
RAS Mediates BET Inhibitor-Endued Repression of Lymphoma Migration and Prognosticates a Novel Proteomics-Based Subgroup of DLBCL through Its Negative Regulator IQGAP3


 , and
, and
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
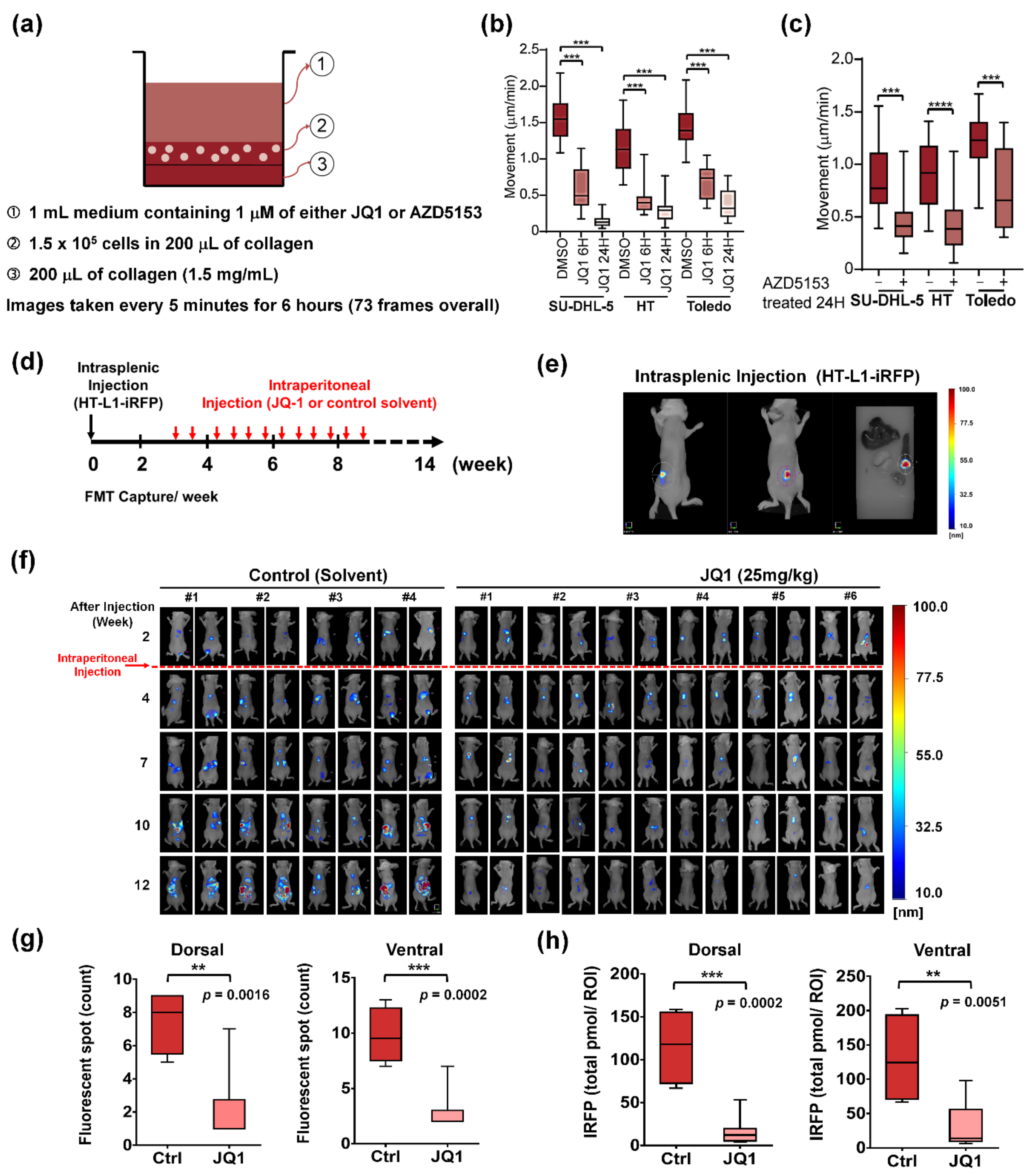
2.1. Trajectory Tracking of DLBCL Cell Migration
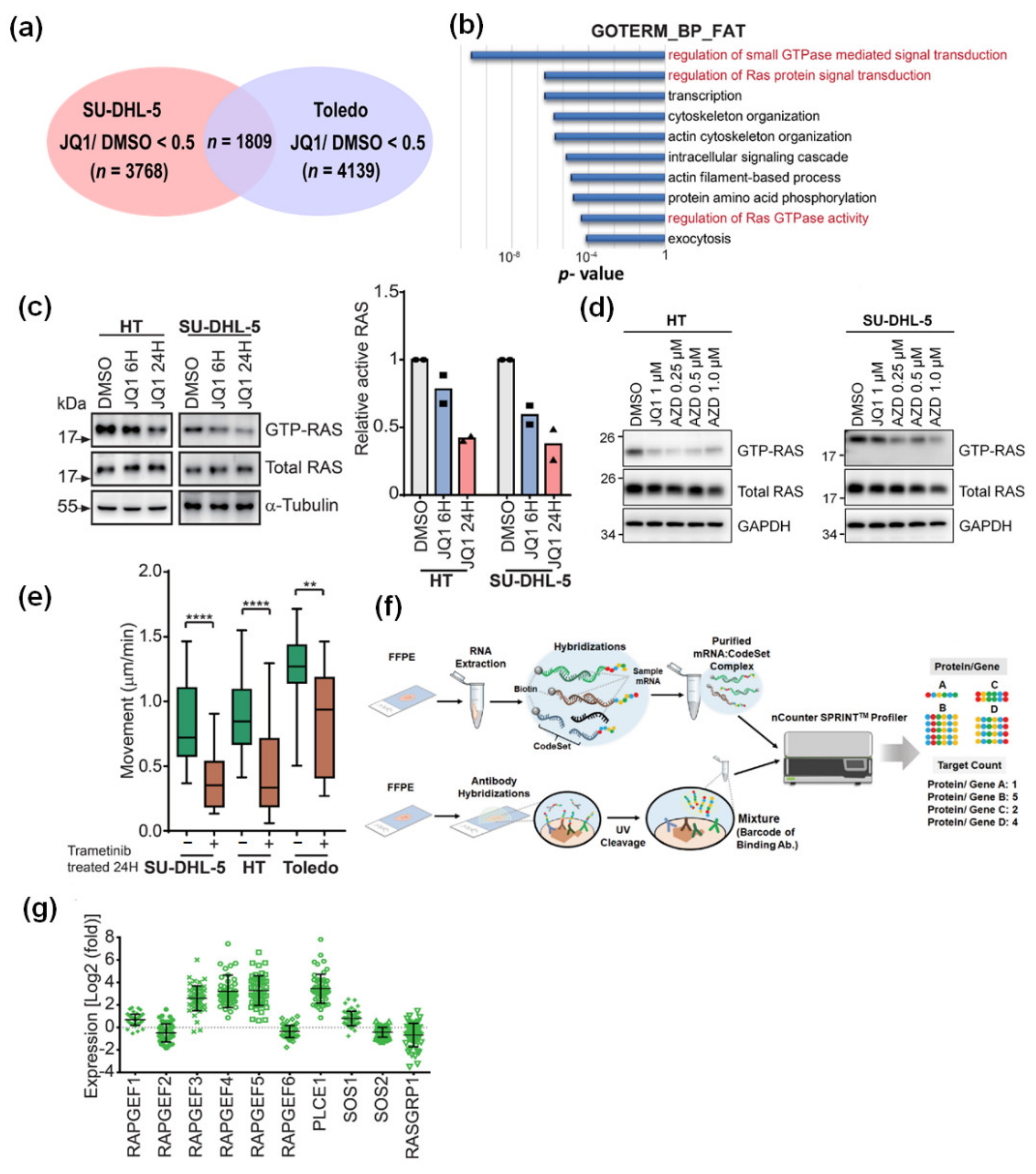
2.2. Small GTPase Activity Assay and Immunoblotting
2.3. Mouse Experiments
2.4. Patients and Sample Collection
2.5. Barcoded mRNA and Protein Profiling with the nCounter Technology
2.6. External Validation
2.7. Others
3. Results
3.1. BET Inhibitors Suppresses Amoeboid Movement and Dissemination of DLBCL Cells
3.2. JQ1-Mediated Restraint of DLBCL Migration Is Partially Attributable to the Inactivated MYC-RhoA Loop
3.3. BET Inhibition Also Leads to Attenuated RAS Activity in DLBCL Cells
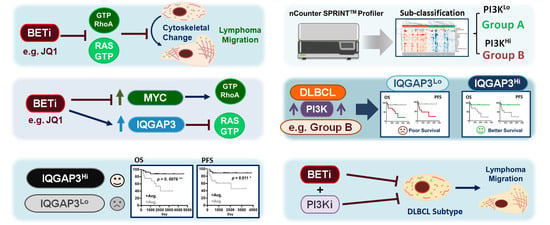
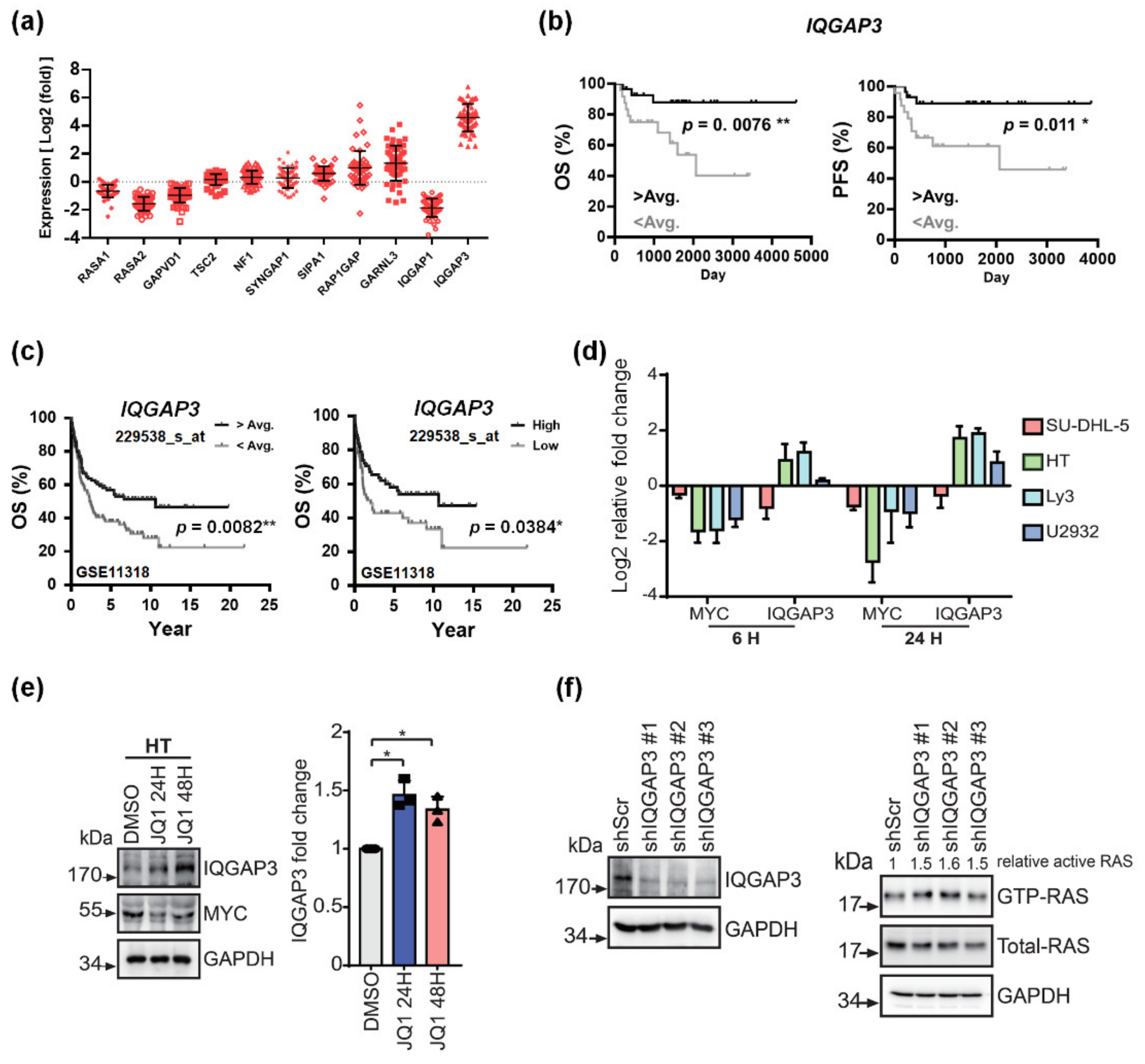
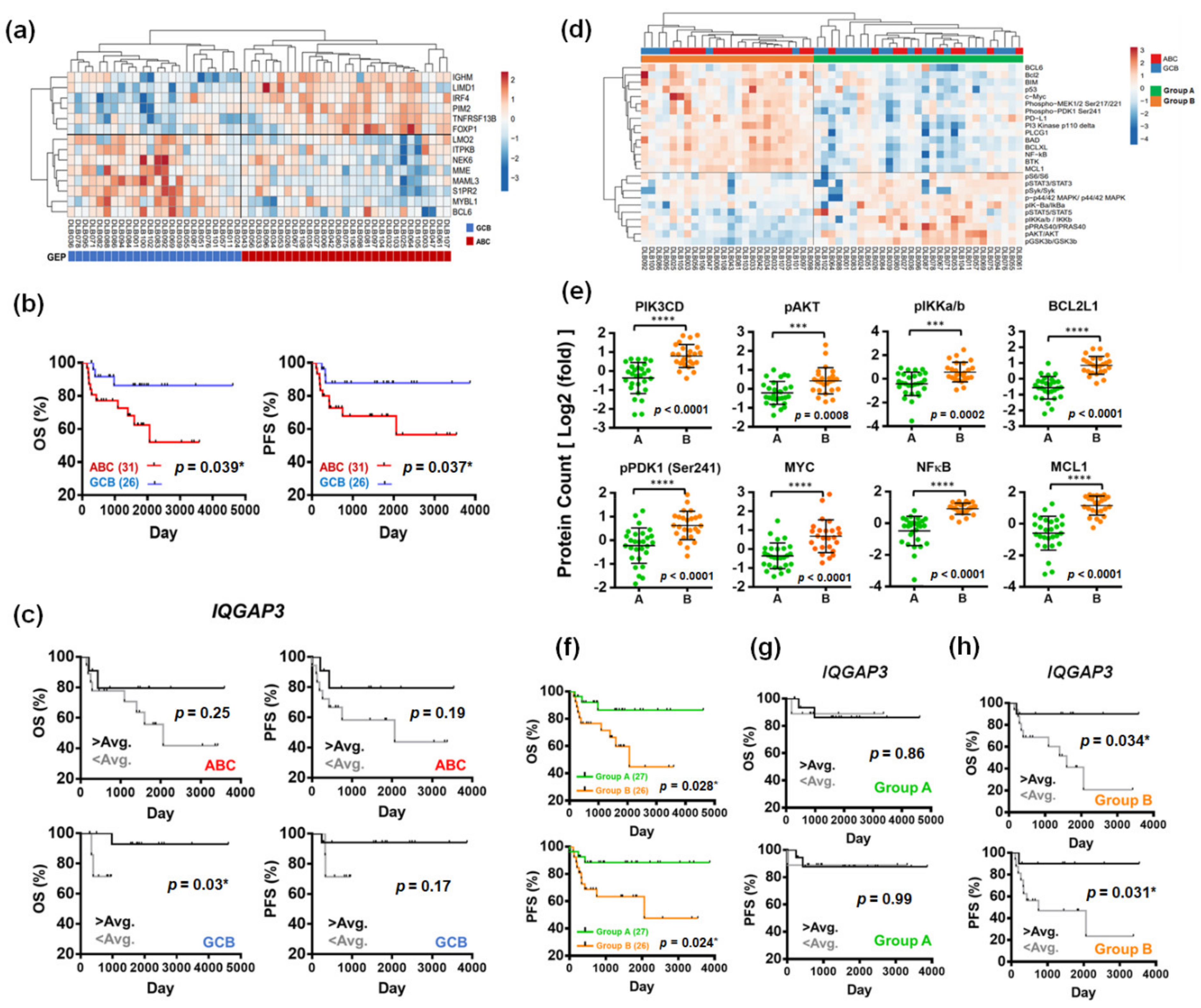
3.4. IQGAP3 Induces RAS Inactivation and Holds Prognostic Relevance in DLBCL
3.5. Proteomics-Based Re-Categorization Identified a Subgroup of Patients with PI3K-Activated DLBCL Whose Prognosis Could Be Further Defined by IQGAP3
3.6. Inhibition of PI3K Signaling Similarly Represses In Vitro Migration and In Vivo Dissemination of DLBCL
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roschewski, M.; Staudt, L.M.; Wilson, W.H. Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nat. Rev. Clin. Oncol. 