
Characteristics of Nephroblastoma/Nephroblastomatosis in Children with a Clinically Reported Underlying Malformation or Cancer Predisposition Syndrome


 , , , and
, , , and
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
3. Results
3.1. Characteristics of Study Population
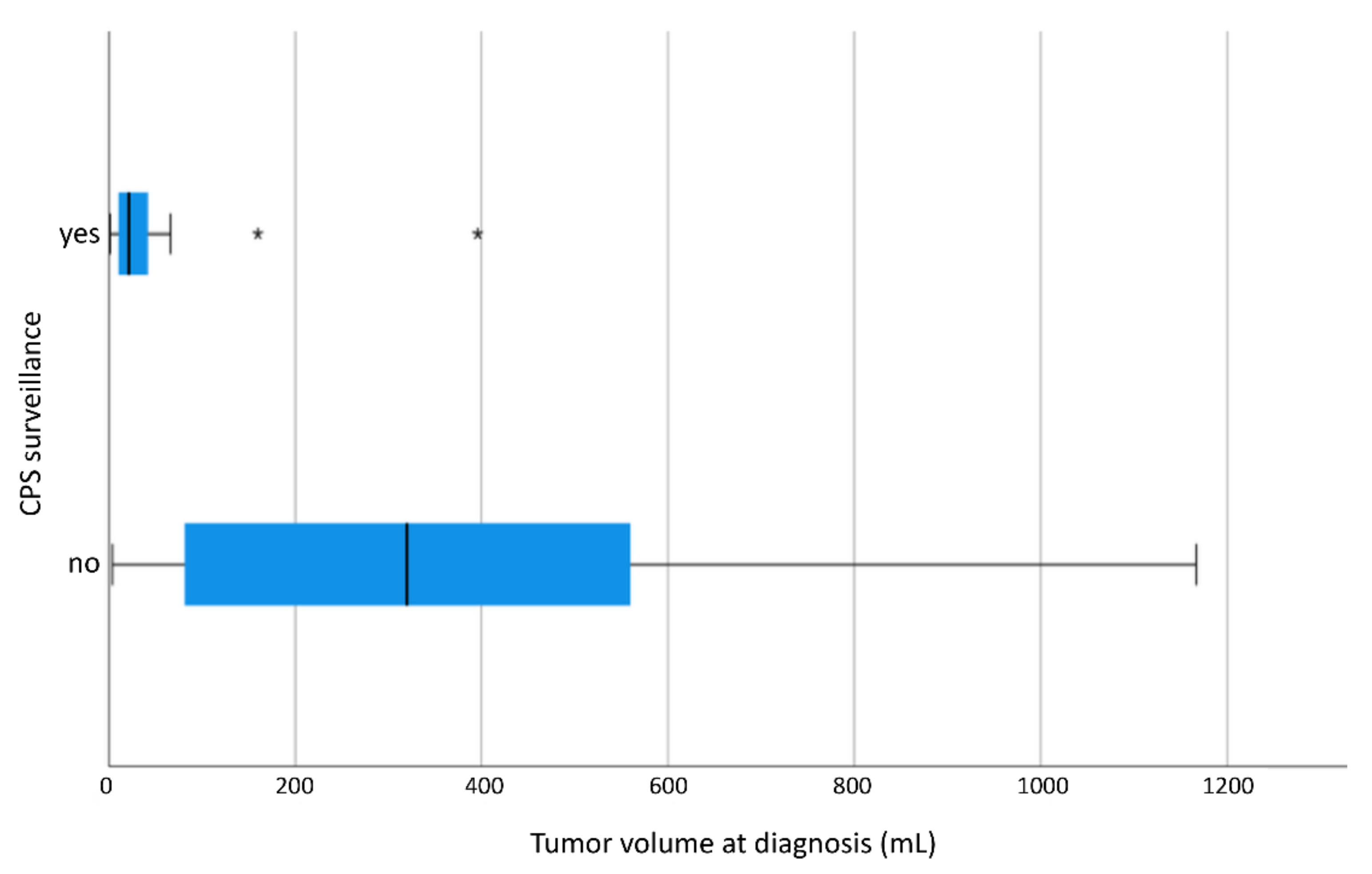
3.2. Ultrasound Surveillance Every 3 Months
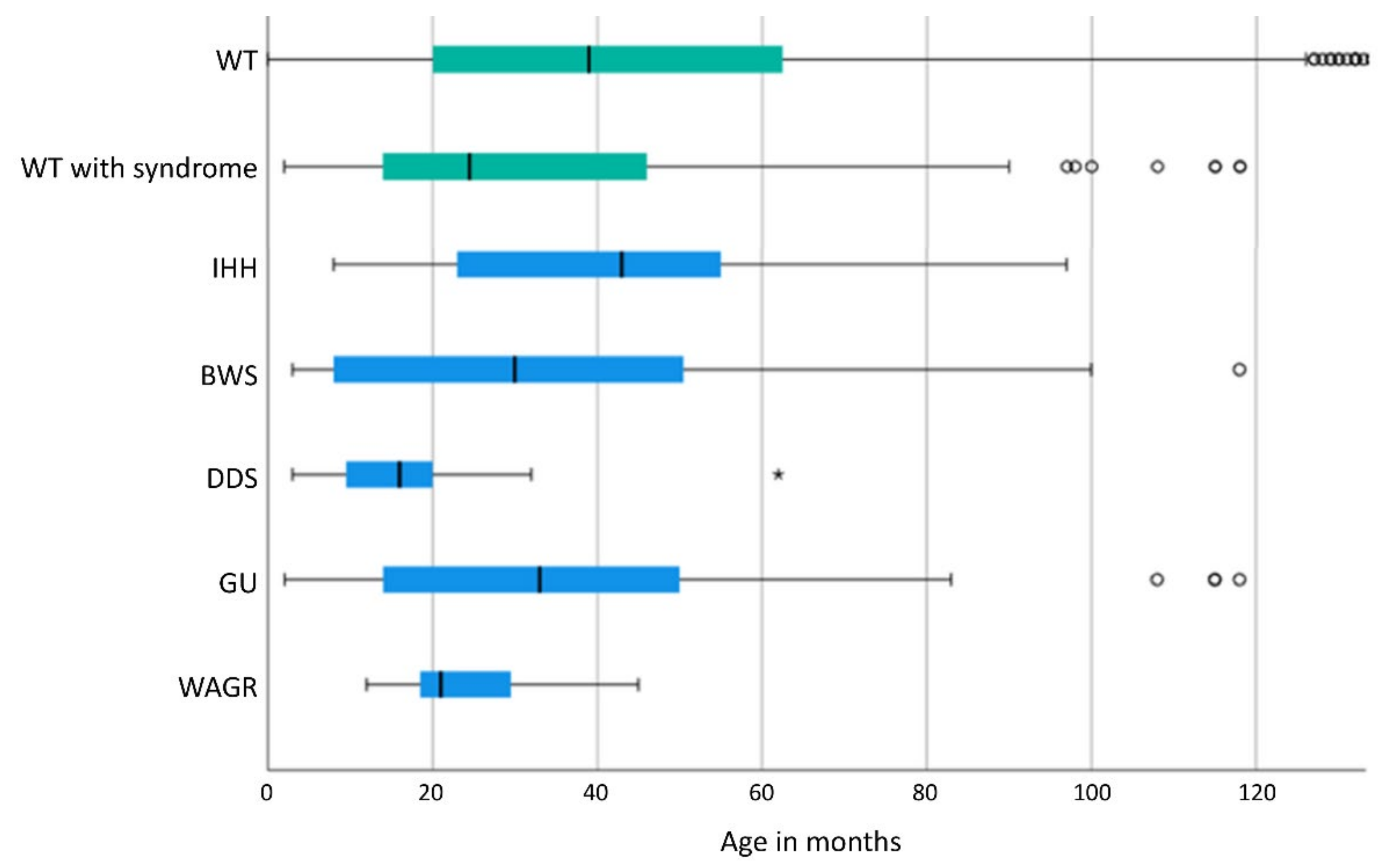
3.3. Gender Distribution and Age at Diagnosis of WT/Nephroblastomatosis
3.4. Bilaterality
3.5. Metastatic Disease in Patients with CPS or Malformations
3.6. Histology
3.7. Tumor Volume
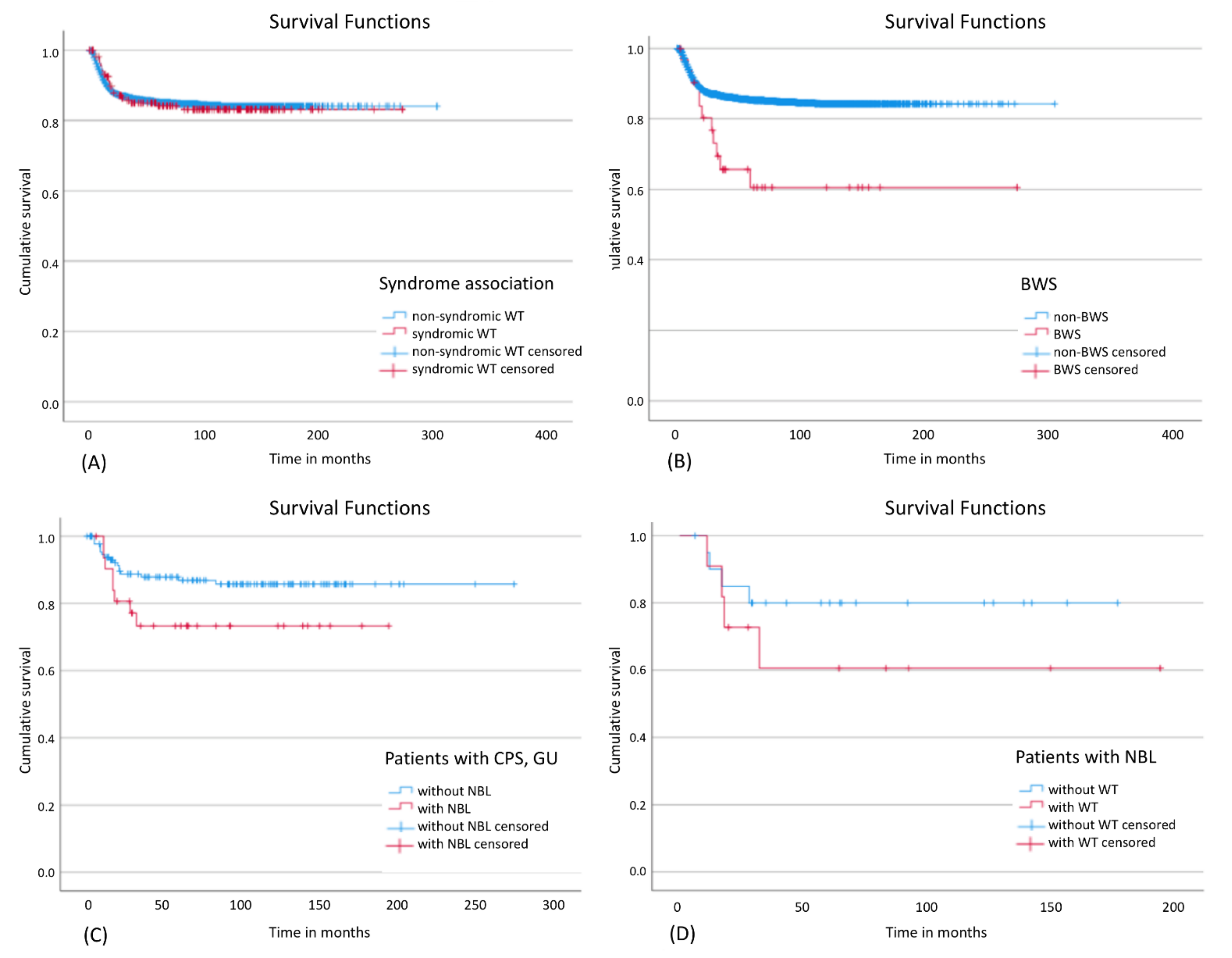
3.8. Outcome
4. Discussion
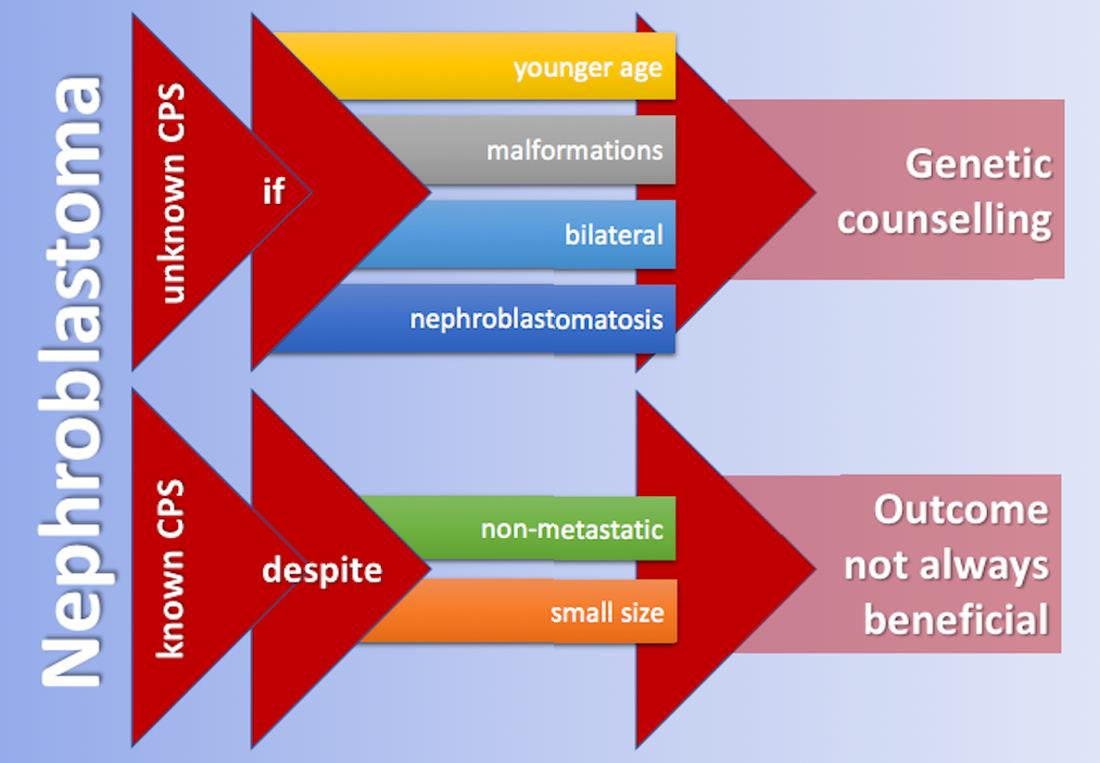
4.1. Prevalence and Surveillance
4.2. Age at Diagnosis of WT/Nephroblastomatosis
4.3. Tumor Volume and Response to Preoperative Chemotherapy
4.4. Bilaterality
4.5. Metastatic Disease
4.6. Histology
4.7. Outcome
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stiller, C.A.; Parkint, D.M. International variations in the incidence of childhood soft-tissue sarcomas. Paediatr. Perinat. Epidemiol. 1994, 8, 107–119. [Google Scholar] [CrossRef]
- Pritchard-Jones, K.; Moroz, V.; Vujanić, G.; Powis, M.; Walker, J.; Messahel, B.; Hobson, R.; Levitt, G.; Kelsey, A.; Mitchell, C. Treatment and outcome of Wilms’ tumour patients: An analysis of all cases registered in the UKW3 trial. Ann. Oncol. 2012, 23, 2457–2463. [Google Scholar] [CrossRef]
- Dome, J.S.; Graf, N.; Geller, J.I.; Fernandez, C.V.; Mullen, E.A.; Spreafico, F.; Van Den Heuvel-Eibrink, M.; Pritchard-Jones, K. Advances in wilms tumor treatment and biology: Progress through international collaboration. J. Clin. Oncol. 2015, 33, 2999–3007. [Google Scholar] [CrossRef]
- van den Heuvel-Eibrink, M.M.; Hol, J.A.; Pritchard-Jones, K.; Tinteren, H.V.; Furtwängler, R.; Verschuur, A.C.; Vujanic, G.M.; Leuschner, I.; Brok, J.; Rübe, C.; et al. Position Paper: Rationale for the treatment of Wilms tumour in the UMBRELLA SIOP-RTSG 2016 protocol. Nat. Rev. Urol. 2017, 14, 743–752. [Google Scholar] [CrossRef]
