
Poly(ADP)-Ribosylation Inhibition: A Promising Approach for Clear Cell Renal Cell Carcinoma Therapy

 ,
,
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cells and Culture Conditions
2.3. PARP-1 Inhibitory Assay in Human Cell Culture
2.4. Cell Proliferation Assay
2.5. Cell Survival Assay under PARP-1 Inhibitors Treatment
2.6. Measurement of Cell Viability under PARP-1 Inhibitors Treatment
2.7. Assessment of In Vivo Tumor Growth
2.8. Tumor Immunohistochemistry
2.9. Apoptosis Detection in Tumors
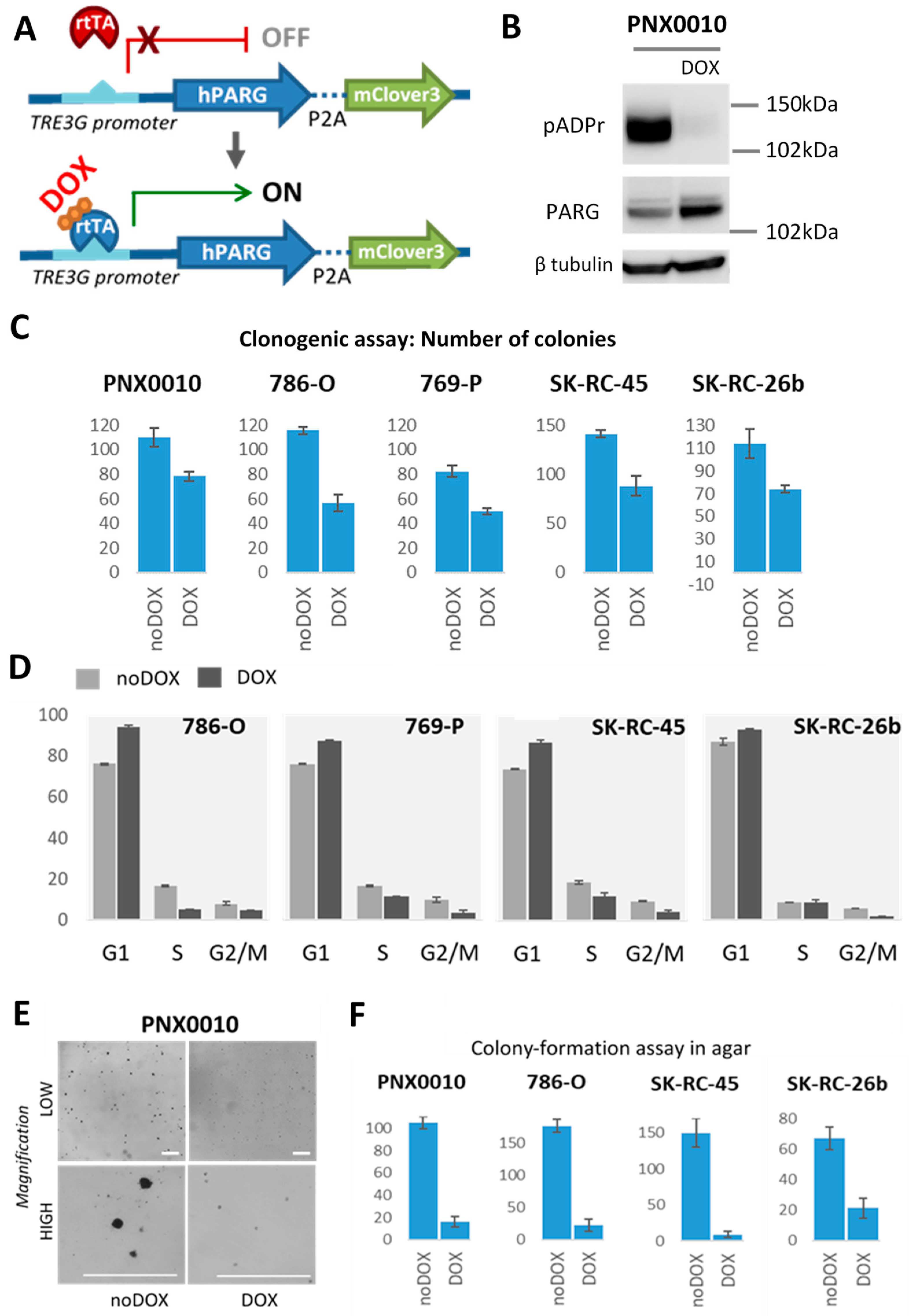
2.10. Generation of Stable ccRCC Cell Lines with Human PARG Overexpression Lentivirus Construct under Doxycycline Inducible Promoter
2.11. Western Blotting
2.12. Cell Cycle Analysis
2.13. Apoptosis Level Evaluation in ccRCC Cells
2.14. Clonogenic Assay
2.15. Colony Formation Assay in Soft Agar
2.16. Immunofluorescence Staining
2.17. RNA-Seq Analysis
2.18. Quantitative PCR (qPCR)
2.19. Statistical Analysis
3. Results
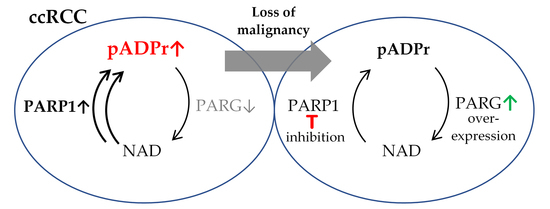
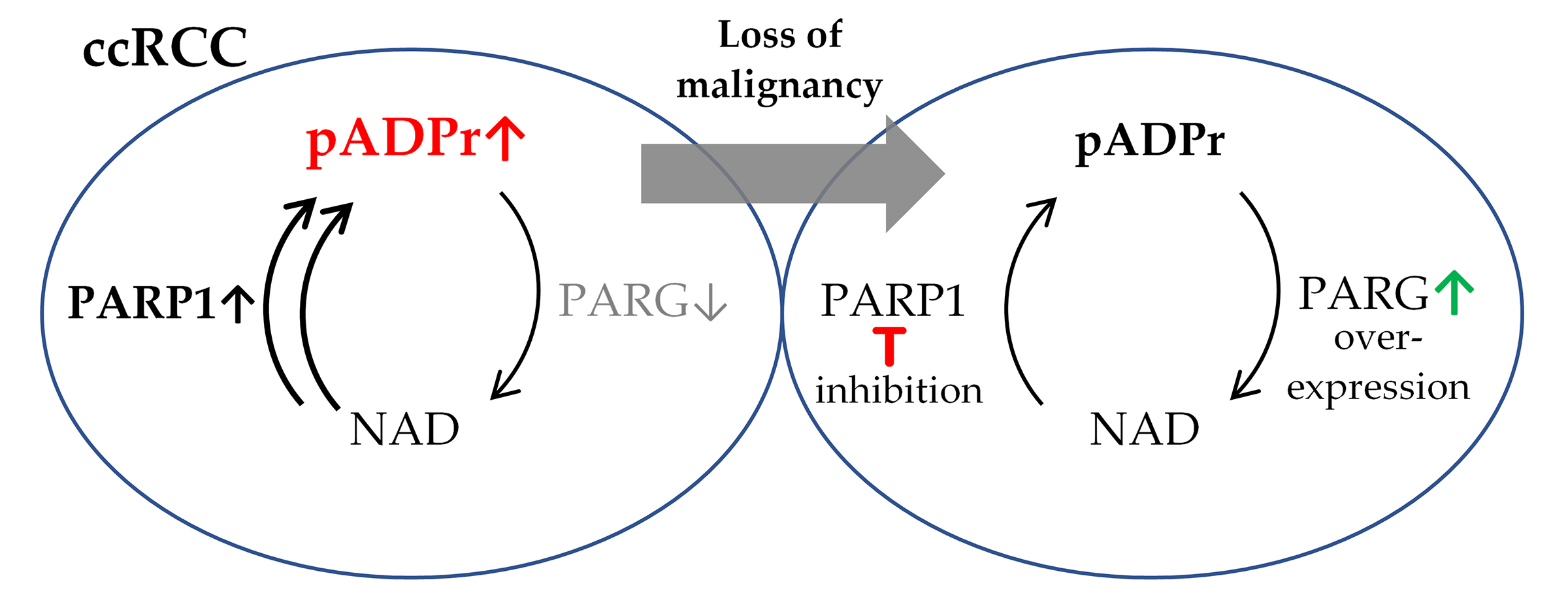
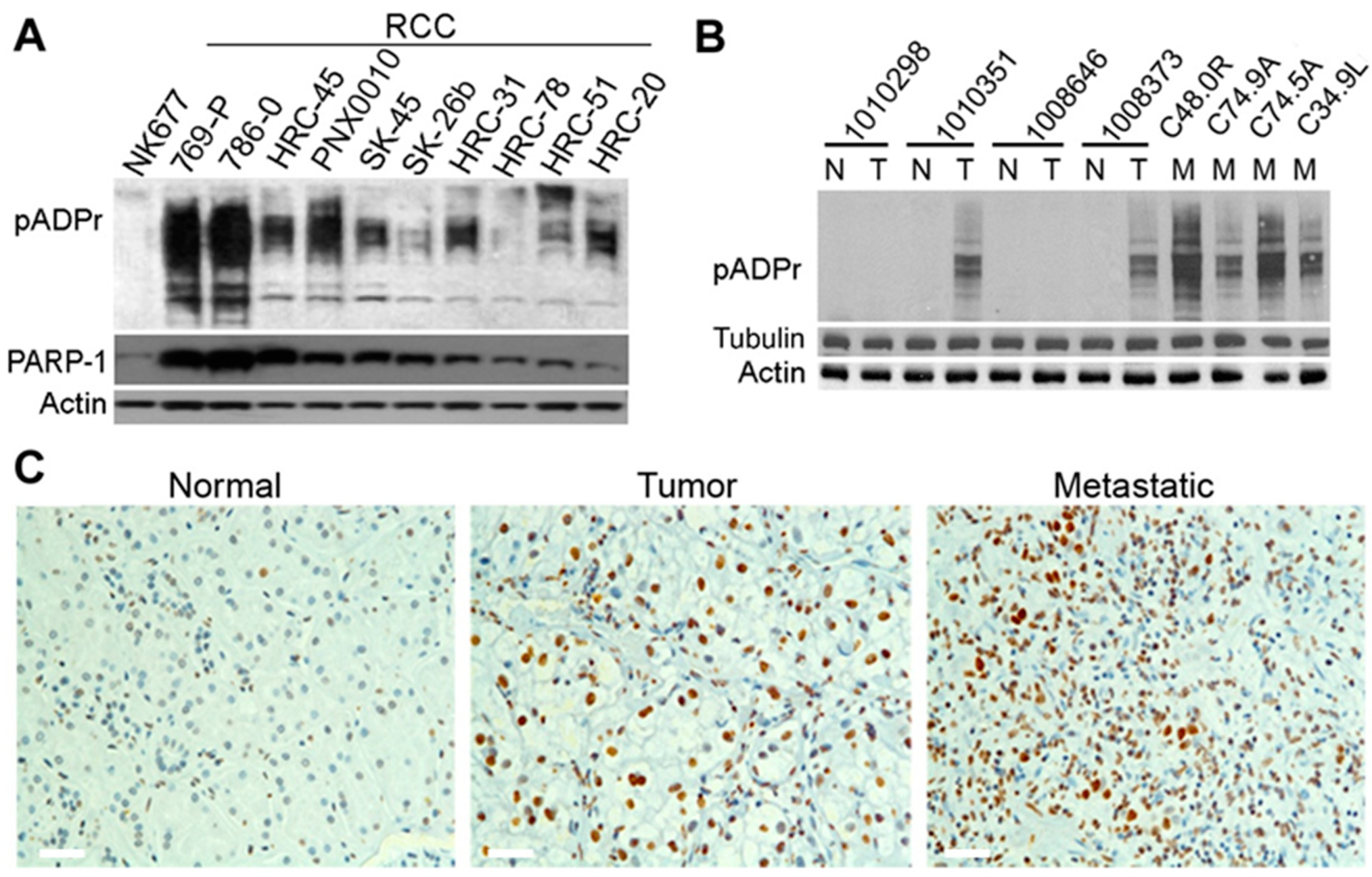
3.1. ccRCC Malignancy Is Associated with Aberrations in the Regulation of pADPr Turnover
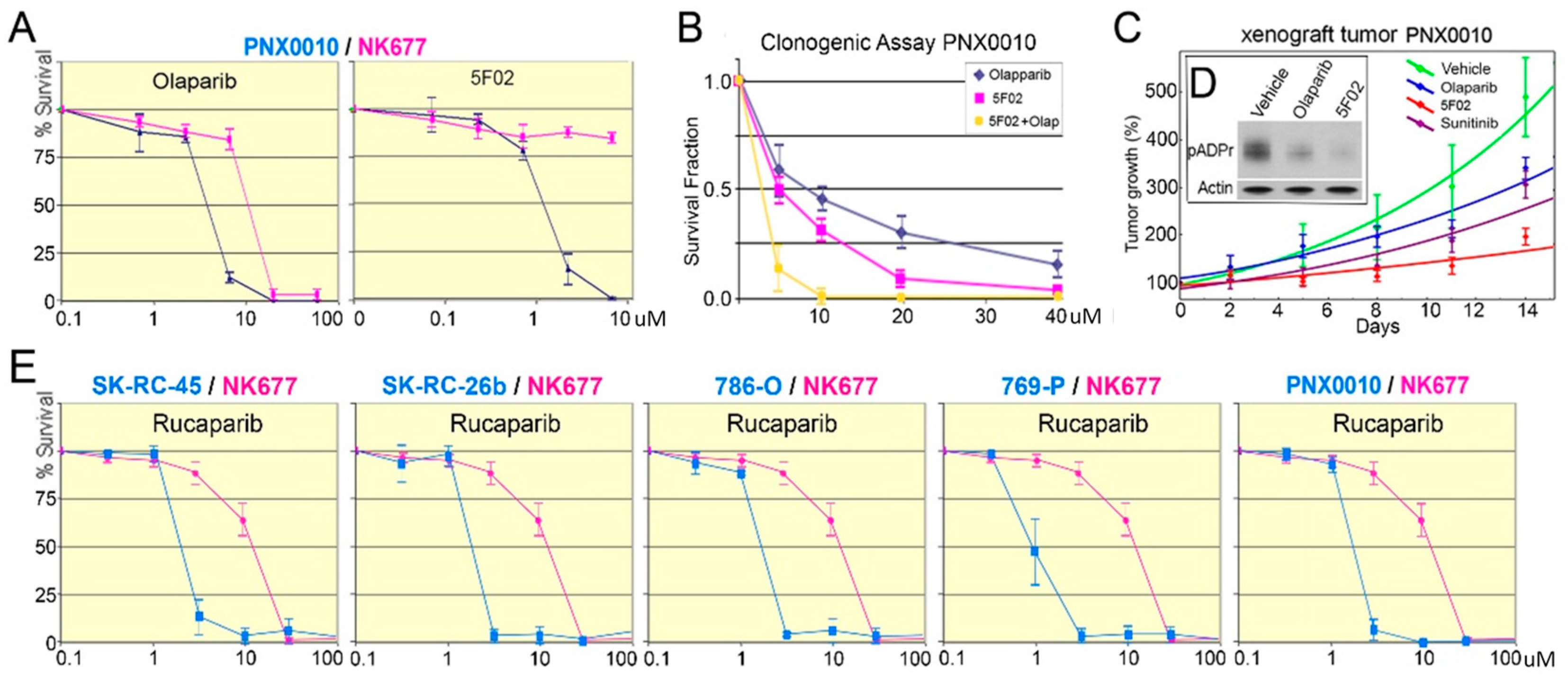
3.2. pADPr Reduction by PARP-1 Inhibitors Suppresses the Growth of ccRCC Tumors In Vitro and In Vivo
