
Immune Therapies for Myelodysplastic Syndromes and Acute Myeloid Leukemia


Abstract
:Simple Summary
Abstract
1. Introduction
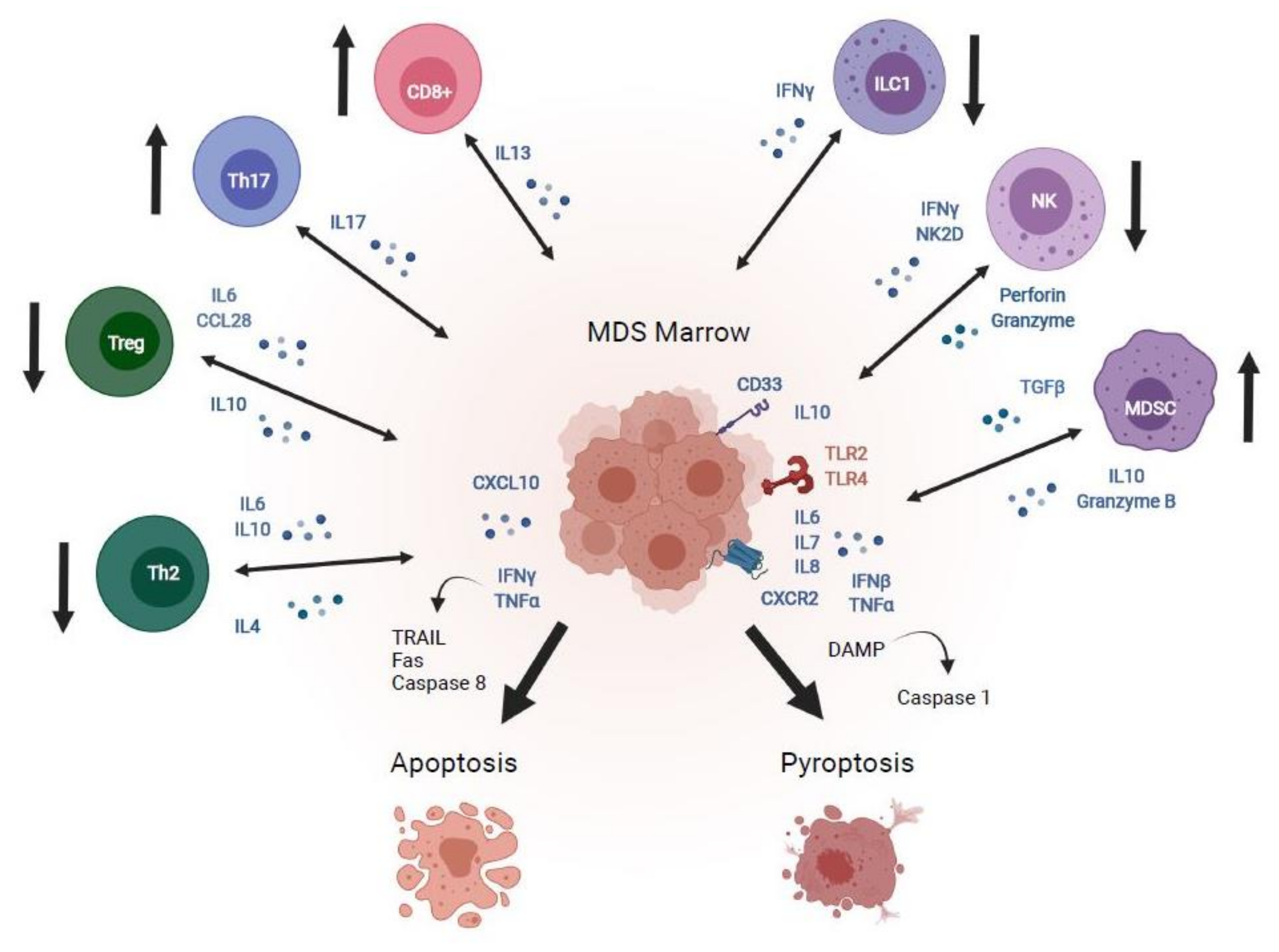
2. Immune Dysregulation in MDS and AML

3. Immunosuppression Trials for MDS
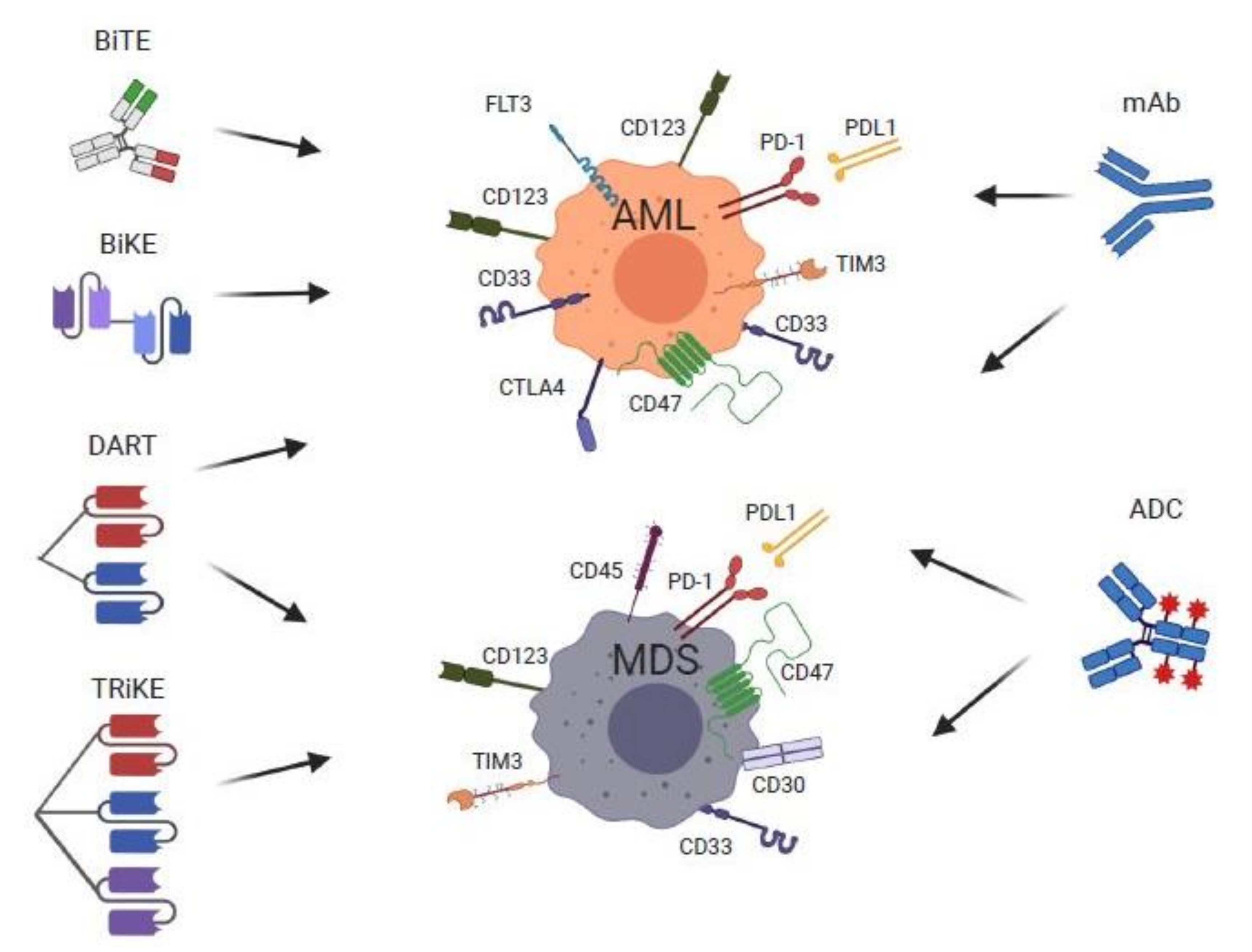
4. Monoclonal Antibody Therapy for MDS and AML
4.1. Anti CD-47 (Macrophage Checkpoint Inhibitor) Therapies

4.2. Immune Checkpoint Inhibitor Therapies
4.2.1. Anti-CTLA-4 Therapies
4.2.2. Anti-PD-1 and Anti-TIM-3 Therapies
4.3. Antibody-Dependent Cellular Cytotoxicity (Anti-CD33 and CD123 Fc-Optimized Antibodies)
5. Bispecific T-Cell Engagers, Dual-Affinity Re-Targeting Molecules, Bi- and Tri-SPECIFIC Killer Cell Engager Therapies for MDS and AML

{kind=link}
{kind=link}
{kind=link}
| Study | Trial Identification | Mechanism | Study Population | Study Status | Results | MRD Status | Adverse Effects | Ref |
|---|---|---|---|---|---|---|---|---|
| Bispecific T-Cell Engagers (BiTE) | ||||||||
| Phase 1 dose escalation (AMG 330) | NCT02520427 | Bispecific mAb w/ specificity to CD33 and CD3ε | R/R AML | Recruiting | CR/CRi: 16% | N/A | CRS: 67% | [92] |
| Phase 1b dose escalation, AMG 330+ pembrolizumab | NCT04478695 | Bispecific mAb w/ specificity to CD33 and CD3ε + PD-L inhibitor | R/R AML | Terminated | N/A | N/A | N/A | N/A |
| Phase 1 dose escalation (AMG 427) | NCT03541369 | Bispecific mAb w/ specificity to CD3ε and FLT3 scFv | R/R AML | Active, Recruiting | N/A | N/A | N/A | [95] |
| Phase 1 dose escalation (AMV 564) | NCT03144245 | Bispecific mAb w/ specificity to CD33 and CD3ε- tetravalent | R/R AML | Active, Not Recruiting | Reductions in BM blasts ranging from 13 to 38% in 6 of 9 evaluable patients | N/A | Febrile neutropenia in 25% | [90] |
| Phase 1 dose escalation Vibecotamab (XmAb14045) | NCT02730312 | Bispecific mAb w/ specificity to CD123/CD3ε | R/R AML | Active, Recruiting | ORR: CR/CRi/MLFS: 14% SD:71% Low disease burden: RR 26% | N/A | CRS: 58% | [96] |
| Phase 1 dose escalation (AMG 673) | NCT03224819 | Half-life extended bispecific T-cell engager—HLE BiTE to CD33 and CD3 | R/R AMl | Active, Not Recruiting | ORR 44% decrease in BM blasts, 22% with >50% reduction and 3% with >85% reduction in BM blasts | N/A | CRS: 50%, | [93] |
| Dual Affinity Re-Targeting Molecules DART (BiTE) | ||||||||
| Phase 1/2 Flotetuzumab (MGD006) | NCT02152956 | Bispecific mAb w/ specificity to CD123/CD3ε | PIF or R/R AML or Intermediate-2/High Risk MDS | Active, Recruiting | CR/CRh: 24% Median OS (in patients that achieved CR/CRh): 10.2 m | NA | IRR/CRS in PIF/ER population (n = 30): 100% (all grades), Gr > 3: 3.3% | [97] |
| Tri-Specific Killer Engager (TriKE) | ||||||||
| Phase 1—GTB-3550 TriKE | NCT03214666 | Tri-Specific Killer Engager-CD16/IL-15/CD33 | PIF/ R/R AML, High risk MDS | Active, recruiting | 4 pts enrolled, 3 completed, 2 w/SD, 1 w/POD. | NA | No toxicity reported. | [98] |
6. Antibody Drug Conjugate Therapies for MDS and AML
6.1. Anti-CD33 ADC Therapies
6.1.1. Gemtuzumab Ozogamicin
6.1.2. Other CD33-Directed ADCs: Vadastuximab Talirine, IMGN779 and AVE9633
6.2. CD123-Directed ADCs: IMGN632, SGN-CD123A, SL-101, and SL-401
6.3. Anti-CD45 and -CD30 Directed ADCs for MDS and AML
7. Cellular and Vaccine Therapies for MDS and AML
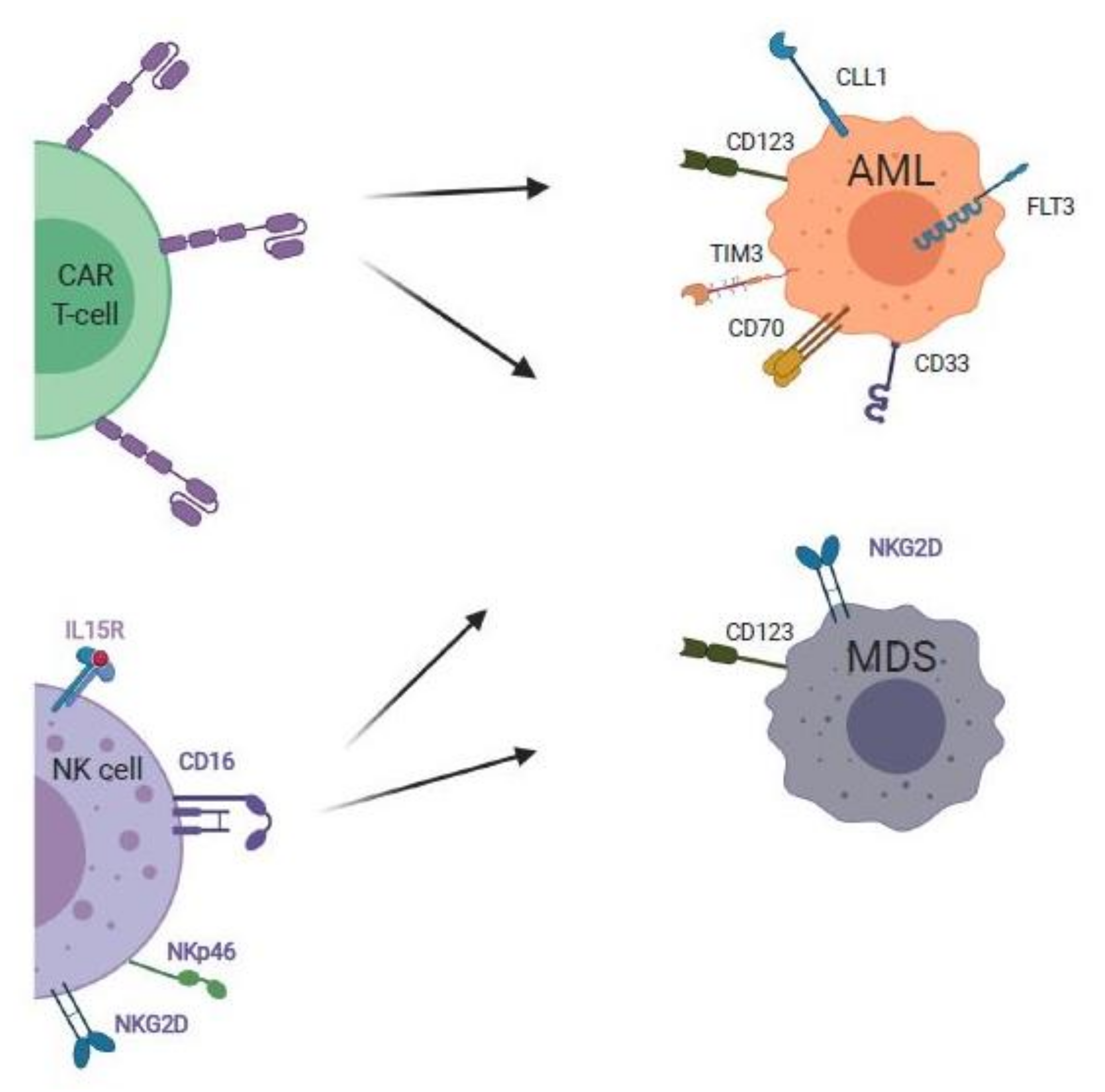
7.1. CAR T-Cell Therapies for MDS and AML
7.2. NK Cell Therapies for AML and MDS
7.3. Vaccine Therapies for AML and MDS
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised International prognostic scoring system for myelodysplastic syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Dinardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable remissions with ivosidenib inIDH1-mutated relapsed or refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Stein, E.M.; Dinardo, C.D.; Fathi, A.T.; Pollyea, D.A.; Stone, R.M.; Altman, J.K.; Roboz, G.J.; Patel, M.R.; Collins, R.; Flinn, I.W.; et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood 2019, 133, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Glenthøj, A.; Ørskov, A.D.; Hansen, J.W.; Hadrup, S.R.; O’Connell, C.; Grønbæk, K. Immune mechanisms in myelodysplastic syndrome. Int. J. Mol. Sci. 2016, 17, 944. [Google Scholar] [CrossRef] [Green Version]
- Fozza, C.; Crobu, V.; Isoni, M.A.; Dore, F. The immune landscape of myelodysplastic syndromes. Crit. Rev. Oncol. 2016, 107, 90–99. [Google Scholar] [CrossRef]
- Ivy, K.S.; Brent Ferrell, P., Jr. Disordered immune regulation and its therapeutic targeting in myelodysplastic syndromes. Curr. Hematol. Malig. Rep. 2018, 13, 244–255. [Google Scholar] [CrossRef]
- Wang, C.; Yang, Y.; Gao, S.; Chen, J.; Yu, J.; Zhang, H.; Li, M.; Zhan, X.; Li, W. Immune dysregulation in myelodysplastic syndrome: Clinical features, pathogenesis and therapeutic strategies. Crit. Rev. Oncol. Hematol. 2018, 122, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Barreyro, L.; Chlon, T.; Starczynowski, D.T. Chronic immune response dysregulation in MDS pathogenesis. Blood 2018, 132, 1553–1560. [Google Scholar] [CrossRef] [Green Version]
- Sallman, D.A.; List, A. The central role of inflammatory signaling in the pathogenesis of myelodysplastic syndromes. Blood 2019, 133, 1039–1048. [Google Scholar] [CrossRef] [Green Version]
- Mufti, G.J.; Figes, A.; Hamblin, T.J.; Oscier, D.G.; Copplestoni, J.A. Immunological abnormalities in myelodysplastic syndromes I. Serum immunoglobulins and autoantibodies. Br. J. Haematol. 1986, 63, 143–147. [Google Scholar] [CrossRef]
- Hamblin, T.J. Immunological abnormalities in myelodysplastic syndromes. Semin. Hematol. 1996, 33, 150–162. [Google Scholar]
- Komrokji, R.S.; Kulasekararaj, A.; Al Ali, N.H.; Kordasti, S.Y.; Bart-Smith, E.; Craig, B.M.; Padron, E.; Zhang, L.; Lancet, J.E.; Pinilla-Ibarz, J.; et al. Autoimmune diseases and myelodysplastic syndromes. Am. J. Hematol. 2016, 91, E280–E283. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Park, J.K.; Lee, E.Y.; Joo, S.H.; Jung, K.C.; Lee, E.B.; Song, Y.W.; Yoon, S.S. Certain autoimmune manifestations are associated with distinctive karyotypes and outcomes in patients with myelodysplastic syndrome: A retrospective cohort study. Medicine 2016, 95, e3091. [Google Scholar] [CrossRef]
- Kristinsson, S.Y.; Björkholm, M.; Hultcrantz, M.; Derolf, Å.R.; Landgren, O.; Goldin, L.R. Chronic immune stimulation might act as a trigger for the development of acute myeloid leukemia or myelodysplastic syndromes. J. Clin. Oncol. 2011, 29, 2897–2903. [Google Scholar] [CrossRef] [Green Version]
- Beck, D.B.; Ferrada, M.A.; Sikora, K.A.; Ombrello, A.K.; Collins, J.C.; Pei, W.; Balanda, N.; Ross, D.L.; Cardona, D.O.; Wu, Z.; et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N. Engl. J. Med. 2020, 383, 2628–2638. [Google Scholar] [CrossRef]