- Narod, S.A.; Hawkins, M.M.; Robertson, C.M.; Stiller, C.A. Congenital anomalies and childhood cancer in Great Britain. Am.J.Hum.Genet. 1997, 60, 474–485. [Google Scholar]
- Merks, J.H.M.; Caron, H.N.; Hennekam, R.C.M. High Incidence of Malformation Syndromes in a Series of 1, 073 Child. Cancer 2005, 143, 132–143. [Google Scholar]
- Shannon, R.S.; Mann, J.R.; Harper, E.; Harnden, D.G.; Morten, J.E.N.; Herbert, A. Wilms’s tumour and aniridia: Clinical and cytogenetic features. Arch. Dis. Child. 1982, 57, 685–690. [Google Scholar]
- Hol, J.A.; Jongmans, M.C.J.; Sudour-Bonnange, H.; Ramírez-Villar, G.L.; Chowdhury, T.; Rechnitzer, C.; Pal, N.; Schleiermacher, G.; Karow, A.; Kuiper, R.P.; et al. Clinical characteristics and outcomes of children with WAGR syndrome and Wilms tumor and/or nephroblastomatosis: The 30-year SIOP-RTSG experience. Cancer 2021, 127, 628–638. [Google Scholar] [CrossRef]
- Breslow, N.E.; Norris, R.; Norkool, P.A.; Kang, T.; Beckwith, J.B.; Perlman, E.J.; Ritchey, M.L.; Green, D.M.; Nichols, K.E. Characteristics and outcomes of children with the Wilms tumor-aniridia syndrome: A report from the National Wilms Tumor Study Group. J. Clin. Oncol. 2003, 21, 4579–4585. [Google Scholar] [CrossRef]
- Drash, A.; Sherman, F.; Hartmann, W.H.; Blizzard, R.M. A syndrome of pseudohermaphroditism, Wilms’ tumor, hypertension, and degenerative renal disease. J. Pediatr. 1970, 76, 585–593. [Google Scholar] [CrossRef]
- Mueller, R.F. the month The Denys-Drash syndrome. J. Med Genet. 1994, 36, 471–477. [Google Scholar] [CrossRef]
- Scott, R.H.; Stiller, C.A.; Walker, L.; Rahman, N. Syndromes and constitutional chromosomal abnormalities associated with Wilms tumour. J. Med. Genet. 2006, 43, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Pritchard-Jones, K.; Fleming, S.; Davidson, D.; Bickmore, W.; Porteous, D.; Gosden, C.; Bard, J.; Buckler, A.; Pelletier, J.; Housman, D.; et al. The candidate Wilms’ tumour gene is involved in genitourinary development. Nature 1990, 346, 194–197. [Google Scholar] [CrossRef]