3.3. pADPr Reduction by PARG Overexpression Suppresses the Growth of ccRCC Tumors In Vitro
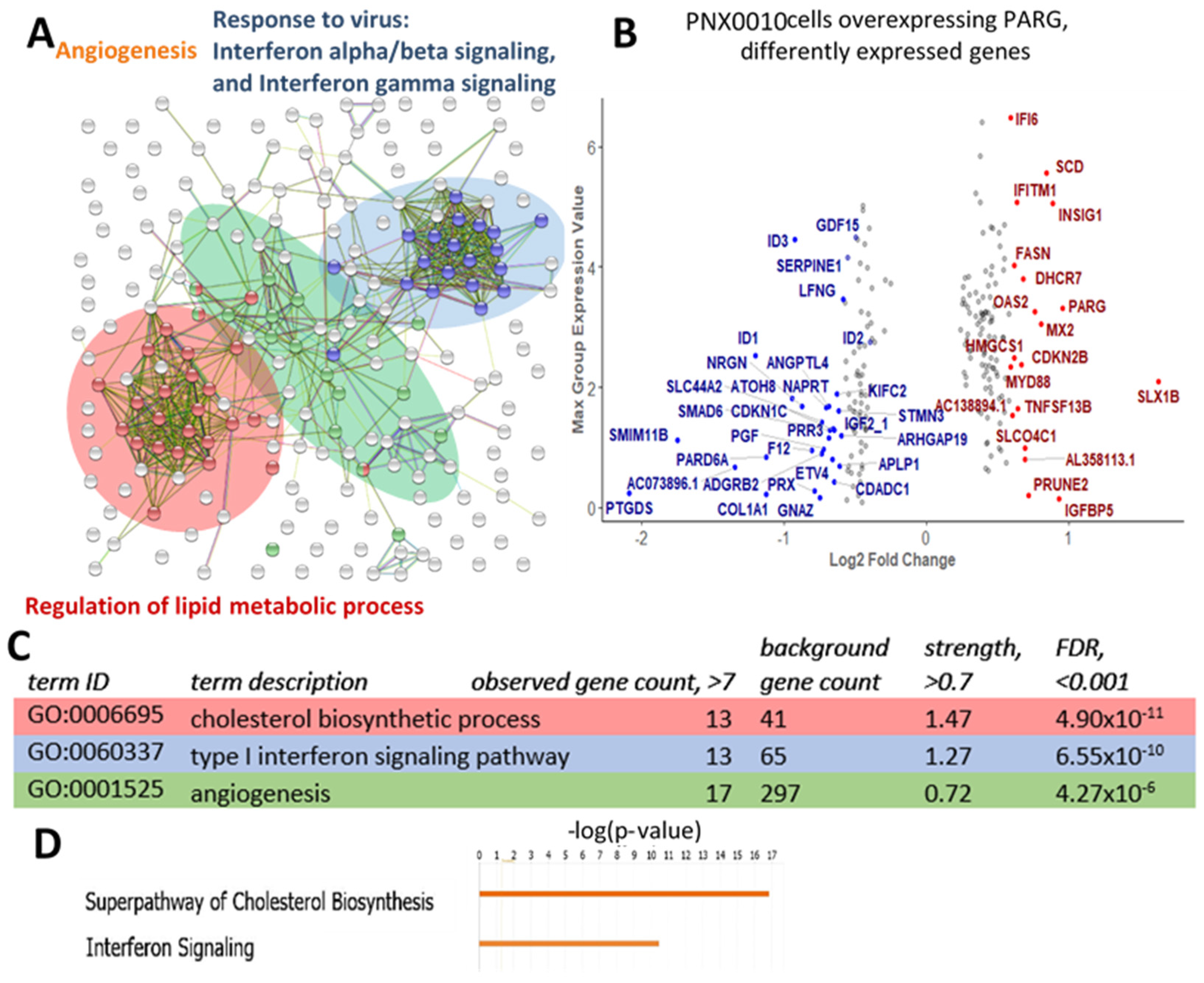
3.4. Lipid/Cholesterol Biosynthesis and Interferon Signaling Pathways Are Up-Regulatedand Angiogenesis and Tumor Growth-Related Genes Are Down-Regulated under PARG Overexpression in PNX0010 ccRCC Cells
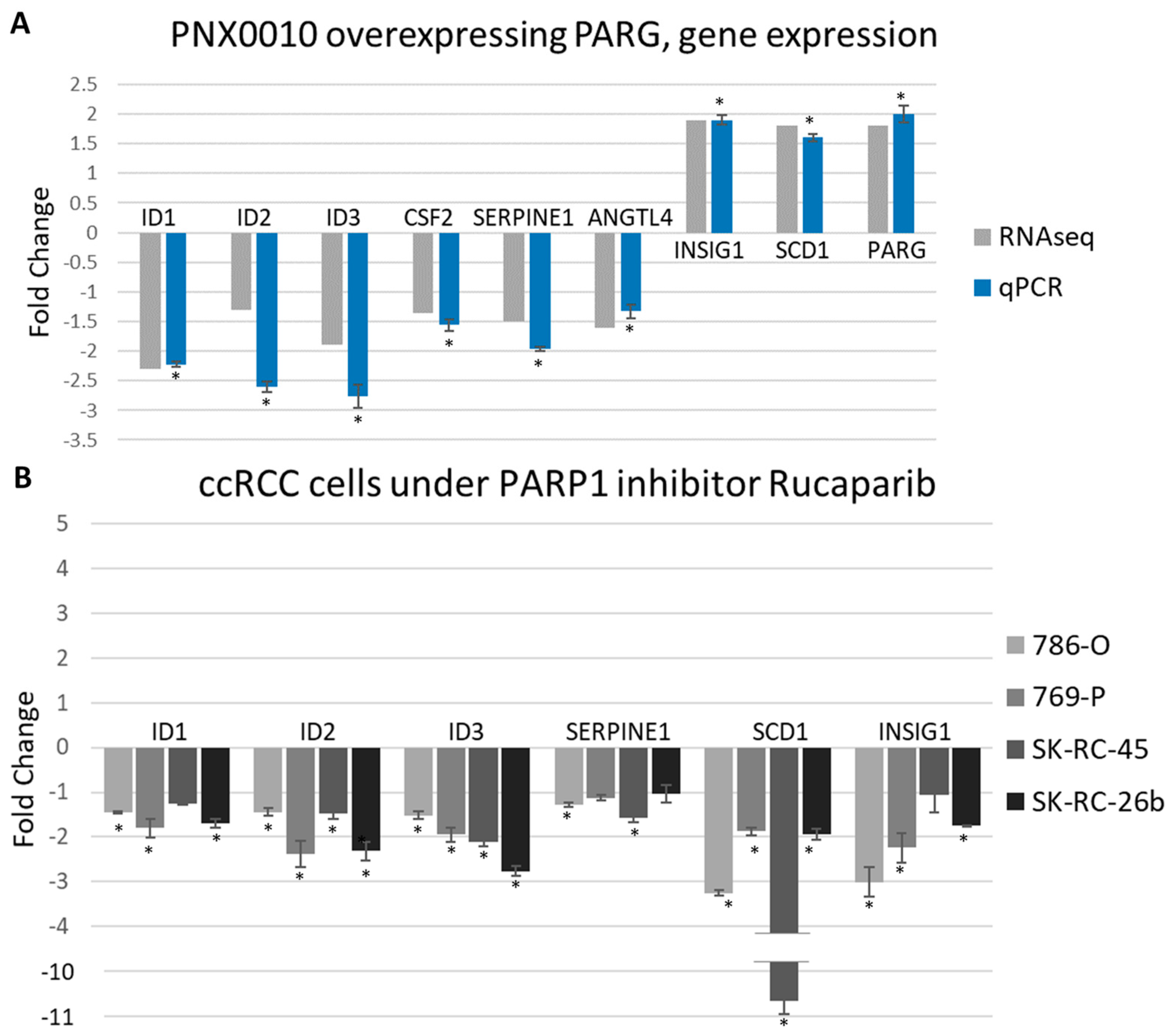
3.5. PARG Overexpression and PARPs Inhibition Similarly Affect Gene Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shuch, B.; Amin, A.; Armstrong, A.J.; Eble, J.N.; Ficarra, V.; Lopez-Beltran, A.; Martignoni, G.; Rini, B.I.; Kutikov, A. Understanding Pathologic Variants of Renal Cell Carcinoma: Distilling Therapeutic Opportunities from Biologic Comple×ity. Eur. Urol. 2015, 67, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Frew, I.J.; Moch, H. A Clearer View of the Molecular Complexity of Clear Cell Renal Cell Carcinoma. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 263–289. [Google Scholar] [CrossRef] [PubMed]
- Gossage, L.; Eisen, T.; Maher, E.R. VHL, the Story of a Tumour Suppressor Gene. Nat. Rev. Cancer 2015, 15, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Hughson, M.D.; He, Z.; Liu, S.; Coleman, J.; Shingleton, W.B. Expression of HIF-1 and Ubiquitin in Conventional Renal Cell Carcinoma: Relationship to Mutations of the von Hippel–Lindau Tumor Suppressor Gene. Cancer Genet. Cytogenet. 