Functional T Cell Reactivity to Melanocyte Antigens Is Lost during the Progression of Malignant Melanoma, but Is Restored by Immunization

 ,
,  , and
, and 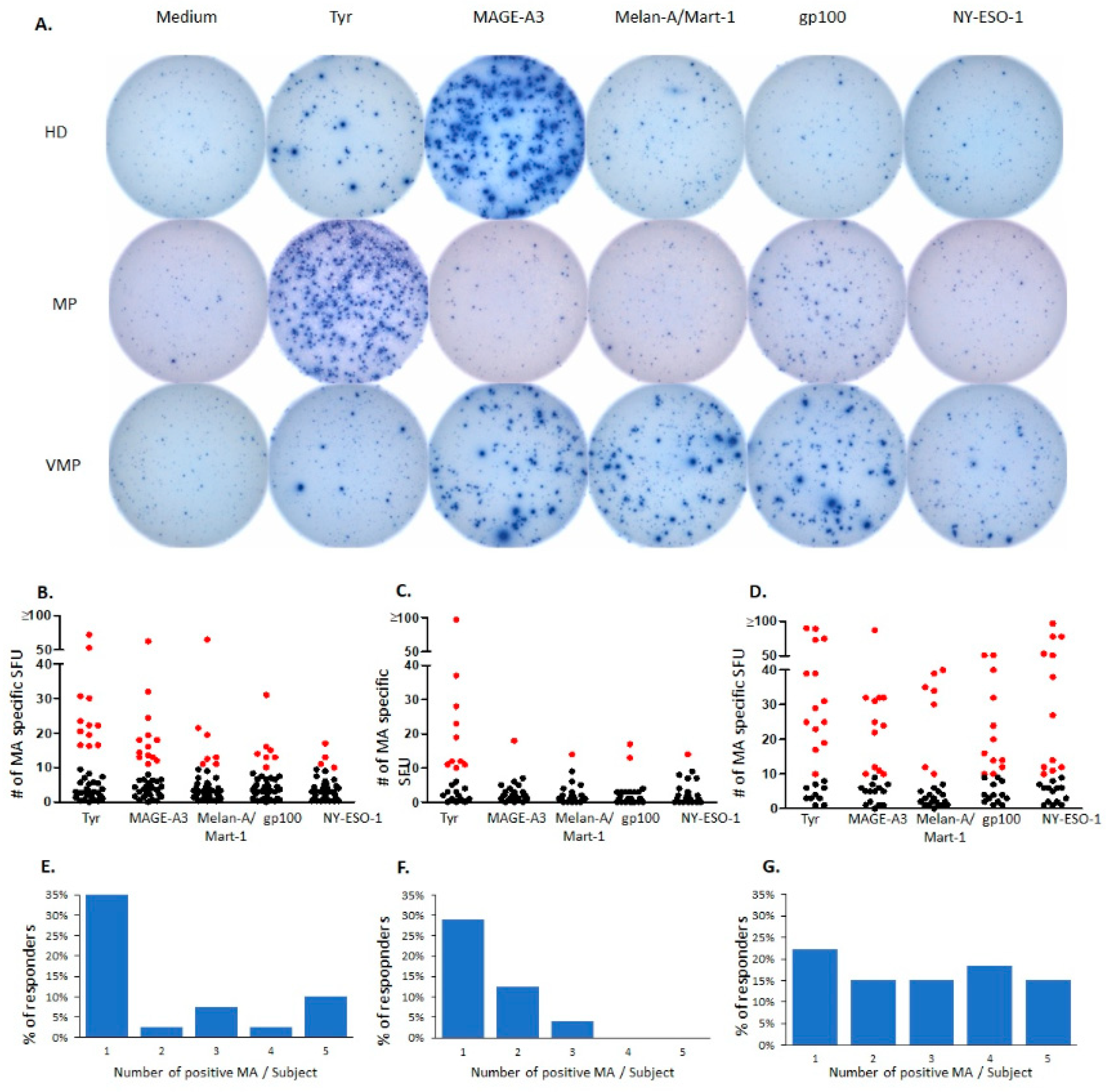{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Natural T Cell Reactivity to Melanocyte Antigens in Healthy Donors
2.2. Natural Melanocyte Antigen-Specific T Cell Immunity Is Deficient in Patients with Untreated Malignant Melanoma
2.3. Successful Vaccination Restores Natural T Cell Autoimmunity in Melanoma Patients
3. Discussion
4. Materials and Methods
4.1. Human Subjects
4.1.1. Healthy Controls
4.1.2. Untreated Melanoma Patients
4.1.3. Melanoma Patients Treated with AGI-101H Vaccine
4.2. Peripheral Blood Mononuclear Cell Preparation
4.3. Antigens
4.4. IFN-γ ImmunoSpot Assay
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Passarelli, A.; Mannavola, F.; Stucci, L.S.; Tucci, M.; Silvestris, F. Immune system and melanoma biology: A balance between immunosurveillance and immuneescape. Oncotarget 2017, 31, 106132–106142. [Google Scholar] [CrossRef]
- Ko, J.S. The immunology of melanoma. Clin. Lab. Med. 2017, 37, 449–471. [Google Scholar] [CrossRef]
- Lehmann, P.V.; Forsthuber, T.; Miller, A.; Sercarz, E.E. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature 1992, 358, 155–157. [Google Scholar] [CrossRef]
- Schlingmann, T.R.; Rininsland, F.H.; Bartholomae, W.C.; Kuekrek, H.; Lehmann, P.V.; Tary-Lehmann, M. Vaccination with tumor cells pulsed with foreign peptide induces immunity to the tumor itself. Clin. Immunol. 2009, 133, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Corbiere, V.; Chapiro, J.; Stroobant, V.; Ma, W.; Lurquin, C.; Lethe, B.; Van Baren, N.; Eynde, B.J.V.D.; Boon, T.; Coulie, P.G. Antigen spreading contributes to MAGE vaccination-induced regression of melanoma metastases. Cancer Res. 2011, 71, 1253–1262. [Google Scholar] [CrossRef] [Green Version]
- Kreiter, S.; Vormehr, M.; van de Roemer, N.; Diken, M.; Lower, M.; Diekmann, J.; Boegel, S.; Schrörs, B.; Vascotto, F.; Castle, J.C.; et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015, 520, 692–696. [Google Scholar] [CrossRef] [Green Version]
- Ott, P.A.; Hu-Lieskovan, S.; Chmielowski, B.; Govindan, R.; Naing, A.; Bhardwaj, N.; Margolin, K.; Awad, M.M.; Hellmann, M.D.; Lin, J.J.; et al. A Phase Ib trial of personalized neoantigen therapy plus anti-PD-1 in patients with advanced melanoma, non-small cell Lung cancer, or bladder cancer. Cell 2020, 183, 347–362.e24. [Google Scholar] [CrossRef]
- Boon, T.C.J.; Van den Eynde, B.; van der Bruggen, P.; Van Pel, A. Tumor antigens recognized by T lymphocytes. Annu. Rev. Immunol. 1994, 12, 337–365. [Google Scholar] [CrossRef]