- Royer-Pokora, B.; Ragg, S.; Heckl-Östreicher, B.; Held, M.; Loos, U.; Call, K.; Glaser, T.; Housman, D.; Saunders, G.; Zabel, B.; et al. Direct pulsed field gel electrophoresis of Wilms’ tumors shows that dna deletions in 11 p 13 are rare. Genes Chromosom. Cancer 1991, 3, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Hoyme, H.E.; Seaver, L.H.; Jones, K.L.; Procopio, F.; Crooks, W.; Feingold, M. Isolated hemihyperplasia (hemihypertrophy): Report of a prospective multicenter study of the incidence of neoplasia and review. Am. J. Med. Genet. 1998, 79, 274–278. [Google Scholar] [CrossRef]
- Grundy, P.E.; Feinberg, A.P.; Niemitz, E.L.; Brandenburg, S.A.; DeBaun, M.R. Children with Idiopathic Hemihypertrophy and Beckwith-Wiedemann Syndrome Have Different Constitutional Epigenotypes Associated with Wilms Tumor. Am. J. Hum. Genet. 2005, 77, 887–891. [Google Scholar]
- Pettenati, M.J.; Haines, J.L.; Higgins, R.R.; Wappner, R.S.; Palmer, C.G.; Weaver, D.D. Wiedemann-Beckwith syndrome: Presentation of clinical and cytogenetic data on 22 new cases and review of the literature. Hum. Genet. 1986, 74, 143–154. [Google Scholar] [CrossRef]
- Brioude, F.; Toutain, A.; Giabicani, E.; Cottereau, E.; Cormier-Daire, V.; Netchine, I. Overgrowth syndromes—Clinical and molecular aspects and tumour risk. Nat. Rev. Endocrinol. 2019, 15, 299–311. [Google Scholar] [CrossRef]
- Dome, J.S.; Perlman, E.J.; Graf, N. Risk Stratification for Wilms Tumor: Current Approach and Future Directions. Am. Soc. Clin. Oncol. Educ. B. 2014, 34, 215–223. [Google Scholar] [CrossRef]
- Mussa, A.; Duffy, K.A.; Carli, D.; Griff, J.R.; Fagiano, R.; Kupa, J.; Brodeur, G.M.; Ferrero, G.B.; Kalish, J.M. The effectiveness of Wilms tumor screening in Beckwith–Wiedemann spectrum. J. Cancer Res. Clin. Oncol. 2019, 145, 3115–3123. [Google Scholar] [CrossRef]
- Beckwith, J.B. Nephrogenic rests and the pathogenesis of Wilms tumor: Developmental and clinical considerations. Am. J. Med. Genet. 1998, 79, 268–273. [Google Scholar] [CrossRef]