2014, 11, 12–23. [Google Scholar] [CrossRef]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Dominguez-Sola, D.; Chiarenza, A.; Fabbri, G.; Grunn, A.; Trifonov, V.; Kasper, L.H.; Lerach, S.; Tang, H.; Ma, J.; et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 2011, 471, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-Andre, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [Green Version]
- Dawson, M.A.; Kouzarides, T.; Huntly, B.J. Targeting epigenetic readers in cancer. N. Engl. J. Med. 2012, 367, 647–657. [Google Scholar] [CrossRef] [Green Version]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henssen, A.; Althoff, K.; Odersky, A.; Beckers, A.; Koche, R.; Speleman, F.; Schafers, S.; Bell, E.; Nortmeyer, M.; Westermann, F.; et al. Targeting MYCN-Driven Transcription By BET-Bromodomain Inhibition. Clin. Cancer Res. 2016, 22, 2470–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapuy, B.; McKeown, M.R.; Lin, C.Y.; Monti, S.; Roemer, M.G.; Qi, J.; Rahl, P.B.; Sun, H.H.; Yeda, K.T.; Doench, J.G.; et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 2013, 24, 777–790. [Google Scholar] [CrossRef] [Green Version]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceribelli, M.; Kelly, P.N.; Shaffer, A.L.; Wright, G.W.; Xiao, W.; Yang, Y.; Mathews Griner, L.A.; Guha, R.; Shinn, P.; Keller, J.M.; et al. Blockade of oncogenic IkappaB kinase activity in diffuse large B-cell lymphoma by bromodomain and extraterminal domain protein inhibitors. Proc. Natl. Acad. Sci. USA 2014, 111, 11365–11370. [Google Scholar] [CrossRef] [Green Version]
