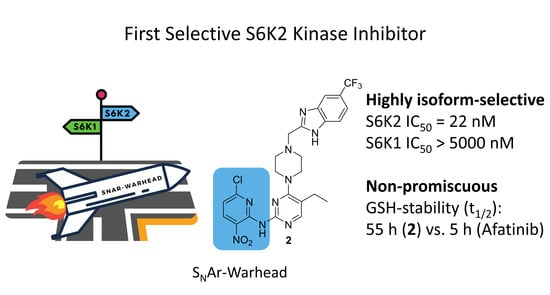
1. Introduction
2. Materials and Methods
2.1. Molecular Modeling
2.2. Chemistry
2.3. Glutathione (GSH) Stability Assay
2.4. Nα-Acetyl Lysine Stability Assay
2.5. Biochemical Assays
2.6. Microsomal Stability
2.7. Differential Scanning Fluorimetry (DSF) Assay
3. Results and Discussion
3.1. Design
3.2. Synthesis
3.3. Biochemical Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021. [Google Scholar] [CrossRef]
- Berginski, M.E.; Moret, N.; Liu, C.; Goldfarb, D.; Sorger, P.K.; Gomez, S.M. The Dark Kinase Knowledgebase: An online compendium of knowledge and experimental results of understudied kinases. Nucleic Acids Res. 2021, 49, D529–D535. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, O.; Müller, S.; Knapp, S. The (un)targeted cancer kinome. Nat. Chem. Biol. 2010, 6, 166–169. [Google Scholar] [CrossRef]
- Serafim, R.A.M.; Elkins, J.M.; Zuercher, W.J.; Laufer, S.A.; Gehringer, M. Chemical Probes for Understudied Kinases: Challenges and Opportunities. J. Med. Chem. 2021. [Google Scholar] [CrossRef]
- Edwards, A.M.; Isserlin, R.; Bader, G.D.; Frye, S.V.; Willson, T.M.; Yu, F.H. Too many roads not taken. Nature 2011, 470, 163–165. [Google Scholar] [CrossRef] [Green Version]
- Carter, A.J.; Kraemer, O.; Zwick, M.; Mueller-Fahrnow, A.; Arrowsmith, C.H.; Edwards, A.M. Target 2035: Probing the human proteome. Drug Discov. Today 2019, 24, 2111–2115. [Google Scholar] [CrossRef] [PubMed]
- Pardo, O.E.; Seckl, M.J. S6K2: The Neglected S6 Kinase Family Member. Front. Oncol. 2013, 3, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridharan, S.; Basu, A. Distinct Roles of mTOR Targets S6K1 and S6K2 in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, C.L.; Freitas, L.B.; Tamura, R.E.; Tavares, M.R.; Pavan, I.C.; Bajgelman, M.C.; Simabuco, F.M. S6Ks isoforms contribute to viability, migration, docetaxel resistance and tumor formation of prostate cancer cells. BMC Cancer 2016, 16, 602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Tenorio, G.; Karlsson, E.; Waltersson, M.A.; Olsson, B.; Holmlund, B.; Nordenskjöld, B.; Fornander, T.; Skoog, L.; Stål, O. Clinical potential of the mTOR targets S6K1 and S6K2 in breast cancer. Breast Cancer Res. Treat. 2011, 128, 713–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo, O.E.; Wellbrock, C.; Khanzada, U.K.; Aubert, M.; Arozarena, I.; Davidson, S.; Bowen, F.; Parker, P.J.; Filonenko, V.V.; Gout, I.T.; et al. FGF-2 protects small cell lung cancer cells from apoptosis through a complex involving PKCepsilon, B-Raf and S6K2. EMBO J. 2006, 25, 3078–3088. [Google Scholar] [CrossRef] [PubMed]
- Mizuarai, S.; Kawagishi, A.; Kotani, H. Inhibition of p70S6K2 down-regulates Hedgehog/GLI pathway in non-small cell lung cancer cell lines. Mol. Cancer 2009, 8, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, A.; Sridharan, S. Regulation of anti-apoptotic Bcl-2 family protein Mcl-1 by S6 kinase 2. PLoS ONE 2017, 12, e0173854. [Google Scholar] [CrossRef] [PubMed]