- Bonaïti-Pellié, C.; Chompret, A.; Tournade, M.-F.; Hochez, J.; Moutou, C.; Zucker, J.-M.; Steschenko, D.; Brunat-Mentigny, M.; Roché, H.; Tron, P.; et al. Genetics and epidemiology of Wilms’ tumor: The French Wilms’ tumor study. Med. Pediatr. Oncol. 1992, 20, 284–291. [Google Scholar] [CrossRef]
- Diller, L.; Ghahremani, M.; Morgan, J.; Grundy, P.; Reeves, C.; Breslow, N.; Green, D.; Neuberg, D.; Pelletier, J.; Li, F.P. Constitutional WT1 mutations in Wilms’ tumor patients. J. Clin. Oncol. 1998, 16, 3634–3640. [Google Scholar] [CrossRef]
- Auber, F.; Jeanpierre, C.; Denamur, E.; Jaubert, F.; Schleiermacher, G.; Patte, C.; Cabrol, S.; Leverger, G.; Nihoul-Fékété, C.; Sarnacki, S. Management of Wilms tumors in Drash and Frasier syndromes. Pediatr. Blood Cancer 2009, 52, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Little, S.E.; Hanks, S.P.; King-Underwood, L.; Jones, C.; Rapley, E.A.; Rahman, N.; Pritchard-Jones, K. Frequency and Heritability of WT1 Mutations in Nonsyndromic Wilms’ Tumor Patients: A UK Children’s Cancer Study Group Study. J. Clin. Oncol. 2004, 22, 4140–4146. [Google Scholar] [CrossRef]
- Pelletier, J.; Bruening, W.; Kashtan, C.E.; Mauer, S.M.; Manivel, J.C.; Striegel, J.E.; Houghton, D.C.; Junien, C.; Habib, R.; Fouser, L. Germline mutations in the Wilms’ tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell 1991, 67, 437–447. [Google Scholar] [CrossRef]
- Weirich, A.; von Harrach, M.; Royer-Pokora, B.; Schneider, D.; Leuschner, I.; Schumacher, V.; Graf, N.; Autschbach, F.; Uschkereit, C.; Beier, M. Clinical relevance of mutations in the Wilms tumor suppressor 1 gene WT1 and the cadherin-associated protein β1 gene CTNNB1 for patients with Wilms tumors. Cancer 2008, 113, 1080–1089. [Google Scholar]
- Sotelo-Avila, C.; Gonzalez-Crussi, F.; Fowler, J.W. Complete and incomplete forms of Beckwith-Wiedemann syndrome: Their oncogenic potential. J. Pediatr. 1980, 96, 47–50. [Google Scholar] [CrossRef]
- DeBaun, M.R.; Tucker, M.A. Risk of cancer during the first four years of life in children from The Beckwith-Wiedemann Syndrome Registry. J. Pediatr. 1998, 132, 398–400. [Google Scholar] [CrossRef]