- Boi, M.; Gaudio, E.; Bonetti, P.; Kwee, I.; Bernasconi, E.; Tarantelli, C.; Rinaldi, A.; Testoni, M.; Cascione, L.; Ponzoni, M.; et al. The BET Bromodomain Inhibitor OTX015 Affects Pathogenetic Pathways in Preclinical B-cell Tumor Models and Synergizes with Targeted Drugs. Clin. Cancer Res. 2015, 21, 1628–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernasconi, E.; Gaudio, E.; Lejeune, P.; Tarantelli, C.; Cascione, L.; Kwee, I.; Spriano, F.; Rinaldi, A.; Mensah, A.A.; Chung, E.; et al. Preclinical evaluation of the BET bromodomain inhibitor BAY 1238097 for the treatment of lymphoma. Br. J. Haematol. 2017, 178, 936–948. [Google Scholar] [CrossRef] [Green Version]
- Mensah, A.A.; Cascione, L.; Gaudio, E.; Tarantelli, C.; Bomben, R.; Bernasconi, E.; Zito, D.; Lampis, A.; Hahne, J.C.; Rinaldi, A.; et al. Bromodomain and extra-terminal domain inhibition modulates the expression of pathologically relevant microRNAs in diffuse large B-cell lymphoma. Haematologica 2018, 103, 2049–2058. [Google Scholar] [CrossRef] [PubMed]
- Amorim, S.; Stathis, A.; Gleeson, M.; Iyengar, S.; Magarotto, V.; Leleu, X.; Morschhauser, F.; Karlin, L.; Broussais, F.; Rezai, K.; et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: A dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016, 3, e196–e204. [Google Scholar] [CrossRef]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.R.; Chen, C.C.; Chan, Y.T.; Wang, H.J.; Chien, F.T.; Chen, Y.L.; Liu, J.L.; Yang, M.H. STAT3-coordinated migration facilitates the dissemination of diffuse large B-cell lymphomas. Nat. Commun. 2018, 9, 3696. [Google Scholar] [CrossRef]