2003, 143, 145–153. [Google Scholar] [CrossRef]
- Raval, R.R.; Lau, K.W.; Tran, M.G.B.; Sowter, H.M.; Mandriota, S.J.; Li, J.-L.; Pugh, C.W.; Maxwell, P.H.; Harris, A.L.; Ratcliffe, P.J. Contrasting Properties of Hypoxia-Inducible Factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-Associated Renal Cell Carcinoma. Mol. Cell Biol. 2005, 25, 5675–5686. [Google Scholar] [CrossRef]
- Schödel, J.; Grampp, S.; Maher, E.R.; Moch, H.; Ratcliffe, P.J.; Russo, P.; Mole, D.R. Hypoxia, Hypoxia-Inducible Transcription Factors, and Renal Cancer. Eur. Urol. 2016, 69, 646–657. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.-M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-α to the von Hippel-Lindau Ubiquitylation Complex by O2-Regulated Prolyl Hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.-W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The Tumour Suppressor Protein VHL Targets Hypoxia-Inducible Factors for Oxygen-Dependent Proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Lupo, B.; Trusolino, L. Inhibition of Poly(ADP-Ribosyl)Ation in Cancer: Old and New Paradigms Revisited. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2014, 1846, 201–215. [Google Scholar] [CrossRef]