- Grayson, P.C.; A Patel, B.; Young, N.S. VEXAS syndrome. Blood 2021, 137, 3591–3594. [Google Scholar]
- Bourbon, E.; Heiblig, M.; Valentin, M.; Barba, T.; Durel, C.A.; Lega, J.C.; Barraco, F.; Sève, P.; Jamilloux, Y.; Sujobert, P. Therapeutic options in VEXAS syndrome: Insights from a retrospective series. Blood 2021, 137, 3682–3684. [Google Scholar] [CrossRef]
- Verhoef, G.E.; De Schouwer, P.; Ceuppens, J.L.; Van Damme, J.; Goossens, W.; A Boogaerts, M.A. Measurement of serum cytokine levels in patients with myelodysplastic syndromes. Leukemia 1992, 6, 1268–1272. [Google Scholar]
- Kitagawa, M.; Saito, I.; Kuwata, T.; Yoshida, S.; Yamaguchi, S.; Takahashi, M.; Tanizawa, T.; Kamiyama, R.; Hirokawa, K. Overexpression of tumor necrosis factor (TNF)-alpha and interferon (IFN)-gamma by bone marrow cells from patients with myelodysplastic syndromes. Leukemia 1997, 11, 2049–2054. [Google Scholar] [CrossRef] [Green Version]
- Kordasti, S.Y.; Afzali, B.; Lim, Z.; Ingram, W.; Hayden, J.; Barber, L.; Matthews, K.; Chelliah, R.; Guinn, B.; Lombardi, G.; et al. IL-17-producing CD4+T cells, pro-inflammatory cytokines and apoptosis are increased in low risk myelodysplastic syndrome. Br. J. Haematol. 2009, 145, 64–72. [Google Scholar] [CrossRef]
- Stifter, G.; Heiss, S.; Gastl, G.; Tzankov, A.; Stauder, R. Over-expression of tumor necrosis factor-alpha in bone marrow biopsies from patients with myelodysplastic syndromes: Relationship to anemia and prognosis. Eur. J. Haematol. 2005, 75, 485–491. [Google Scholar] [CrossRef]
- Pardanani, A.; Finke, C.; Lasho, T.L.; Al-Kali, A.; Begna, K.H.; Hanson, C.A.; Tefferi, A. IPSS-independent prognostic value of plasma CXCL10, IL-7 and IL-6 levels in myelodysplastic syndromes. Leukemia 2012, 26, 693–699. [Google Scholar] [CrossRef]
- Gersuk, G.M.; Beckham, C.; Loken, M.R.; Kiener, P.; Anderson, J.E.; Farrand, A.; Troutt, A.B.; Ledbetter, J.A.; Deeg, J. A role for tumour necrosis factor-α, Fas and Fas-Ligand in marrow failure associated with myelodysplastic syndrome. Br. J. Haematol. 1998, 103, 176–188. [Google Scholar] [CrossRef]
- Zeng, W.; Miyazato, A.; Chen, G.; Kajigaya, S.; Young, N.S.; Maciejewski, J.P. Interferon-gamma-induced gene expression in CD34 cells: Identification of pathologic cytokine-specific signature profiles. Blood 2006, 107, 167–175. [Google Scholar] [CrossRef] [Green Version]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Shastri, A.; Will, B.; Steidl, U.; Verma, A. Stem and progenitor cell alterations in myelodysplastic syndromes. Blood 2017, 129, 1586–1594. [Google Scholar] [CrossRef]
- Maratheftis, C.I.; Andreakos, E.; Moutsopoulos, H.M.; Voulgarelis, M. Toll-like receptor-4 is up-regulated in hematopoietic progenitor cells and contributes to increased apoptosis in myelodysplastic syndromes. Clin. Cancer Res. 2007, 13, 1154–1160. [Google Scholar] [CrossRef] [Green Version]
- Pellagatti, A.; Cazzola, M.; Giagounidis, A.; Perry, J.; Malcovati, L.; Della Porta, M.G.; Jädersten, M.; Killick, S.; Verma, A.; Norbury, C.J.; et al. Deregulated gene expression pathways in myelodysplastic syndrome hematopoietic stem cells. Leukemia 2010, 24, 756–764. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Dimicoli, S.; Bueso-Ramos, C.; Chen, R.; Yang, H.; Neuberg, D.; Pierce, S.; Jia, Y.; Zheng, H.; Wang, H.; et al. Toll-like receptor alterations in myelodysplastic syndrome. Leukemia 2013, 27, 1832–1840. [Google Scholar] [CrossRef] [Green Version]
- Starczynowski, D.T.; Kuchenbauer, F.; Argiropoulos, B.; Sung, S.; Morin, R.; Muranyi, A.; Hirst, M.; Hogge, D.; Marra, M.; A Wells, R.; et al. Identification of miR-145 and miR-146a as mediators of the 5q– syndrome phenotype. Nat. Med. 2010, 16, 49–58. [Google Scholar] [CrossRef]
- Rhyasen, G.W.; Bolanos, L.; Fang, J.; Jerez, A.; Wunderlich, M.; Rigolino, C.; Mathews, L.; Ferrer, M.; Southall, N.; Guha, R.; et al. Targeting IRAK1 as a therapeutic approach for myelodysplastic syndrome. Cancer Cell 2013, 24, 90–104. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Eksioglu, E.A.; Zhou, J.; Zhang, L.; Djeu, J.; Fortenbery, N.; Epling-Burnette, P.; Van Bijnen, S.; Dolstra, H.; Cannon, J.; et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J. Clin. Investig. 2013, 123, 4595–4611. [Google Scholar] [CrossRef]
- Giudice, V.; Wu, Z.; Kajigaya, S.; Fernandez Ibanez, M.D.P.; Rios, O.; Cheung, F.; Ito, S.; Young, N.S. Circulating S100A8 and S100A9 protein levels in plasma of patients with acquired aplastic anemia and myelodysplastic syndromes. Cytokine 2019, 113, 462–465. [Google Scholar] [CrossRef]
- Basiorka, A.A.; McGraw, K.L.; Eksioglu, E.A.; Chen, X.; Johnson, J.; Zhang, L.; Zhang, Q.; Irvine, B.A.; Cluzeau, T.; Sallman, D.A.; et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 2016, 128, 2960–2975. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Q.; Ding, Y.; Liu, Y.; Zhao, D.; Zhao, K.; Shen, Q.; Liu, X.; Zhu, X.; Li, N.; et al. Methyltransferase Dnmt3a upregulates HDAC9 to deacetylate the kinase TBK1 for activation of antiviral innate immunity. Nat. Immunol. 2016, 17, 806–815. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Cabrero, M.; Wei, Y.; Yang, H.; Ganan-Gomez, I.; Bohannan, Z.; Colla, S.; Marchesini, M.; Montalban Bravo, G.; Takahashi, K.; Bueso-Ramos, C.; et al. Down-regulation of EZH2 expression in myelodys-plastic syndromes. Leuk. Res. 2016, 44, 1–7. [Google Scholar] [CrossRef]
- Wei, Y.; Chen, R.; Dimicoli, S.; Bueso-Ramos, C.; Neuberg, D.; Pierce, S.; Wang, H.; Yang, H.; Jia, Y.; Zheng, H.; et al. Global H3K4me3 genome mapping reveals alterations of innate immunity signaling and overexpression of JMJD3 in human myelodysplastic syndrome CD34+ cells. Leukemia 2013, 27, 2177–2186. [Google Scholar] [CrossRef] [Green Version]
- Chiossone, L.; Dumas, P.Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688, Erratum in 2018, 18, 726. [Google Scholar] [CrossRef]
- Lordo, M.; Scoville, S.; Goel, A.; Yu, J.; Freud, A.; Caligiuri, M.; Mundy-Bosse, B. Unraveling the role of innate lymphoid cells in acute myeloid leukemia. Cancers 2021, 13, 320. [Google Scholar] [CrossRef]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate lymphoid cells: 10 years on. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epling-Burnette, P.K.; Bai, F.; Painter, J.S.; Rollison, D.E.; Salih, H.R.; Krusch, M.; Zou, J.; Ku, E.; Zhong, B.; Boulware, D.; et al. Reduced natural killer (NK) function associated with high-risk myelodysplastic syndrome (MDS) and reduced expression of activating NK receptors. Blood 2007, 109, 4816–4824. [Google Scholar] [CrossRef] [PubMed]