- Lenz, G.; Wright, G.W.; Emre, N.C.; Kohlhammer, H.; Dave, S.S.; Davis, R.E.; Carty, S.; Lam, L.T.; Shaffer, A.L.; Xiao, W.; et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 13520–13525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.H.; Lee, S.W.; Li, C.F.; Wang, J.; Yang, W.L.; Wu, C.Y.; Wu, J.; Nakayama, K.I.; Kang, H.Y.; Huang, H.Y.; et al. Deciphering the transcriptional complex critical for RhoA gene expression and cancer metastasis. Nat. Cell Biol. 2010, 12, 457–467. [Google Scholar] [CrossRef]
- Li, W.; Gupta, S.K.; Han, W.; Kundson, R.A.; Nelson, S.; Knutson, D.; Greipp, P.T.; Elsawa, S.F.; Sotomayor, E.M.; Gupta, M. Targeting MYC activity in double-hit lymphoma with MYC and BCL2 and/or BCL6 rearrangements with epigenetic bromodomain inhibitors. J. Hematol. Oncol. 2019, 12, 73. [Google Scholar] [CrossRef] [Green Version]
- Ueyama, T. Rho-Family Small GTPases: From Highly Polarized Sensory Neurons to Cancer Cells. Cells 2019, 8, 92. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Wolf, K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2016, 109, 314–341. [Google Scholar] [CrossRef] [PubMed]
- Tajiri, H.; Uruno, T.; Shirai, T.; Takaya, D.; Matsunaga, S.; Setoyama, D.; Watanabe, M.; Kukimoto-Niino, M.; Oisaki, K.; Ushijima, M.; et al. Targeting Ras-Driven Cancer Cell Survival and Invasion through Selective Inhibition of DOCK1. Cell Rep. 2017, 19, 969–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.M.; Hedman, A.C.; Sacks, D.B. IQGAPs choreograph cellular signaling from the membrane to the nucleus. Trends Cell Biol. 2015, 25, 171–184. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Zhang, X.; Li, F.; Xiao, D.; Hou, Y.; Zhu, S.; Liu, D.; Ye, X.; Ye, M.; Yang, J.; et al. Frequent alterations in cytoskeleton remodelling genes in primary and metastatic lung adenocarcinomas. Nat. Commun. 2015, 6, 10131. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Liu, Y.; Ding, X.; Ke, Q.; Shi, J.; Ma, Z.; Gu, H.; Wang, H.; Zhang, C.; Yang, C.; et al. E2F1 transactivates IQGAP3 and promotes proliferation of hepatocellular carcinoma cells through IQGAP3-mediated PKC-alpha activation. Ame. J. Cancer Res. 2019, 9, 285–299. [Google Scholar]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef]
- Lacy, S.E.; Barrans, S.L.; Beer, P.A.; Painter, D.; Smith, A.G.; Roman, E.; Cooke, S.L.; Ruiz, C.; Glover, P.; Van Hoppe, S.J.L.; et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: A Haematological Malignancy Research Network report. Blood 2020, 135, 1759–1771. [Google Scholar] [CrossRef]
- Wright, G.W.; Huang, D.W.; Phelan, J.D.; Coulibaly, Z.A.; Roulland, S.; Young, R.M.; Wang, J.Q.; Schmitz, R.; Morin, R.D.; Tang, J.; et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 2020, 37, 551–568.e514. [Google Scholar] [CrossRef]
- Scott, D.W.; Wright, G.W.; Williams, P.M.; Lih, C.J.; Walsh, W.; Jaffe, E.S.; Rosenwald, A.; Campo, E.; Chan, W.C.; Connors, J.M.; et al. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood 2014, 123, 1214–1217. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Dalla-Favera, R. Genetics of diffuse large B-cell lymphoma. Blood 2018, 131, 2307–2319. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [Green Version]