- Lee, P.P.; Yee, C.; Savage, P.A.; Fong, L.; Brockstedt, D.; Weber, J.S.; Johnson, D.; Swetter, S.; Thompson, J.; Greenberg, P.D.; et al. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat. Med. 1999, 5, 677–685. [Google Scholar] [CrossRef]
- Van Oijen, M.; Bins, A.; Elias, S.; Sein, J.; Weder, P.; de Gast, G.; Mallo, H.; Gallee, M.; Van Tinterten, H.; Schumacher, T.; et al. On the role of melanoma-specific CD8+ T cell immunity in disease progression of advanced-stage melanoma patients. Clin. Cancer Res. 2004, 10, 4754–4760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Van Der Leun, A.M.; Yofe, I.; Lubling, Y.; Gelbard-Solodkin, D.; Van Akkooi, A.C.; Braber, M.V.D.; Rozeman, E.A.; Haanen, J.B.; Blank, C.U.; et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell 2019, 176, 775–789.e18. [Google Scholar] [CrossRef] [PubMed]
- Kurachi, M. CD8+ T cell exhaustion. Semin. Immunopathol. 2019, 41, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Pauken, K.E.; Wherry, E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Rubio-Godoy, V.; Dutoit, V.; Rimoldi, D.; Lienard, D.; Lejeune, F.; Speiser, D.E.; Guillaume, P.; Cerottini, J.-C.; Romero, P.; Valmori, D. Discrepancy between ELISPOT IFN-gamma secretion and binding of A2/peptide multimers to TCR reveals interclonal dissociation of CTL effector function from TCR-peptide/MHC complexes half-life. Proc. Natl. Acad. Sci. USA 2001, 98, 10302–10307. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, M.; Kamphorst, A.O.; Im, S.J.; Kissick, H.T.; Pillai, R.N.; Ramalingam, S.S.; Araki, K.; Ahmed, R. CD8 T cell exhaustion in chronic infection and cancer: Opportunities for interventions. Annu. Rev. Med. 2018, 69, 301–318. [Google Scholar] [CrossRef]
- Lehmann, P.V.; Suwansaard, M.; Zhang, T.; Roen, D.R.; Kirchenbaum, G.A.; Karulin, A.Y.; Lehmann, A.; Reche, P.A. Comprehensive evaluation of the expressed CD8+ T cell epitope space using high-throughput epitope mapping. Front. Immunol. 2019, 10, 655. [Google Scholar] [CrossRef]
- Palermo, B.; Campanelli, R.; Montovani, S.; Lantelme, E.; Manganoni, A.M.; Carella, G.; Da Prada, G.A.; della Cuna, G.R.; Romagne, F.; Gauthier, L.; et al. Diverse expansion potential and heterogeneous avidity in tumor-associated antigen-specific T lymphocytes from primary melanoma patients. Eur. J. Immunol. 2001, 31, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Przybyla, A.; Zhang, T.; Li, R.; Roen, D.R.; Mackiewicz, A.; Lehmann, P.V. Natural T cell autoreactivity to melanoma antigens: Clonally expanded melanoma-antigen specific CD8+ memory T cells can be detected in healthy humans. Cancer Immunol. Immunother. 