- Hejazi, M.; Manser, A.R.; Fröbel, J.; Kündgen, A.; Zhao, X.; Schönberg, K.; Germing, U.; Haas, R.; Gattermann, N.; Uhrberg, M. Impaired cytotoxicity associated with defective natural killer cell differentiation in myelodysplastic syndromes. Haematologica 2015, 100, 643–652. [Google Scholar] [CrossRef] [Green Version]
- Costello, R.T.; Sivori, S.; Marcenaro, E.; Lafage-Pochitaloff, M.; Mozziconacci, M.-J.; Reviron, D.; Gastaut, J.-A.; Pende, D.; Olive, D.; Moretta, A. Defective expression and function of natural killer cell–triggering receptors in patients with acute myeloid leukemia. Blood 2002, 99, 3661–3667. [Google Scholar] [CrossRef]
- Chretien, A.S.; Fauriat, C.; Orlanducci, F.; Galseran, C.; Rey, J.; Bouvier Borg, G.; Gautherot, E.; Granjeaud, S.; Hamel-Broza, J.-F.; Demerle, C.; et al. Natural killer defective maturation is associated with adverse clinical outcome in patients with acute myeloid leukemia. Front. Immunol. 2017, 8, 573. [Google Scholar] [CrossRef] [PubMed]
- Sendker, S.; Reinhardt, D.; Niktoreh, N. Redirecting the immune microenvironment in acute myeloid leukemia. Cancers 2021, 13, 1423. [Google Scholar] [CrossRef] [PubMed]
- Barrett, A.J. Acute myeloid leukaemia and the immune system: Implications for immunotherapy. Br. J. Haematol. 2020, 188, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Sloand, E.M.; Mainwaring, L.; Fuhrer, M.; Ramkissoon, S.; Risitano, A.M.; Keyvanafar, K.; Lu, J.; Basu, A.; Barrett, A.J.; Young, N.S. Preferential suppression of trisomy 8 compared with normal hematopoietic cell growth by autologous lymphocytes in patients with trisomy 8 myelodysplastic syndrome. Blood 2005, 106, 841–851. [Google Scholar] [CrossRef]
- Fozza, C.; Contini, S.; Galleu, A.; Simula, M.P.; Virdis, P.; Bonfigli, S.; Longinotti, M. Patients with myelodysplastic syndromes display several T-cell expansions, which are mostly polyclonal in the CD4+ subset and oligoclonal in the CD8+ subset. Exp. Hematol. 2009, 37, 947–955. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, X.; Guo, J.; Xu, F.; He, Q.; Zhao, Y.; Yang, Y.; Gu, S.; Zhang, Y.; Wu, L.; et al. Interleukin-17 enhances the production of interferon-γ and tumour necrosis factor-α by bone marrow T lymphocytes from patients with lower risk myelodysplastic syndromes. Eur. J. Haematol. 2013, 90, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Wlodarski, M.W.; Gondek, L.P.; Nearman, Z.P.; Plasilova, M.; Kalaycio, M.; Hsi, E.D.; Maciejewski, J.P. Molecular strategies for detection and quantitation of clonal cytotoxic T-cell responses in aplastic anemia and myelodysplastic syndrome. Blood 2006, 108, 2632–2641. [Google Scholar] [CrossRef]
- Kotsianidis, I.; Bouchliou, I.; Nakou, E.; Spanoudakis, E.; Margaritis, D.; Christophoridou, A.V.; Anastasiades, A.; Tsigalou, C.; Bourikas, G.; Karadimitris, A.; et al. Kinetics, function and bone marrow trafficking of CD4+CD25+FOXP3+ regulatory T cells in myelodysplastic syndromes (MDS). Leukemia 2009, 23, 510–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fozza, C.; Contini, S.; Virdis, P.; Galleu, A.; Massa, A.; Bonfigli, S.; Longinotti, M. Patients with myelodysplastic syndromes show reduced frequencies of CD4+ CD8+ double-positive T cells. Eur. J. Haematol. 2012, 88, 89–90. [Google Scholar] [CrossRef] [PubMed]
- Parikh, A.R.; Olnes, M.J.; Barrett, A.J. Immunomodulatory treatment of myelodysplastic syndromes: Antithymocyte globulin, cyclosporine, and alemtuzumab. Semin. Hematol. 2012, 49, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Stahl, M.; Bewersdorf, J.P.; Giri, S.; Wang, R.; Zeidan, A.M. Use of immunosuppressive therapy for management of myelodysplastic syndromes: A systematic review and meta-analysis. Haematologica 2020, 105, 102–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steensma, D.P.; Dispenzieri, A.; Moore, S.B.; Schroeder, G.; Tefferi, A. Antithymocyte globulin has limited efficacy and substantial toxicity in unselected anemic patients with myelodysplastic syndrome. Blood 2003, 101, 2156–2158. [Google Scholar] [CrossRef]
- Xiao, L.; Qi, Z.; Qiusheng, C.; Li, X.; Luxi, S.; Lingyun, W. The use of selective immunosuppressive therapy on myelodysplastic syn-dromes in targeted populations results in good response rates and avoids treatment-related disease progression. Am. J. Hematol. 2012, 87, 26–31. [Google Scholar] [CrossRef]
- Saunthararajah, Y.; Nakamura, R.; Wesley, R.; Wang, Q.J.; Barrett, A.J. A simple method to predict response to immunosuppressive therapy in patients with myelodysplastic syndrome. Blood 2003, 102, 3025–3027. [Google Scholar] [CrossRef] [Green Version]
- Stahl, M.; Deveaux, M.; de Witte, T.; Neukirchen, J.; Sekeres, M.A.; Brunner, A.M.; Roboz, G.J.; Steensma, D.P.; Bhatt, V.R.; Platzbecker, U.; et al. The use of immunosuppressive therapy in MDS: Clinical outcomes and their predictors in a large international patient cohort. Blood Adv. 2018, 2, 1765–1772. [Google Scholar] [CrossRef] [Green Version]
- Sloand, E.M.; Olnes, M.J.; Shenoy, A.; Weinstein, B.; Boss, C.; Loeliger, K.; Wu, C.O.; More, K.; Barrett, A.J.; Scheinberg, P.; et al. Alemtuzumab treatment of intermediate-1 myelodys-plasia patients is associated with sustained improvement in blood counts and cytogenetic remissions. J. Clin. Oncol. 2010, 28, 5166–5173. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.; Ranpura, V.; Wu, C.; Olnes, M.J.; Parikh, A.R.; Shenoy, A.; Thompson, J.; Weinstein, B.; Scheinberg, P.; Barrett, A.J.; et al. Long-term outcomes in myelodysplastic syndrome patients treated with alemtuzumab. Blood Adv. 2019, 3, 980–983. [Google Scholar] [CrossRef]
- Pietsch, E.C.; Dong, J.; Zhang, X.; Chin, D.; Hawkins, R.; Dinh, T.; Zhou, M.; Strake, B.; Feng, P.-H.; Rocca, M.; et al. Anti-leukemic activity and tolerability of anti-human CD47 monoclonal antibodies. Blood Cancer J. 2017, 7, e536. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.; Al Malki, M.M.; Asch, A.; Lee, D.; Kambhampati, S.; Donnellan, W.; Vyas, P.; Pollyea, D.; Bradley, T.; Jeyakumar, D.; et al. The first- in- class anti- CD47 antibody magrolimab combined with azacytidine is well-tolerated and effective in MDS patients: Phase 1B study results. Blood 2020, 136, 330. [Google Scholar]
- Sallman, D.; Asch, A.; Al Malki, M.M.; J Lee, D.; Donnellan, W.; Marcucci, G.; Kambhampati, S.; Daver, N.G.; Garcia-Manero, G.; komrokji, R.S.; et al. The first- in- class anti- CD47 antibody magrolimab combined with azacytidine is well-tolerated and effective in MDS patients: Phase 1B study results. Blood 2019, 134, 569. [Google Scholar] [CrossRef]
- Dorahy, D.J.; Thorne, R.F.; Fecondo, J.V.; Burns, G.F. Stimulation of platelet activation and aggregation by a carboxyl-terminal peptide from thrombospondin binding to the integrin-associated protein receptor. J. Biol. Chem. 1997, 272, 1323–1330. [Google Scholar] [CrossRef] [Green Version]