- Bouhaddou, M.; Memon, D.; Meyer, B.; White, K.M.; Rezelj, V.V.; Correa Marrero, M.; Polacco, B.J.; Melnyk, J.E.; Ulferts, S.; Kaake, R.M.; et al. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell 2020, 182, 685–712.e19. [Google Scholar] [CrossRef]
- Lee-Fruman, K.K.; Kuo, C.J.; Lippincott, J.; Terada, N.; Blenis, J. Characterization of S6K2, a novel kinase homologous to S6K1. Oncogene 1999, 18, 5108–5114. [Google Scholar] [CrossRef] [Green Version]
- Gout, I.; Minami, T.; Hara, K.; Tsujishita, Y.; Filonenko, V.; Waterfield, M.D.; Yonezawa, K. Molecular Cloning and Characterization of a Novel p70 S6 Kinase, p70 S6 Kinase β Containing a Proline-rich Region. J. Biol. Chem. 1998, 273, 30061–30064. [Google Scholar] [CrossRef] [Green Version]
- Chaikuad, A.; Koch, P.; Laufer, S.A.; Knapp, S. The Cysteinome of Protein Kinases as a Target in Drug Development. Angew. Chem. Int. Ed. 2018, 57, 4372–4385. [Google Scholar] [CrossRef]
- Fenton, T.R.; Gout, I.T. Functions and regulation of the 70 kDa ribosomal S6 kinases. Int. J. Biochem. Cell Biol. 2011, 43, 47–59. [Google Scholar] [CrossRef]
- Sever, N.İ.; Cengiz Şahin, S. S6K2 promises an important therapeutic potential for cancer. Future Oncol. 2018, 15, 95–102. [Google Scholar] [CrossRef]
- Qin, J.; Rajaratnam, R.; Feng, L.; Salami, J.; Barber-Rotenberg, J.S.; Domsic, J.; Reyes-Uribe, P.; Liu, H.; Dang, W.; Berger, S.L.; et al. Development of Organometallic S6K1 Inhibitors. J. Med. Chem. 2015, 58, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Pearce, L.R.; Alton, G.R.; Richter, D.T.; Kath, J.C.; Lingardo, L.; Chapman, J.; Hwang, C.; Alessi, D.R. Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1). Biochem. J. 2010, 431, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Ma, S. Research Progress of 70 kDa Ribosomal Protein S6 Kinase (P70S6K) Inhibitors as Effective Therapeutic Tools for Obesity, Type II Diabetes and Cancer. Curr. Med. Chem. 2020, 27, 4699–4719. [Google Scholar] [CrossRef] [PubMed]
- Gehringer, M.; Laufer, S.A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62, 5673–5724. [Google Scholar] [CrossRef] [PubMed]
- Motiwala, H.F.; Kuo, Y.-H.; Stinger, B.L.; Palfey, B.A.; Martin, B.R. Tunable Heteroaromatic Sulfones Enhance in-Cell Cysteine Profiling. J. Am. Chem. Soc. 2020, 142, 1801–1810. [Google Scholar] [CrossRef] [PubMed]
- Zambaldo, C.; Vinogradova, E.V.; Qi, X.; Iaconelli, J.; Suciu, R.M.; Koh, M.; Senkane, K.; Chadwick, S.R.; Sanchez, B.B.; Chen, J.S.; et al. 2-Sulfonylpyridines as Tunable, Cysteine-Reactive Electrophiles. J. Am. Chem. Soc. 2020, 142, 8972–8979. [Google Scholar] [CrossRef] [PubMed]
- Fairhurst, R.A.; Knoepfel, T.; Leblanc, C.; Buschmann, N.; Gaul, C.; Blank, J.; Galuba, I.; Trappe, J.; Zou, C.; Voshol, J.; et al. Approaches to selective fibroblast growth factor receptor 4 inhibition through targeting the ATP-pocket middle-hinge region. MedChemComm 2017, 8, 1604–1613. [Google Scholar] [CrossRef]