2019, 68, 709–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobo, P.I. Role of natural autoantibodies and natural IgM anti-leucocyte autoantibodies in health and disease. Front. Immunol. 2016, 7, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, A.A.; Lehmann, P.V. Aleatory epitope recognition prevails in human T cell responses? Crit. Rev. Immunol. 2020, 40, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Viola, A.; Lanzavecchia, A. T cell activation determined by T cell receptor number and tunable tresholds. Science 1996, 5, 104–108. [Google Scholar] [CrossRef]
- Matzinger, P. The danger model: A renewed sense of self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef] [Green Version]
- Karulin, A.Y.; Lehmann, P.V. How ELISPOT morphology reflects on the productivity and kinetics of cells’ secretory activity. Methods Mol. Biol. 2012, 792, 125–143. [Google Scholar] [CrossRef]
- Currier, J.R.; Kuta, E.G.; Turk, E.; Earhart, L.B.; Loomis-Price, L.; Janetzki, S.; Ferrari, G.; Birx, D.L.; Cox, J.H. A panel of MHC class I restricted viral peptides for use as a quality control for vaccine trial ELISPOT assays. J. Immunol. Methods 2002, 260, 157–172. [Google Scholar] [CrossRef]
- Mackiewicz, A.; Wysocki, P.J.; Suchorska, W. Vaccine Composition. U.S. Patent US2011002898, 6 January 2011. [Google Scholar]
- Mackiewicz, A.; Mackiewicz, J.; Wysocki, P.J. Long-term survival of high-risk melanoma patients immunized with a Hyper-IL-6-modified allogeneic whole-cell vaccine after complete resection. Expert Opin. Investig. Drugs 2012, 21, 773–783. [Google Scholar] [CrossRef]
- Mackiewicz, J.; Karczewska-Dzionk, A.; Laciak, M.; Kapcinska, M.; Wiznerowicz, M.; Burzykowski, T.; Zakowska, M.; Rose-John, S.; Mackiewicz, A. Whole cell therapeutic vaccine modified with hyper-IL6 for combinational treatment of nonresected advanced melanoma. Medicine 2015, 94, e853. [Google Scholar] [CrossRef]
- Mackiewicz, J.; Burzykowski, T.; Izycki, D.; Mackiewicz, A. Re-induction using whole cell melanoma vaccine genetically modified to melanoma stem cells-like beyond recurrence extends long term survival of high risk resected patients—Updated results. J. Immunother. Cancer 2018, 6, 134. [Google Scholar] [CrossRef]
- Datta, J.; Rosemblit, C.; Berk, E.; Showalter, L.; Namjoshi, P.; Mick, R.; Lee, K.P.; Brod, A.M.; Yang, R.L.; Kelz, R.R.; et al. Progressive loss of anti-HER2 CD4+ T-helper type 1 response in breast tumorigenesis and the potential for immune restoration. Oncoimmunology 2015, 4, e1022301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohashi, P.S.; DeFranco, A.L. Making and breaking tolerance. Curr. Opin. Immunol. 2002, 14, 744–759. [Google Scholar] [CrossRef]