- Brierley, C.; Staves, J.; Roberts, C.; Johnson, H.; Vyas, P.; Goodnough, L.; Murphy, M. The effects of monoclonal anti-CD47 on RBCs, compatibility testing, and transfusion requirements in refractory acute myeloid leukemia. Transfusion 2019, 59, 2248–2254. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, M.A.M.; DeAngelo, D.J.; Palmer, J.M.; Seet, C.S.; Tallman, M.S.; Wei, X.; Li, Y.F.; Hock, R.N.; Burgess, M.R.; Hege, K.; et al. A phase i study of CC-90002, a monoclonal antibody targeting CD47, in patients with relapsed and/or refractory (R/R) Acute Myeloid Leukemia (AML) and High-Risk Myelodysplastic Syndromes (MDS): Final results. Blood 2019, 134, 1320. [Google Scholar] [CrossRef]
- Davids, M.S.; Kim, H.T.; Bachireddy, P.; Costello, C.; Liguori, R.; Savell, A.; Lukez, A.P.; Avigan, D.; Chen, Y.-B.; McSweeney, P.; et al. Ipilimumab for patients with relapse after allogeneic transplantation. N. Engl. J. Med. 2016, 375, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia, T.M.; et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: A nonrandomized, open-label, phase II study. Cancer Discov. 2019, 9, 370–383. [Google Scholar] [CrossRef] [Green Version]
- Reville, P.K.; Kantarjian, H.M.; Ravandi, F.; Jabbour, E.; DiNardo, C.D.; Daver, N.; Pemmaraju, N.; Ohanian, M.; Alvarado, Y.; Xiao, L.; et al. Nivolumab maintenance in high-risk acute myeloid leukemia patients: A single-arm, open-label, phase II study. Blood Cancer J. 2021, 11, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.M.; Esteve, J.; Porkka, K.; Knapper, S.; Vey, N.; Wei, A.; Garcia-Manero, G.; Wermke, M.; Janssen, J.; Traer, E.; et al. Efficacy and safety of sabatolimab (MBG453) in combination with Hypomethylating Agents (HMAs) in patients with Acute Myeloid Leukemia (AML) and High-Risk Myelodysplastic Syndrome (HR-MDS): Updated results from a phase 1b study. Blood 2020, 136, 1–2. [Google Scholar] [CrossRef]
- Chao, M.P.; Weissman, I.L.; Majeti, R. The CD47–SIRPα pathway in cancer immune evasion and potential therapeutic implications. Curr. Opin. Immunol. 2012, 24, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D., Jr.; Van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Bewersdorf, J.P.; Stahl, M.; Zeidan, A.M. Immunotherapy in acute myeloid leukemia and myelodysplastic syndromes: The dawn of a new era? Blood Rev. 2019, 34, 67–83. [Google Scholar] [CrossRef]
- Costello, R.T.; Mallet, F.; Sainty, D.; Maraninchi, D.; Gastaut, J.A.; Olive, D. Regulation of CD80/B7-1 and CD86/B7-2 molecule ex-pression in human primary acute myeloid leukemia and their role in allogenic immune recognition. Eur. J. Immunol. 1998, 28, 90–103. [Google Scholar] [CrossRef]
- Graf, M.; Reif, S.; Hecht, K.; Pelka-Fleischer, R.; Kroell, T.; Pfister, K.; Schmetzer, H. High expression of costimulatory molecules correlates with low relapse-free survival probability in acute myeloid leukemia (AML). Ann. Hematol. 2005, 84, 287–297. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Knaus, H.A.; Robinson, T.M.; Towlerton, A.M.H.; Warren, E.H.; Zeidner, J.F.; Blackford, A.L.; Duffield, A.S.; Rizzieri, D.; Frattini, M.G.; et al. A multi-center phase I trial of ipili-mumab in patients with myelodysplastic syndromes following hypomethylating agent failure. Clin. Cancer Res. 2018, 24, 3519–3527. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Bueso-Ramos, C.; Dinardo, C.D.; Estecio, M.R.; Davanlou, M.; Geng, Q.R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gajewski, T.F.; Kline, J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leu-kemia model. Blood 2009, 114, 1545–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, R.; Rotem-Yehudar, R.; Slama, G.; Landes, S.; Kneller, A.; Leiba, M.; Koren-Michowitz, M.; Shimoni, A.; Nagler, A. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin. Cancer Res. 2008, 14, 3044–3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daver, N.; Basu, S.; Garcia-Manero, G.; Abbas, H.A.; Konopleva, M.; Kadia, T.M.; Dinardo, M.C.D.; Borthakur, G.; Alotaibi, A.S.; Pemmaraju, N.; et al. Azacitidine (AZA) with Nivolumab (Nivo), and AZA with Nivo + Ipilimumab (Ipi) in relapsed/refractory (R/R) acute myeloid leukemia: Clinical and immune biomarkers of response. Blood 2020, 136, 43–45. [Google Scholar] [CrossRef]
- Zeidan, A.; Cavenagh, J.; Voso, M.; Taussig, D.; Tormo, M.; Boss, I.; Copeland, W.B.; Gray, V.E.; Previtali, A.; O’Connor, T.; et al. Efficacy and safety of azacitidine (AZA) in combination with the anti-PD-L1 durvalumab (durva) for the front-line treatment of older patients (pts) with Acute Myeloid Leukemia (AML) who are unfit for intensive chemotherapy (IC) and Pts with Higher-Risk Myelodysplastic Syndromes (HR-MDS): Results from a large, international, randomized phase 2 study. Blood 2019, 134, 829. [Google Scholar]
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 2020, 20, 173–185. [Google Scholar] [CrossRef]
- Stringaris, K.; Sekine, T.; Khoder, A.; Alsuliman, A.; Razzaghi, B.; Sargeant, R.; Pavlu, J.; Brisley, G.; de Lavallade, H.; Sarvaria, A.; et al. Leukemia-induced phenotypic and functional defects in natural killer cells predict failure to achieve remission in acute myeloid leukemia. Haematologica 2014, 99, 836–847. [Google Scholar] [CrossRef] [Green Version]
- Vasu, S.; Karl-Heinz, H.; Cheney, C.; Gopalakrishnan, B.; Mani, R.; Lozanski, G.; Mo, X.; Groh, V.; Whitman, S.P.; Konopitzky, R.; et al. Decitabine enhances anti-CD33 monoclonal antibody BI 836858–mediated natural killer ADCC against AML blasts. Blood 2016, 127, 2879–2889. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; He, S.; Ritchie, D.; Hertzberg, M.S.; Kerridge, I.; Durrant, S.T.; Kennedy, G.; Lewis, I.D.; Marlton, P.; McLachlan, A.J.; et al. A phase I study of anti-CD123 monoclonal antibody (mAb) CSL360 targeting leukemia stem cells (LSC) in AML. J. Clin. Oncol. 2010, 28, e13012. [Google Scholar] [CrossRef]
- Syed, K.; Pietsch, C.; Axel, A.; Forslund, A.; Sasser, K.; Salvati, M. Preclinical evaluation of CSL362/JNJ-56022473 in combination with decitabine or azacitidine in in vitro Assays. Blood 2015, 126, 1370. [Google Scholar] [CrossRef]
- Smith, B.D.; Roberts, A.W.; Roboz, G.J.; DeWitte, M.; Ferguson, A.; Garrett, L.; Curcio, T.; Orlowski, K.F.; Dasen, S.; Bensen-Kennedy, D.M.; et al. Minimal Residual Disease (MRD) As exploratory endpoint in a phase 1 study of the anti-CD123 Mab CSL362 given as post-remission therapy in adult Acute Myeloid Leukemia (AML). Blood 2015, 126, 3819. [Google Scholar] [CrossRef]