- Green, D.M.; Breslow, N.E.; Beckwith, J.B.; Norkool, P. Screening of children with hemihypertrophy, aniridia, and Beckwith-Wiedemann syndrome in patients with wilms tumor: A report from the national Wilms tumor study. Med. Pediatr. Oncol. 1993, 21, 188–192. [Google Scholar] [CrossRef]
- Beckwith, J.B.; Kiviat, N.B.; Bonadio, J.F. Nephrogenic rests, nephroblastomatosis, and the pathogenesis of Wilms’ tumor. Pediatr. Pathol. 1990, 10, 1–36. [Google Scholar] [CrossRef]
- MacFarland, S.P.; Duffy, K.A.; Bhatti, T.R.; Bagatell, R.; Balamuth, N.J.; Brodeur, G.M.; Ganguly, A.; Mattei, P.A.; Surrey, L.F.; Balis, F.M.; et al. Diagnosis of Beckwith-Wiedemann syndrome in children presenting with Wilms tumor. Pediatr. Blood Cancer 2018, e27296. [Google Scholar] [CrossRef]
- Brioude, F.; Kalish, J.M.; Mussa, A.; Foster, A.C.; Bliek, J.; Ferrero, G.B.; Boonen, S.E.; Cole, T.; Baker, R.; Bertoletti, M.; et al. Expert consensus document: Clinical and molecular diagnosis, screening and management of Beckwith–Wiedemann syndrome: An international consensus statement. Nat. Rev. Endocrinol. 2018, 14, 229–249. [Google Scholar] [CrossRef] [PubMed]
- Ripperger, T.; Bielack, S.S.; Borkhardt, A.; Brecht, I.B.; Burkhardt, B.; Calaminus, G.; Debatin, K.-M.; Deubzer, H.; Dirksen, U.; Eckert, C.; et al. Childhood cancer predisposition syndromes-A concise review and recommendations by the Cancer Predisposition Working Group of the Society for Pediatric Oncology and Hematology. Am. J. Med. Genet. Part A 2017, 173, 1017–1037. [Google Scholar] [CrossRef]
- Royer-Pokora, B.; Weirich, A.; Schumacher, V.; Uschkereit, C.; Beier, M.; Leuschner, I.; Graf, N.; Autschbach, F.; Schneider, D.; von Harrach, M. Clinical relevance of mutations in the Wilms tumor suppressor 1 gene WT1 and the cadherin-associated protein beta1 gene CTNNB1 for patients with Wilms tumors: Results of long-term surveillance of 71 patients from International Society of Pediatric Oncolog. Cancer 2008, 113, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Verschuur, A.C.; Vujanic, G.M.; Van Tinteren, H.; Jones, K.P.; de Kraker, J.; Sandstedt, B. Stromal and epithelial predominant Wilms tumours have an excellent outcome: The SIOP 93 01 experience. Pediatr. Blood Cancer 2010, 55, 233–238. [Google Scholar] [CrossRef]