- Keeley, A.; Abranyi-Balogh, P.; Keserű, G.M. Design and characterization of a heterocyclic electrophilic fragment library for the discovery of cysteine-targeted covalent inhibitors. MedChemComm 2019, 10, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Dahal, U.P.; Gilbert, A.M.; Obach, R.S.; Flanagan, M.E.; Chen, J.M.; Garcia-Irizarry, C.; Starr, J.T.; Schuff, B.; Uccello, D.P.; Young, J.A. Intrinsic reactivity profile of electrophilic moieties to guide covalent drug design: N-α-acetyl-l-lysine as an amine nucleophile. MedChemComm 2016, 7, 864–872. [Google Scholar] [CrossRef]
- Fedorov, O.; Niesen, F.H.; Knapp, S. Kinase inhibitor selectivity profiling using differential scanning fluorimetry. Methods Mol. Biol. 2012, 795, 109–118. [Google Scholar] [CrossRef]
- Niwa, H.; Mikuni, J.; Sasaki, S.; Tomabechi, Y.; Honda, K.; Ikeda, M.; Ohsawa, N.; Wakiyama, M.; Handa, N.; Shirouzu, M.; et al. Crystal structures of the S6K1 kinase domain in complexes with inhibitors. J. Struct. Funct. Genomics 2014, 15, 153–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wydra, V.; Gerstenecker, S.; Schollmeyer, D.; Andreev, S.; Dimitrov, T.; Massarico Serafim, R.A.; Laufer, S.; Gehringer, M. N-(6-Chloro-3-nitropyridin-2-yl)-5-(1-methyl-1H-pyrazol-4-yl)isoquinolin-3-amine. Molbank 2021, 2021, M1181. [Google Scholar] [CrossRef]
- Dermatakis, A.; Kabat, M.M.; Luk, K.-C.; Rosmann, P.L.; So, S.-S. Pyrimido[4,5-D]pyrimidine Derviatives with Anticancer Activity. Patent WO2004041822A1, 21 May 2004. [Google Scholar]
- Anastassiadis, T.; Deacon, S.W.; Devarajan, K.; Ma, H.; Peterson, J.R. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1039–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terrier, F. Modern Nucleophilic Aromatic Substitution, 1st ed.; Wiley-VCH: Weinheim, Germany, 2013; pp. 1–84. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | S6K2 IC50 [nM] | S6K1 IC50 [nM] | MAPKAPK2 IC50 [nM] | MAPKAPK3 IC50 [nM] | FGFR4 IC50 [nM] | TTK IC50 [nM] |
|---|---|---|---|---|---|---|
| 2 | 22 ± 1.6 | >5000 | >5000 | >5000 | 216 | >5000 |
| 21 | >5000 | ND | ND | ND | ND | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerstenecker, S.; Haarer, L.; Schröder, M.; Kudolo, M.; Schwalm, M.P.; Wydra, V.; Serafim, R.A.M.; Chaikuad, A.; Knapp, S.; Laufer, S.; et al. Discovery of a Potent and Highly Isoform-Selective Inhibitor of the Neglected Ribosomal Protein S6 Kinase Beta 2 (S6K2). Cancers 2021, 13, 5133. https://doi.org/10.3390/cancers13205133
Gerstenecker S, Haarer L, Schröder M, Kudolo M, Schwalm MP, Wydra V, Serafim RAM, Chaikuad A, Knapp S, Laufer S, et al. Discovery of a Potent and Highly Isoform-Selective Inhibitor of the Neglected Ribosomal Protein S6 Kinase Beta 2 (S6K2). Cancers. 2021; 13(20):5133. https://doi.org/10.3390/cancers13205133
Chicago/Turabian StyleGerstenecker, Stefan, Lisa Haarer, Martin Schröder, Mark Kudolo, Martin P. Schwalm, Valentin Wydra, Ricardo A. M. Serafim, Apirat Chaikuad, Stefan Knapp, Stefan Laufer, and et al. 2021. "Discovery of a Potent and Highly Isoform-Selective Inhibitor of the Neglected Ribosomal Protein S6 Kinase Beta 2 (S6K2)" Cancers 13, no. 20: 5133. https://doi.org/10.3390/cancers13205133