- Ravandi, F.; Walter, R.B.; Subklewe, M.; Buecklein, V.; Jongen-Lavrencic, M.; Paschka, P.; Ossenkoppele, G.J.; Kantarjian, H.M.; Hindoyan, A.; Agarwal, S.K.; et al. Updated results from phase I dose-escalation study of AMG 330, a bispecific T-cell engager molecule, in patients with relapsed/refractory Acute Myeloid Leukemia (R/R AML). J. Clin. Oncol. 2020, 38, 7508. [Google Scholar] [CrossRef]
- Subklewe, M.; Stein, A.; Walter, R.; Bhatia, R.; Wei, A.; Ritchie, D.; Bucklein, V.; Vachhani, P.; Dai, T.; Hindoyan, A.; et al. Preliminary results from a phase 1 first-in-human study of AMG 673, a Novel Half Life (HLE) anti-CD33/CD3 BiTE (bispecific T-cell engager) in patients with Relapsed/Refractory ( R/R) Acute Myeloid Leukemia ( AML). Blood 2019, 134, 833. [Google Scholar] [CrossRef]
- Krupka, C.; Kufer, P.; Kischel, R.; Zugmaier, G.; Lichtenegger, F.S.; Köhnke, T.; Vick, B.; Jeremias, I.; Metzeler, K.H.; Altmann, T.; et al. Blockade of the PD-1/PD-L1 axis augments lysis of AML cells by the CD33/CD3 BiTE antibody construct AMG 330: Reversing a T-cell-induced immune escape mechanism. Leukemia 2016, 30, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Brauchle, B.; Goldstein, R.L.; Karbowski, C.M.; Henn, A.; Li, C.-M.; Bücklein, V.L.; Krupka, C.; Boyle, M.C.; Koppikar, P.; Haubner, S.; et al. Characterization of a Novel FLT3 BiTE Molecule for the Treatment of Acute Myeloid Leukemia. Mol. Cancer Ther. 2020, 19, 1875–1888. [Google Scholar] [CrossRef] [PubMed]
- Westervelt, P.; Cortes, J.E.; Altman, J.K.; Long, M.; Oehler, V.G.; Gojo, I.; Guenot, J.; Chun, P.; Roboz, G.J. Phase 1 first-in-human trial of AMV564, a bivalent bispecific (2:2) CD33/CD3 T-cell engager, in patients with relapsed/refractory Acute Myeloid Leukemia (AML). Blood 2019, 134, 834. [Google Scholar] [CrossRef]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef]
- Warlick, E.D.; Weisdorf, D.J.; Vallera, D.A.; Wangen, R.; Lewis, D.; Knox, J. GTB-3550 TriKE™ for the Treatment of High-Risk Myel-odysplastic Syndromes (MDS) and refractory/relapsed Acute Myeloid Leukemia (AML) safely drives natural killer (NK) cell proliferation at initial dose cohorts. Blood 2020, 136, 7–8. [Google Scholar] [CrossRef]
- Ravandi, F.; Bashey, A.; Foran, J.M.; Stock, W.; Mawad, R.; Blum, W. Complete responses in relapsed/refractory Acute Myeloid Leukemia (AML) patients on a weekly dosing schedule of XmAb14045, a CD123 x CD3 T cell-engaging bispecific antibody: Initial results of a phase 1 study. Blood 2018, 132, 763. [Google Scholar] [CrossRef]
- Gleason, M.K.; Ross, J.A.; Warlick, E.D.; Lund, T.C.; Verneris, M.R.; Wiernik, A.; Spellman, S.; Haagenson, M.D.; Lenvik, A.J.; Litzow, M.R.; et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood 2014, 123, 3016–3026. [Google Scholar] [CrossRef]
- Davis, Z.B.; Vallera, D.A.; Miller, J.S.; Felices, M. Natural killer cells unleashed: Checkpoint receptor blockade and BiKE/TriKE utilization in NK-mediated anti-tumor immunotherapy. Semin. Immunol. 2017, 31, 64–75. [Google Scholar] [CrossRef]
- Miller, J.S.; Felice, M.; McElmurry, R.; McCullar, V.; Zhou, X.; Tolar, J.; Schmohl, J.U.; Panoskaltsis-Mortari, A.; Zhang, B.; Taras, E.; et al. Trispecific killer engagers (TriKEs) that contain IL-15 to make NK cells antigen specific and to sustain their persistence and expansion. Blood 2015, 126, 232. [Google Scholar] [CrossRef]
- Thomas, A.; Teicher, B.A.; Hassan, R. Antibody-drug conjugates for cancer therapy. Lancet Oncol. 2016, 17, e254–e262. [Google Scholar] [CrossRef]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar]
- Fathi, A.T.; Erba, H.P.; Lancet, J.E.; Stein, E.M.; Ravandi, F.; Faderl, S.; Walter, R.B.; Advani, A.S.; DeAngelo, D.; Kovacsovics, T.J.; et al. A phase 1 trial of vadastuximab talirine combined with hypomethylating agents in patients with CD33-positive AML. Blood 2018, 132, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; DeAngelo, D.J.; Erba, H.P.; Traer, E.; Papadantonakis, N.; Arana-Yi, C.; Blum, W.; Sloss, C.M.; Culm-Merdek, K.; Zweidler-McKay, P.A.; et al. Maturing clinical profile of IMGN779, a next-generation CD33-targeting antibody-drug conjugate, in patients with relapsed or refractory Acute Myeloid Leukemia. Blood 2018, 132, 26. [Google Scholar] [CrossRef]
- Daver, N.G.; Erba, H.P.; Papadantonakis, N.; DeAngelo, D.J.; Wang, E.S.; Konopleva, M.Y.; Sloss, C.M.; Culm-Merdek, K.; Zweidler-McKay, P.A.; Kantarjian, H.M. A Phase I, first-in-human study evaluating the safety and preliminary antileukemia activity of IMGN632, a novel CD123-targeting antibody-drug conjugate, in patients with relapsed/refractory Acute Myeloid Leukemia and other CD123-positive hematologic malignancies. Blood 2018, 132, 27. [Google Scholar]
- Pagel, J.M.; Gooley, T.A.; Rajendran, J.; Fisher, D.R.; Wilson, W.A.; Sandmaier, B.M.; Matthews, D.C.; Deeg, H.J.; Gopal, A.; Martin, P.J.; et al. Allogeneic hematopoietic cell transplantation after conditioning with 131I–anti-CD45 antibody plus fludarabine and low-dose total body irradiation for elderly patients with advanced acute myeloid leukemia or high-risk myelodysplastic syndrome. Blood 2009, 114, 5444–5453. [Google Scholar] [CrossRef] [PubMed]
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860. [Google Scholar] [CrossRef] [Green Version]
- Castaigne, S.; Pautas, C.; Terré, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.-N.; Legrand, O.; Thomas, X.; Turlure, P.; Reman, O.; et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet 2012, 379, 1508–1516. [Google Scholar] [CrossRef]
- Baron, J.; Wang, E.S. Gemtuzumab ozogamicin for the treatment of acute myeloid leukemia. Expert Rev. Clin. Pharmacol. 2018, 11, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Hills, R.K.; Castaigne, S.; Appelbaum, F.R.; Delaunay, J.; Petersdorf, S.; Othus, M.; Estey, E.H.; Dombret, H.; Chevret, S.; Ifrah, N.; et al. Addition of gemtuzumab ozogamicin to in-duction chemotherapy in adult patients with acute myeloid leukaemia: A meta-analysis of individual patient data from ran-domised controlled trials. Lancet Oncol. 2014, 15, 986–996. [Google Scholar] [CrossRef] [Green Version]
- Amadori, S.; Suciu, S.; Selleslag, D.; Aversa, F.; Gaidano, G.; Musso, M.; Annino, L.; Venditti, A.; Voso, M.T.; Mazzone, C.; et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed Acute Myeloid Leukemia unsuitable for intensive chemotherapy: Results of the randomized phase III EORTC-GIMEMA AML-19 trial. J. Clin. Oncol. 2016, 34, 972–979. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, K.R.; Noordhuis, P.; Walker, R.; Watkins, K.; Kovtun, Y.; Harvey, L.; Wilhelm, A.; Johnson, H.; Schuurhuis, G.J.; Ossenkoppele, G.J.; et al. The Antibody-Drug Conjugate (ADC) IMGN779 is highly active in vitro and in vivo against Acute Myeloid Leukemia (AML) with FLT3-ITD mutations. Blood 2014, 124, 2321. [Google Scholar] [CrossRef]