- Li, N.; Chen, J. ADP-Ribosylation: Activation, Recognition, and Removal. Mol. Cells 2014, 37, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Domagala, P.; Huzarski, T.; Lubinski, J.; Gugala, K.; Domagala, W. PARP-1 Expression in Breast Cancer Including BRCA1-Associated, Triple Negative and Basal-like Tumors: Possible Implications for PARP-1 Inhibitor Therapy. Breast Cancer Res. Treat 2011, 127, 861–869. [Google Scholar] [CrossRef]
- Kirsanov, K.I.; Kotova, E.; Makhov, P.; Golovine, K.; Lesovaya, E.A.; Kolenko, V.M.; Yakubovskaya, M.G.; Tulin, A.V. Minor Grove Binding Ligands Disrupt PARP-1 Activation Pathways. Oncotarget 2014, 5, 428–437. [Google Scholar] [CrossRef]
- Masutani, M.; Nakagama, H.; Sugimura, T. Poly-ADP-Ribosylation in Health and Disease. CMLS Cell. Mol. Life Sci. 2005, 62, 769–783. [Google Scholar] [CrossRef]
- Prasad, S.C.; Thraves, P.J.; Bhatia, K.G.; Smulson, M.E.; Dritschilo, A. Enhanced Poly(Adenosine Diphosphate Ribose) Polymerase Activity and Gene Expression in Ewing’s Sarcoma Cells. Cancer Res 1990, 50, 38–43. [Google Scholar] [PubMed]