- Targoni, O.S.; Lehmann, P.V. Endogenous myelin basic protein inactivates the high avidity T cell repertoire. J. Exp. Med. 1998, 187, 2055–2063. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Fröhlich, A.; Kisielow, J.; Schmitz, I.; Freigang, S.; Shamshiev, A.T.; Weber, J.; Marsland, B.J.; Oxenius, A.; Kopf, M. IL-21R on T cells is critical for sustained functionality and control of chronic viral infection. Science 2009, 324, 1576–1580. [Google Scholar] [CrossRef] [Green Version]
- Elsaesser, H.; Sauer, K.; Brooks, D.G. IL-21 is required to control chronic viral infection. Science 2009, 324, 1569–1572. [Google Scholar] [CrossRef] [Green Version]
- Yi, J.S.; Du, M.; Zajac, A.J. A vital role for interleukin-21 in the control of a chronic viral infection. Science 2009, 324, 1572–1576. [Google Scholar] [CrossRef]
- Jiang, T.; Zhou, C.; Ren, S. Role of IL-2 in cancer immunotherapy. Oncoimmunology 2016, 5, e1163462. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, H.; Laux, J.; Moldovan, I.; Caspell, R.; Lehmann, P.V.; Subbramanian, R.A. Optimal thawing of cryopreserved peripheral blood mononuclear cells for use in high-throughput human immune monitoring studies. Cells 2012, 1, 313–324. [Google Scholar] [CrossRef]
- Zhang, W.; Lehmann, P.V. Objective, user-independent ELISPOT data analysis based on scientifically validated principles. Methods Mol. Biol. 2012, 792, 155–171. [Google Scholar] [CrossRef]
- Karulin, A.Y.; Caspell, R.; Dittrich, M.; Lehmann, P.V. Normal Distribution of CD8+ T-Cell-Derived ELISPOT Counts within replicates justifies the reliance on parametric statistics for identifying positive responses. Cells 2015, 4, 96–111. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Przybyla, A.; Lehmann, A.A.; Zhang, T.; Mackiewicz, J.; Galus, Ł.; Kirchenbaum, G.A.; Mackiewicz, A.; Lehmann, P.V. Functional T Cell Reactivity to Melanocyte Antigens Is Lost during the Progression of Malignant Melanoma, but Is Restored by Immunization. Cancers 2021, 13, 223. https://doi.org/10.3390/cancers13020223
Przybyla A, Lehmann AA, Zhang T, Mackiewicz J, Galus Ł, Kirchenbaum GA, Mackiewicz A, Lehmann PV. Functional T Cell Reactivity to Melanocyte Antigens Is Lost during the Progression of Malignant Melanoma, but Is Restored by Immunization. Cancers. 2021; 13(2):223. https://doi.org/10.3390/cancers13020223
Chicago/Turabian StylePrzybyla, Anna, Alexander A. Lehmann, Ting Zhang, Jacek Mackiewicz, Łukasz Galus, Greg A. Kirchenbaum, Andrzej Mackiewicz, and Paul V. Lehmann. 2021. "Functional T Cell Reactivity to Melanocyte Antigens Is Lost during the Progression of Malignant Melanoma, but Is Restored by Immunization" Cancers 13, no. 2: 223. https://doi.org/10.3390/cancers13020223