- Lapusan, S.; Vidriales, M.B.; Thomas, X.; DE Botton, S.; Vekhoff, A.; Tang, R.; Dumontet, C.; Morariu-Zamfir, R.; Lambert, J.M.; Ozoux, M.-L.; et al. Phase I studies of AVE9633, an anti-CD33 antibody-maytansinoid conjugate, in adult patients with relapsed/refractory acute myeloid leukemia. Investig. New Drugs 2012, 30, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Sutherland, M.K.; Yu, C.; Walter, R.B.; Westendorf, L.; Valliere-Douglass, J.; Pan, L.; Cronkite, A.; Sussman, D.; Klussman, K.; et al. Characterization of SGN-CD123A, A potent CD123-directed antibody–drug conjugate for Acute Myeloid Leukemia. Mol. Cancer Ther. 2018, 17, 554–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Rowinsky, E.; Brooks, C.; Zal, T.; Zal, M.A.; Burks, J.K.; Zhou, J.; Ciurea, S.O.; Alatrash, G.; Cortes, J.E.; et al. Anti-leukemia efficacy and mechanisms of action of SL-101, a novel anti-CD123 antibody-conjugate in Acute Myeloid Leukemia. Blood 2014, 124, 981. [Google Scholar] [CrossRef]
- Pemmaraju, N.; Lane, A.A.; Sweet, K.L.; Stein, A.S.; Vasu, S.; Blum, W.; Rizzieri, D.A.; Wang, E.S.; Duvic, M.; Sloan, J.M.; et al. Tagraxofusp in blastic plasmacytoid dendritic-cell neoplasm. N. Engl. J. Med. 2019, 380, 1628–1637. [Google Scholar] [CrossRef]
- Frankel, A.E.; Weir, M.A.; Hall, P.D.; Holguin, M.; Cable, C.; Rizzieri, D.A.; Hogge, D.E. Induction of remission in patients with acute myeloid leukemia without prolonged myelosuppression using diphtheria toxin-interleukin 3 fusion protein. J. Clin. Oncol. 2007, 25, 7068. [Google Scholar] [CrossRef]
- Lane, A.; Sweet, K.; Wang, E.; Donnellan, W.; Walter, R.; Stein, A.; Rizzieri, D.A.; Carraway, H.E.; Mantzaris, I.; Prebet, T.; et al. Results from ongoing phase 2 trial of SL-401 as con-solidation therapy in patients with Acute Myeloid Leukemia (AML) in remission with high relapse risk including Minimal Residual Disease (MRD). Blood 2016, 128, 215. [Google Scholar] [CrossRef]
- Saint-Paul, L.; Nguyen, C.; Buffière, A.; Pais de Barros, J.; Hammann, A.; Landras-Guetta, C.; Filomenko, R.; Chretien, M.-L.; Johnson, P.; Bastie, J.-N.; et al. CD45 phosphatase is crucial for human and murine acute myeloid leukemia maintenance through its localization in lipid rafts. Oncotarget 2016, 7, 64785–64797. [Google Scholar] [CrossRef] [Green Version]
- Glatting, G.; Müller, M.; Koop, B.; Hohl, K.; Friesen, C.; Neumaier, B.; Berrie, E.; Bird, P.; Hale, G.; Blumstein, N.M.; et al. Anti-CD45 monoclonal antibody YAML568: A promising radioimmunoconjugate for targeted therapy of acute leukemia. J. Nucl. Med. 2006, 47, 1335–1341. [Google Scholar]
- Zheng, W.; Medeiros, L.J.; Hu, Y.; Powers, I.; Cortes, J.E.; Ravandi-Kashani, F.; Kantarjian, H.H.; Wang, S.A. CD30 expression in high-risk acute myeloid leukemia and myelodysplastic syndromes. Clin. Lymphoma Myeloma Leuk. 2013, 13, 307–314. [Google Scholar] [CrossRef] [Green Version]
- Katz, J.; Janik, J.E.; Younes, A. Brentuximab vedotin (SGN-35). Clin. Cancer Res. 2011, 17, 6428–6436. [Google Scholar] [CrossRef] [Green Version]
- Tasian, S.K. Acute myeloid leukemia chimeric antigen receptor T-cell immunotherapy: How far up the road have we traveled? Ther. Adv. Hematol. 2018, 9, 135–148. [Google Scholar] [CrossRef]
- Driouk, L.; Gicobi, J.K.; Kamihara, Y.; Rutherford, K.; Dranoff, G.; Ritz, J.; Baumeister, S.H.C. Chimeric antigen receptor T cells targeting NKG2D-ligands show robust efficacy against Acute Myeloid Leukemia and T-cell Acute Lymphoblastic Leukemia. Front. Immunol. 2020, 11, 580328. [Google Scholar] [CrossRef]
- Lanier, L.L. NKG2D receptor and its ligands in host defense. Cancer Immunol. Res. 2015, 3, 575–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumeister, S.H.; Murad, J.; Werner, L.; Daley, H.; Trebeden-Negre, H.; Gicobi, J.K.; Schmucker, A.; Reder, J.; Sentman, C.L.; Gilham, D.E.; et al. Phase I trial of autologous CAR T cells targeting NKG2D ligands in patients with AML/MDS and multiple myeloma. Cancer Immunol. Res. 2019, 7, 100–112. [Google Scholar] [CrossRef]
- Björklund, A.T.; Carlsten, M.; Sohlberg, E.; Liu, L.L.; Clancy, T.; Karimi, M.; Cooley, S.; Miller, J.S.; Klimkowska, M.; Schaffer, M.; et al. Complete remission with reduction of high-risk clones following haploidentical NK-cell therapy against MDS and AML. Clin. Cancer Res. 2018, 24, 1834–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tawara, I.; Kageyama, S.; Miyahara, Y.; Fujiwara, H.; Nishida, T.; Akatsuka, Y.; Ikeda, H.; Tanimoto, K.; Terakura, S.; Murata, M.; et al. Safety and persistence of WT1-specific T-cell receptor gene−transduced lymphocytes in patients with AML and MDS. Blood 2017, 130, 1985–1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, E.A.; Srivastava, P.; Matsuzaki, J.; Brumberger, Z.; Wang, E.S.; Kocent, J.; Miller, A.; Roloff, G.; Wong, H.Y.; Paluch, B.E.; et al. NY-ESO-1 vaccination in combination with decitabine induces antigen-specific T-lymphocyte Responses in patients with myelodysplastic syndrome. Clin. Cancer Res. 2018, 24, 1019–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Galat, V.; Galat, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK cell-based cancer immunotherapy: From basic biology to clinical development. J. Hematol. Oncol. 2021, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Stevens, B.M.; Zhang, W.; Pullyea, D.A.; Winters, A.; Gutman, J.; Smith, C.; Budde, E.; Forman, S.J.; Jordan, C.T.; Purev, E. CD123 CAR T cells for the treatment of myelodysplastic syndrome. Exp. Hematol. 2019, 74, 52–63. [Google Scholar] [CrossRef] [Green Version]
- Sommer, C.; Cheng, H.Y.; Nguyen, D.; Dettling, D.; Yeung, Y.A.; Sutton, J.; Hamze, M.; Valton, J.; Smith, J.; Djuretic, I.; et al. Allogeneic FLT3 CAR T cells with an off-switch exhibit potent activity against AML and can be depleted to expedite bone marrow recovery. Mol. Ther. 2020, 28, 2237–2251. [Google Scholar] [CrossRef]
- Zhang, H.; Gan, W.-T.; Hao, W.G.; Wang, P.F.; Li, Z.Y.; Chang, L.J. Successful Anti-CLL1 CAR T-cell therapy in secondary Acute Myeloid Leukemia. Front. Oncol. 2020, 10, 685. [Google Scholar] [CrossRef]
- Sauer, T.; Parikh, K.; Sharma, S.; Omer, B.; Sedloev, D.N.; Chen, Q.; Angenendt, L.; Schliemann, C.; Schmitt, M.; Müller-Tidow, C.; et al. CD70-specific CAR T cells have potent activity against acute myeloid leukemia without HSC toxicity. Blood 2021. [Google Scholar] [CrossRef]
- Hodgins, J.J.; Khan, S.T.; Park, M.M.; Auer, R.C.; Ardolino, M. Killers 2.0: NK cell therapies at the forefront of cancer control. J. Clin. Investig. 2019, 129, 3499–3510. [Google Scholar] [CrossRef] [Green Version]