- Furtwängler, R.; Schmolze, M.; Gräber, S.; Leuschner, I.; Amann, G.; Schenk, J.P.; Niggli, F.; Kager, L.; Schweinitz, D.V.; Graf, N. Pretreatment for bilateral nephroblastomatosis is an independent risk factor for progressive disease in patients with stage V nephroblastoma. Klin. Padiatr. 2014, 226, 175–181. [Google Scholar] [CrossRef]
- Furtwängler, R.; Nourkami, N.; Alkassar, M.; von Schweinitz, D.; Stehr, M.; Graf, N. Sydromes and syndrome-like features in bilateral Wilms Tumor are associated with inferior outcome. Pediatr. Blood Cancer 2010, 55, 885. [Google Scholar]
- Furtwängler, R.; Müller, M.; Nourkami-Tutdibi1, N.; Warmann, S.; Hubertus, J.; Vokuhl, C.; Leuschner, I.; Schenk, J.-P.; Kager, L.; Graf, N. Treatment of Nephroblastomatosis: The GPOH Experience 1993–2014. Pediatr. Blood Cancer 2016, 63, S34. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Frequency | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| All Patients with WT and/or NBL | Only Bilateral WT and/or NBL | Only Screened Patients with CPS/Malformation and WT and/or NBL | |||||||
| All WT | 2927 | - | 100% | 253 | 8.6% | ** | 29 *** | **** | ** |
| WAGR | 20 | ∑ = 171 | 0.7% | 6 | 2.4% | 30.0% | 8 | 27.6% | 40.0% |
| GU | 66 | 2.3% | 8 | 3.2% | 12.1% | 1 | 3.4% | 1.5% | |
| DDS | 24 | 0.8% | 7 | 2.8% | 29.2% | 3 | 10.3% | 12.5% | |
| BWS | 32 | 1.1% | 10 | 4.0% | 31.3% | 10 | 34.5% | 31.3% | |
| IHH | 29 | 1.0% | 4 | 1.6% | 13.8% | 4 | 13.8% | 13.8% | |
| Other * | 27 | - | 0.9% | 7 | 2.8% | 25.9% | 3 | 10.3% | 11.1% |
| All | 198 | - | 6.8% | 42 | 16.6% | 21.2% | 29 | 100% | 14.6% |
| Isolated NBL | WT + NBL | WT Only | Total | |||||
|---|---|---|---|---|---|---|---|---|
| Total | 73 | 2.5% | 64 | 2.2% | 2790 | 95.3% | 2927 | 100% |
| Bilateral disease | 31 | 12.3% | 61 | 24.1% | 161 | 63.6% | 253 | 100% |
| Patients with CPS or GU | 22 * | 12.9% * | 11 | 6.4% | 138 | 80.7% | 171 | 100% |
| Patients without CPS or GU | 51 | 1.9% | 53 | 1.9% | 2652 | 96.2% | 2756 | 100% |