- McKeown, M.R.; Bradner, J.E. Therapeutic strategies to inhibit MYC. Cold Spring Harbor Perspect. Med. 2014, 4. [Google Scholar] [CrossRef]
- Derenzini, E.; Mondello, P.; Erazo, T.; Portelinha, A.; Liu, Y.; Scallion, M.; Asgari, Z.; Philip, J.; Hilden, P.; Valli, D.; et al. BET Inhibition-Induced GSK3beta Feedback Enhances Lymphoma Vulnerability to PI3K Inhibitors. Cell Rep. 2018, 24, 2155–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.; Anderson, R.A. And Akt-ion! IQGAP1 in control of signaling pathways. EMBO J. 2017, 36, 967–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Watanabe, T.; Noritake, J.; Fukata, M.; Yoshimura, T.; Itoh, N.; Harada, T.; Nakagawa, M.; Matsuura, Y.; Arimura, N.; et al. IQGAP3, a novel effector of Rac1 and Cdc42, regulates neurite outgrowth. J. Cell Sci. 2007, 120, 567–577. [Google Scholar] [CrossRef] [Green Version]
- Castellano, E.; Molina-Arcas, M.; Krygowska, A.A.; East, P.; Warne, P.; Nicol, A.; Downward, J. RAS signalling through PI3-Kinase controls cell migration via modulation of Reelin expression. Nat. Commun. 2016, 7, 11245. [Google Scholar] [CrossRef] [Green Version]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nature Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Stratikopoulos, E.E.; Parsons, R.E. Molecular Pathways: Targeting the PI3K Pathway in Cancer-BET Inhibitors to the Rescue. Clin. Cancer Res. 2016, 22, 2605–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinsley, S.; Meja, K.; Shepherd, C.; Khwaja, A. Synergistic induction of cell death in haematological malignancies by combined phosphoinositide-3-kinase and BET bromodomain inhibition. Br. J. Haematol. 