- Carlsten, M.; Järås, M. Natural killer cells in myeloid malignancies: Immune surveillance, NK cell dysfunction, and pharmacological opportunities to bolster the endogenous NK cells. Front. Immunol. 2019, 10, 2357. [Google Scholar] [CrossRef] [Green Version]
- Ruggeri, L.; Capanni, M.; Casucci, M.; Volpi, I.; Tosti, A.; Perruccio, K.; Urbani, E.; Negrin, R.S.; Martelli, M.F.; Velardi, A. Role of natural killer cell alloreactivity in HLA-mismatched hematopoietic stem cell transplantation. Blood 1999, 94, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Jou, J.; Harrington, K.J.; Zocca, M.-B.; Ehrnrooth, E.; Cohen, E.E.W. The changing landscape of therapeutic cancer vaccines—Novel platforms and neoantigen identification. Clin. Cancer Res. 2021, 27, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Sloand, E.M.; Melenhorst, J.J.; Tucker, Z.C.; Pfannes, L.; Brenchley, J.M.; Yong, A.; Visconte, V.; Wu, C.; Gostick, E.; Scheinberg, P.; et al. T-cell immune responses to Wilms tumor 1 protein in myelodysplasia responsive to immunosuppressive therapy. Blood 2011, 117, 2691–2699. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.K.; Hassan, R.; Yaacob, N.S. Hypomethylating agents and immunotherapy: Therapeutic synergism in acute myeloid leukemia and myelodysplastic syndromes. Front. Oncol. 2021, 11, 624742. [Google Scholar] [CrossRef] [PubMed]
- Brodská, B.; Otevřelová, P.; Šálek, C.; Fuchs, O.; Gašová, Z.; Kuželová, K. High PD-L1 Expression predicts for worse outcome of leukemia patients with concomitant NPM1 and FLT3 mutations. Int. J. Mol. Sci. 2019, 20, 2823. [Google Scholar] [CrossRef] [Green Version]
- Chasov, V.; Zaripov, M.; Mirgayazova, R.; Khadiullina, R.; Zmievskaya, E.; Ganeeva, I.; Valiullina, A.; Rizvanov, A.; Bulatov, E. Promising new tools for targeting p53 mutant cancers: Humoral and cell-based immunotherapies. Front. Immunol. 2021, 12, 707734. [Google Scholar] [CrossRef] [PubMed]

| Study | Trial Identification | Mechanism | Study Population | Study Status | Results | MRD Status | Adverse Effects | Ref |
|---|---|---|---|---|---|---|---|---|
| Phase 1 non-randomized, open label, magrolimab and azacitidine in MDS/AML | NCT03248479 | Anti-CD47 | R/R AML or unfit treatment naïve AML or high risk MDS | Recruiting | High-risk MDS: 30/39 (91%) ORR with 42% CR, 24% marrow CR. Treatment-naïve AML: 22/34 (65%) ORR, 15 (44%) had CR. 15/21 TP53 mutants (71%) ORR, 10/21 (48%) had CR. | High-risk MDS: 22% patients with CR or CRi or marrow CR were MRD negative Treatment-naïve AML: N/A | Anemia, fatigue neutropenia, thrombocytopenia, infusion reaction, increased serum bilirubin, nausea | [66] |
| Phase 1 dose escalation trial, open label, single arm, magrolimab in hematologic malignancies | NCT02678338 | Anti-CD47 | R/R AML or high risk MDS | Completed | N/A | N/A | Decline in hemoglobin, increase in transfusion requirements, RBC agglutination with hemolysis. 9/19 (47%) with positive antibody screen | [69] |
| Phase 1 dose escalation trial, open label, CC-90002 in AML and high risk MDS | NCT02641002 | Anti-CD47 | R/R AML or high risk MDS | Terminated | Only 2/24 patients had stable disease, no ORR noted. No improvement in RBC transfusion requirements | N/A | Disseminated intravascular coagulation in 4 patients, diarrhea, thrombocytopenia, febrile neutropenia, anemia, increased AST/ALT, cough | [70] |
| Phase 1 dose escalation and expansion trial, open label, non randomized, TTI-621 in patients with hematologic malignancies and selected solid tumors | NCT02663518 | Binds CD47 | Relapsed or refractory hematologic malignancies, selected solid tumors | Recruiting | N/A | N/A | N/A | N/A |
| Phase 1/ 2 study, single arm, open label, ALX148 with venetoclax and azacitidine | NCT04755244 | Fusion protein that blocks CD47-SIRPalpha pathway | R/R AML or unfit treatment naïve AML | Recruiting | N/A | N/A | N/A | N/A |
| Phase 1, dose escalation trial, single arm, open label, ipilimumab or nivolumab in relapsed hematologic malignancies after allogeneic HSCT | NCT01822509 | Anti-CTLA4 monoclonal antibody | Variety of R/R hematologic malignancies | Active, not recruiting | MDS/AML specific results: CR in 4 extramedullary AML and 1 secondary AML patient out of 12 AML patients | N/A | Immune-mediated side effects in 6/28 patients (21%), causing 1 death, GVHD in 14% patients | [71] |
| Phase 2, non-randomized, open label, nivolumab and azacitidine with/without ipilimumab | NCT02397720 | Anti-PD-1 monoclonal antibody | R/R AML, unfit treatment naïve AML | Recruiting | Nivolumab and azacitidine (n = 70): ORR 33% including 22% CR/CRi, 1 PR, 7 with hematologic improvement. ORR 58% in HMA naïve, 22% in HMA pre-treated patients. Nivolumab, azacitidine and ipilimumab (n = 36%): 19% with CR/Cri, 3% with PR, 14% with durable SD. | N/A | Grade 3 or 4 immune-mediated side effects in 11% patients Grade 3 or 4 immune-mediated toxicities in 19% patients | [72] |
| Phase 2, single arm, open label, PD-1 inhibition (nivolumab) in AML at high risk of relapse | NCT02532231 | Anti-PD-1 monoclonal antibody | AML in remission with high risk of relapse | Recruiting | 6 month RFS 57.1%, median RFS 8.48 months | 7/9 (78%) with MRD positive stayed MRD positive and progressed. 2/9 cleared MRD. 1/6 with MRD negative status experienced recurrence | Grade 3 or 4 immune mediated toxicities were seen in 27% patients | [73] |
| Phase 1, non-randomized, open label, pembrolizumab and decitabine in newly diagnosed or R/R AML or MDS | NCT03969446 | Anti-PD-1 monoclonal antibody | R/R AML, MDS | Recruiting | N/A | N/A | N/A | N/A |
| Phase 2, randomized, azacitidine and venetoclax with or without pembrolizumab in unfit AML patients | NCT04284787 | Anti-PD-1 monoclonal antibody | Unfit patients with AML | Recruiting | N/A | N/A | N/A | N/A |
| Phase 2, randomized, intensive chemotherapy with or without pembrolizumab in fit AML patients | NCT04214249 | Anti-PD-1 monoclonal antibody | Fit patients with AML | Recruiting | N/A | N/A | N/A | N/A |
| Phase 1b, single arm, pembrolizumab for graft vs leukemia effect in acute leukemia patients with relapse post allo-HSCT | NCT03286114 | Anti-PD-1 monoclonal antibody | AML, ALL, or MDS in relapse after allo-HSCT | Recruiting | N/A | N/A | N/A | N/A |
| Phase 1b study, single arm, open label, pembrolizumab and entinostat in MDS after HMA failure | NCT02936752 | Anti-PD-1 monoclonal antibody | MDS regardless of risk category or oligoblastic AML after HMA failure | Active, not recruiting | N/A | N/A | N/A | N/A |
| Phase 1, non-randomized, sabatolimab with HMAs in AML and high risk MDS | NCT03066648 | Anti-TIM-3 monoclonal antibody | AML and high risk MDS | Active, not recruiting | AML (n = 34): ORR 41.2%, 12 month PFS was 44% High-risk MDS (n = 35): ORR 62.9%, 12 month PFS 58.1% | N/A | Thrombocytopenia, neutropenia, anemia, pneumonia, 7 immune mediate adverse events | [74] |