| WAGR | 7 * | 35.0% * | 2 | 10.0% | 11 | 55.0% | 20 | 100% |
| BWS | 7 * | 21.9% * | 3 | 9.4% | 22 | 68.8% | 32 | 100% |
| IHH | 5 * | 17.2% * | 0 | 0.0% | 24 | 82.8% | 29 | 100% |
| DDS | 1 | 4.2% | 2 | 8.3% | 21 | 87.5% | 24 | 100% |
| GU | 2 | 3.0% | 4 | 6.1% | 60 | 90.9% | 66 | 100% |
| Stromal Subtype | Blastemal Subtype after Preoperative Chemotherapy | Other Histological Subtypes | ||||
|---|---|---|---|---|---|---|
| All WT | 270 | 9.2% | 215 | 7.3% | 2442 | 83.4% |
| WAGR | 0 | 0.0% | 1 | 5.0% | 19 | 95.0% |
| GU | 4 | 6.1% | 5 | 7.6% | 57 | 86.3% |
| DDS | 9 * | 37.5% * | 0 | 0.0% | 15 | 62.5% |
| BWS | 0 | 0.0% | 3 | 9.4% | 29 | 90.6% |
| IHH | 0 | 0.0% | 5 * | 17.2% * | 24 | 82.8% |
| Mean Tumor Volume (TV) and [SD] | |||||||
|---|---|---|---|---|---|---|---|
| at Diagnosis | after Preop. Chemo | Volume Reduction | |||||
| Patients without CPS or GU (N = 1698) | 487.5 mL * | [383.0] | 228.0 mL | [279.8] | 259.5 mL | [326.7] | 53.2% |
| Patients with CPS or GU (N = 91) | 349.4 mL * | [381.7] | 189.6 mL | [255.7] | 159.8 mL | [315.3] | 45.7% |
| WAGR (N = 10) | 104.9 mL | [179.0] | 84.4 mL | [156.8] | 20.4 mL | [90.1] | 19.4% |
| GU (N = 32) | 464.0 mL | [329.3] | 254.0 mL | [213.2] | 210.0 mL | [281.2] | 45.3% |
| DDS (N = 15) | 379.3 mL | [256.3] | 380.7 mL | [375.0] | −1.4 mL | [194.8] | −0.4% |
| BWS (N = 17) | 292.9 mL | [539.1] | 38.3 mL | [54.2] | 254.5 mL | [514.3] | 86.9% |
| IHH (N = 17) | 307.7 mL | [416.7] | 112.7 mL | [244.7] | 195.0 mL | [226.7] | 63.4% |
| Patients with CPS or GU undergoing surveillance (N = 11) | 62.7 mL | [112.0] | 55.4 mL | [142.9] | 7.3 mL | [36.9] | 11.6% |
| WT and/or Nephroblastomatosis | CPS Patients | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Factor | Relapse | Death | Relapse | Death | ||||||||
| values | p-Value | Hazard Ratio | EFS (%) | p-Value | Hazard Ratio | OS (%) | p-Value | Hazard Ratio | EFS (%) | p-Value | Hazard Ratio | OS (%) |
| CPS patients | 0.594 | 0.931 | 83.2 | 0.139 | 1.422 | 88.1 | ||||||
| bilaterality | 0.000 | 1.579 * | 73.4 | 0.030 | 1.976 * | 88.2 | 0.003 | 3.013 * | 65.4 | 0.639 | 1.861 | 85.6 |