- Bieche, I.; Pennaneach, V.; Driouch, K.; Vacher, S.; Zaremba, T.; Susini, A.; Lidereau, R.; Hall, J. Variations in the MRNA Expression of Poly(ADP-Ribose) Polymerases, Poly(ADP-Ribose) Glycohydrolase and ADP-Ribosylhydrolase 3 in Breast Tumors and Impact on Clinical Outcome. Int. J. Cancer 2013, 133, 2791–2800. [Google Scholar] [CrossRef] [PubMed]
- Munshi, A.; Hobbs, M.; Meyn, R.E. Clonogenic Cell Survival Assay. Methods Mol. Med. 2005, 110, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Makhov, P.; Bychkov, I.; Faezov, B.; Deneka, A.; Kudinov, A.; Nicolas, E.; Brebion, R.; Avril, E.; Cai, K.Q.; Kharin, L.V.; et al. Musashi-2 (MSI2) Regulates Epidermal Growth Factor Receptor (EGFR) Expression and Response to EGFR Inhibitors in EGFR-Mutated Non-Small Cell Lung Cancer (NSCLC). Oncogenesis 2021, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Horibata, S.; Vo, T.V.; Subramanian, V.; Thompson, P.R.; Coonrod, S.A. Utilization of the Soft Agar Colony Formation Assay to Identify Inhibitors of Tumorigenicity in Breast Cancer Cells. J. Vis. Exp. 2015, 99, e52727. [Google Scholar] [CrossRef]
- Gottipati, P.; Vischioni, B.; Schultz, N.; Solomons, J.; Bryant, H.E.; Djureinovic, T.; Issaeva, N.; Sleeth, K.; Sharma, R.A.; Helleday, T. Poly(ADP-Ribose) Polymerase Is Hyperactivated in Homologous Recombination–Defective Cells. Cancer Res. 2010, 70, 5389–5398. [Google Scholar] [CrossRef]