| Study | Trial Identification | Mechanism | Study Population | Study Status | Results | MRD Status | Adverse Effects | Ref |
|---|---|---|---|---|---|---|---|---|
| Phase 1 trial of vadastuximab talirine monotherapy (VT) with HMA in patients with CD 33-positive AML patients. | NCT01902329 | Anti-CD33 | AML-CR with initial induction/consolidation or R/R-AML or declined treatment with high dose induction/consolidation | Completed | The composite remission rate (CR + CRi) was 70%. Median PFS and OS were 7.7 and 11.3 months, respectively | 51% were MRD negative | Increased hematological toxicity | [105] |
| Phase 3 Study of VT versus placebo in combination with azacitidine or decitabine in the treatment of older patients with newly diagnosed AML | NCT02785900 | Anti-CD33 | Newly diagnosed AML | Terminated | N/A | N/A | Higher rate of death, including fatal infections in VT versus control arm | N/A |
| Phase 1, open label study of IMGN779 in adult patient with R/R CD 33 positive AML | NCT02674763 | Anti-CD33 | R/R AML | Completed | 11/27 patients (41%) who received IMGN779 had a >30% reduction in bone marrow blasts | N/A | Febrile neutropenia, epitaxis, nausea, diarrhea, fatigue, abdominal pain and hypokalemia. Grade 3+ adverse events, most frequent: febrile neutropenia, bactermia, pneumonia, and anemia | [106] |
| Dose-escalation safety and pharmacokinetic study of AVE9633 | NCT00543972 | Anti-CD33 | R/R AML | Terminated | Terminated due to absence of evidence of clinical activity to toxic doses | N/A | N/A | N/A |
| Phase 1/2 multicenter, open label study of IMGN632 monotherapy sdminstered intravenously in patients with CD 123-positive hematological malignancies | NCT03386513 | Anti-CD 123 | CD 123-postiive AML and other CD123-positive hematological malignancies | Recruiting | Four (33%) achieved an OR including one CR and three CRi | N/A | Decreased appetite, diarrhea, nausea, febrile neturopenia, peripheral edema, hypotension, sinus tachycardia | [107] |
| Phase 1, dose escalation and dose expansion trial of SGN-CD123A to evaluate safety, tolerability, and anti-tumor efficacy | NCT02848248 | Anti-CD 123 | R/R AML | Terminated | NA | N/A | Terminated due to safety concern | N/A |
| A Phase 1/2 study of SL-401 as consolidation therapy for adults with adverse risk AML in first CR and/or evidence of MRD in first CR | NCT02270463 | Anti-CD 123 | Adverse risk AML | Completed | N/A | N/A | N/A | N/A |
| Phase 1 trial of SL-401 in combination with azacitidine or azacitidine/venetoclax in AML, high-risk MDS or blastic plasmacytoid dendritic cell neoplasm | NCT03113643 | Anti-CD123 | AML, high-risk MDS, blastic plasmacytoid dendritic neoplasm | Recruiting | N/A | N/A | N/A | N/A |
| Phase 1 study combining escalating doses of radiolabeled BC8 antibody with fludarabine, and low-dose total-body irradiation followed by donor stem cell transplant and immunosuppresion therapy in treating older patients with advanced AML or high-risk MDS | NCT00008177 | Anti-CD45 | AML/High Risk MDS | Recruiting | CR in all patients and engraftment by day 28 of transplant. Median OS and DFS among all 58 patients—199 days and 159 days, respectively, and among the 21 patients treated at the MTD, 206 and 189 days, respectively | N/A | Infusion toxicites, chills, nausea, vomiting, respiratory symptoms such as throat or chest tightness, and hypotension | [108] |
| Phase 3 study of I 131 monoclonal antibody prior to allogeneic HSCT versus conventional care in older subjects with active, R/R AML | NCT02665065 | Anti-CD45 | Older patients with R/R AML | Recruiting | N/A | N/A | N/A | N/A |
| Phase 1 trial of brentuximab vedotin with re-induction chemotherapy with relapsed, CD30-positive AML | NCT01830777 | Anti-CD30 | R/R AML | Completed | N/A | N/A | N/A | N/A |
| Phase 2, open-label study of brentuximab vedotin in patients with CD30-positive nonlymphomatous malignancies | NCT01461538 | Anti-CD30 | AML, ALL or MDS | Completed | N/A | N/A | N/A | N/A |
| Phase 1/2 study of weekly schedule of brentuximab vedotin alone and in combination with azacytidine in CD 30 positive R/R AML | NCT01830777 | Anti-CD30 | R/R AML | Terminated due to slow accrual | N/A | N/A | N/A | N/A |
| Study | Trial Identification | Mechanism | Study Population | Study Status | Results | MRD Status | Adverse Effects | Ref |
|---|---|---|---|---|---|---|---|---|
| CAR T-Cells | ||||||||
| Phase 1 dose-escalation trial | NCT02203825 | NKG2D CAR T-Cells | AML, MDS, or R/R MM without prior lymphodepleting conditioning | Completed | Robust efficacy not observed; response % not reported | N/A | No adverse events related to NKG2D CAR-T cells | [128] |
| NK Cells | ||||||||
| Phase 1/2 adoptive immunotherapy trial | EudraCT number 2011-003181-32 | IL-2 activated haploidentical NK cells | R/R high-risk myeloid malignancies | Completed | Objective response: 6/16 (38%) in patients with MDS and AML | N/A | Transient, treatable grade 3–4 toxicities including chills and nausea in 2/6 patients | [129] |
| Vaccine-based therapy | ||||||||
| Phase 1 dose-escalation trial | UMIN000011519. | WT1-specific TCR-T cell transfer | R/R AML and high-risk MDS expressing WT1 antigen | Completed | WT1 specific TCR T-cells survived in AML and MDS and displayed reactivity to WT1; hematologic efficacy was not established. | N/A | No dose-limiting toxicities | [130] |
| Single-center, phase 1 study | NCT01834248 | NY-ESO-1 vaccine administered with standard dose decitabine induced NY-ESO-1 expression in circulating blasts. | MDS or low blast count AML | Completed | 7 patients reached the end of the study, 7/7 (100%) demonstrated induction of NY-ESO-1 expression, 6/7 patients (86%) and 4/7 patients (57%) demonstrated NY-ESO-1 specific CD4+ and CD8+ T-lymphocyte response | N/A | Related to decitabine: cytopenias, elevated liver enzymes, fatigue, edema, diarrhea. Majority of patients developed localized skin reaction to the vaccine. | [131] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapoor, S.; Champion, G.; Basu, A.; Mariampillai, A.; Olnes, M.J. Immune Therapies for Myelodysplastic Syndromes and Acute Myeloid Leukemia. Cancers 2021, 13, 5026. https://doi.org/10.3390/cancers13195026
Kapoor S, Champion G, Basu A, Mariampillai A, Olnes MJ. Immune Therapies for Myelodysplastic Syndromes and Acute Myeloid Leukemia. Cancers. 2021; 13(19):5026. https://doi.org/10.3390/cancers13195026
Chicago/Turabian StyleKapoor, Sargam, Grace Champion, Aparna Basu, Anu Mariampillai, and Matthew J. Olnes. 2021. "Immune Therapies for Myelodysplastic Syndromes and Acute Myeloid Leukemia" Cancers 13, no. 19: 5026. https://doi.org/10.3390/cancers13195026