| nephro- blastomatosis | 0.005 | 1.220 | 72.1 | 0.074 | 0.266 * | 96.2 | 0.032 | 1.264 | 73.3 | 0.167 | 0.225 | 96.7 |
| Prevalence(%) | Median Age at Diagnosis, [SD] (Month) | Gender | Characteristic Histology | Bilaterality (%) | Average Volume Reduction by Preoperative Chemotherapy | 5y-EFS (%), {SE} | Confirmed by | |||
|---|---|---|---|---|---|---|---|---|---|---|
| WAGR | 0.7 | 21.0 * | [9.0] | m < f | NBL ** | 30.0 * | 19.4 % | 87.5 | {0.1} | [9,21,22] |
| GU | 2.3 | 33.0 | [47.3] | m > f * | - | 12.1 | 45.3 % | 87.6 | {0.4} | [23] |
| DDS | 0.8 | 16.0 * | [12.2] | m > f | stromal type | 29.2 * | −0.4 % | 94.7 | {0.5} | [24,25,26,27] |
| BWS | 1.1 | 30.0 | [29.7] | m < f | NBL ** | 31.3 * | 86.9 % | 60.6 * | {0.1} | [28,29] |
| IHH | 1.0 | 43.0 | [34.2] | m < f | NBL **/blastemal type after preop. chemo | 13.8 | 63.4 % | 84.6 | {0.1} | [6,15,30,31] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Welter, N.; Wagner, A.; Furtwängler, R.; Melchior, P.; Kager, L.; Vokuhl, C.; Schenk, J.-P.; Meier, C.M.; Siemer, S.; Gessler, M.; et al. Characteristics of Nephroblastoma/Nephroblastomatosis in Children with a Clinically Reported Underlying Malformation or Cancer Predisposition Syndrome. Cancers 2021, 13, 5016. https://doi.org/10.3390/cancers13195016
Welter N, Wagner A, Furtwängler R, Melchior P, Kager L, Vokuhl C, Schenk J-P, Meier CM, Siemer S, Gessler M, et al. Characteristics of Nephroblastoma/Nephroblastomatosis in Children with a Clinically Reported Underlying Malformation or Cancer Predisposition Syndrome. Cancers. 2021; 13(19):5016. https://doi.org/10.3390/cancers13195016
Chicago/Turabian StyleWelter, Nils, Angelo Wagner, Rhoikos Furtwängler, Patrick Melchior, Leo Kager, Christian Vokuhl, Jens-Peter Schenk, Clemens Magnus Meier, Stefan Siemer, Manfred Gessler, and et al. 2021. "Characteristics of Nephroblastoma/Nephroblastomatosis in Children with a Clinically Reported Underlying Malformation or Cancer Predisposition Syndrome" Cancers 13, no. 19: 5016. https://doi.org/10.3390/cancers13195016