- Swindall, A.F.; Stanley, J.A.; Yang, E.S. PARP-1: Friend or Foe of DNA Damage and Repair in Tumorigenesis? Cancers 2013, 5, 943–958. [Google Scholar] [CrossRef]
- Makhov, P.; Naito, S.; Haifler, M.; Kutikov, A.; Boumber, Y.; Uzzo, R.G.; Kolenko, V.M. The Convergent Roles of NF-ΚB and ER Stress in Sunitinib-Mediated Expression of pro-Tumorigenic Cytokines and Refractory Phenotype in Renal Cell Carcinoma. Cell Death Dis. 2018, 9, 374. [Google Scholar] [CrossRef]
- Thomas, H.D.; Calabrese, C.R.; Batey, M.A.; Canan, S.; Hostomsky, Z.; Kyle, S.; Maegley, K.A.; Newell, D.R.; Skalitzky, D.; Wang, L.-Z.; et al. Preclinical Selection of a Novel Poly(ADP-Ribose) Polymerase Inhibitor for Clinical Trial. Mol. Cancer Ther. 2007, 6, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Kotova, E.; Pinnola, A.D.; Tulin, A.V. Small-Molecule Collection and High-Throughput Colorimetric Assay to Identify PARP1 Inhibitors. Methods Mol. Biol. 2011, 780, 491–516. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Ji, Y.; Lodhi, N.; Kotova, E.; Pinnola, A.D.; Golovine, K.; Makhov, P.; Pechenkina, K.; Kolenko, V.; Tulin, A.V. Non-NAD-Like Poly(ADP-Ribose) Polymerase-1 Inhibitors Effectively Eliminate