2015, 170, 275–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratikopoulos, E.E.; Dendy, M.; Szabolcs, M.; Khaykin, A.J.; Lefebvre, C.; Zhou, M.M.; Parsons, R. Kinase and BET Inhibitors Together Clamp Inhibition of PI3K Signaling and Overcome Resistance to Therapy. Cancer Cell 2015, 27, 837–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-C.; Hsu, C.-C.; Chen, S.-L.; Lin, P.-H.; Chen, J.-P.; Pan, Y.-R.; Huang, C.-E.; Chen, Y.-J.; Chen, Y.-Y.; Wu, Y.-Y.; et al. RAS Mediates BET Inhibitor-Endued Repression of Lymphoma Migration and Prognosticates a Novel Proteomics-Based Subgroup of DLBCL through Its Negative Regulator IQGAP3. Cancers 2021, 13, 5024. https://doi.org/10.3390/cancers13195024
Chen C-C, Hsu C-C, Chen S-L, Lin P-H, Chen J-P, Pan Y-R, Huang C-E, Chen Y-J, Chen Y-Y, Wu Y-Y, et al. RAS Mediates BET Inhibitor-Endued Repression of Lymphoma Migration and Prognosticates a Novel Proteomics-Based Subgroup of DLBCL through Its Negative Regulator IQGAP3. Cancers. 2021; 13(19):5024. https://doi.org/10.3390/cancers13195024
Chicago/Turabian StyleChen, Chih-Cheng, Chia-Chen Hsu, Sung-Lin Chen, Po-Han Lin, Ju-Pei Chen, Yi-Ru Pan, Cih-En Huang, Ying-Ju Chen, Yi-Yang Chen, Yu-Ying Wu, and et al. 2021. "RAS Mediates BET Inhibitor-Endued Repression of Lymphoma Migration and Prognosticates a Novel Proteomics-Based Subgroup of DLBCL through Its Negative Regulator IQGAP3" Cancers 13, no. 19: 5024. https://doi.org/10.3390/cancers13195024