Cancer in Vivo. EBioMedicine 2016, 13, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Karpova, Y.; Wu, C.; Divan, A.; McDonnell, M.E.; Hewlett, E.; Makhov, P.; Gordon, J.; Ye, M.; Reitz, A.B.; Childers, W.E.; et al. Non-NAD-like PARP-1 Inhibitors in Prostate Cancer Treatment. Biochem. Pharmacol. 2019, 167, 149–162. [Google Scholar] [CrossRef]
- Divan, A.; Sibi, M.P.; Tulin, A. Structurally Unique PARP-1 Inhibitors for the Treatment of Prostate Cancer. Pharmacol. Res. Perspect. 2020, 8, e00586. [Google Scholar] [CrossRef]
- Lee, C.-H.; Motzer, R.J. Sunitinib as a Paradigm for Tyrosine Kinase Inhibitor Development for Renal Cell Carcinoma. Urol. Oncol. Semin. Orig. Investig. 2015, 33, 275–279. [Google Scholar] [CrossRef]
- Madison, B.B. Srebp2: A Master Regulator of Sterol and Fatty Acid Synthesis1. J. Lipid. Res. 2016, 57, 333–335. [Google Scholar] [CrossRef]
- Yang, T.; Espenshade, P.J.; Wright, M.E.; Yabe, D.; Gong, Y.; Aebersold, R.; Goldstein, J.L.; Brown, M.S. Crucial Step in Cholesterol Homeostasis: Sterols Promote Binding of SCAP to INSIG-1, a Membrane Protein That Facilitates Retention of SREBPs in ER. Cell 2002, 110, 489–500. [Google Scholar] [CrossRef]
- Li, J.; Takaishi, K.; Cook, W.; McCorkle, S.K.; Unger, R.H. Insig-1 “Brakes” Lipogenesis in Adipocytes and Inhibits Differentiation of Preadipocytes. Proc. Natl. Acad. Sci. USA 2003, 100, 9476–9481. [Google Scholar] [CrossRef]
- Qiu, X.; Gu, Y.; Ni, Y.; Li, Q. ID1 As a Prognostic Biomarker and Promising Drug Target Plays a Pivotal Role in Deterioration of Clear Cell Renal Cell Carcinoma. BioMed Res. Int. 2020, 2020, e2064582. [Google Scholar] [CrossRef]
- Ljungberg, B.; Campbell, S.C.; Cho, H.Y.; Jacqmin, D.; Lee, J.E.; Weikert, S.; Kiemeney, L.A. The Epidemiology of Renal Cell Carcinoma. Eur. Urol. 2011, 60, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.L.; Swisher, E.M.; Kaufmann, S.H. Poly (ADP-Ribose) Polymerase Inhibitors: Recent Advances and Future Development. J. Clin. Oncol. 2015, 33, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Tulin, A. Re-Evaluating PARP1 Inhibitor in Cancer. Nat. Biotechnol. 2011, 29, 1078–1079. [Google Scholar] [CrossRef] [PubMed]
- Nieborowska-Skorska, M.; Maifrede, S.; Ye, M.; Toma, M.; Hewlett, E.; Gordon, J.; Le, B.V.; Sliwinski, T.; Zhao, H.; Piwocka, K.; et al. Non-NAD-like PARP1 Inhibitor Enhanced Synthetic Lethal Effect of NAD-like PARP Inhibitors against BRCA1-Deficient Leukemia. Leuk. Lymphoma 2019, 60, 1098–1101. [Google Scholar] [CrossRef]
- Roschger, C.; Cabrele, C. The Id-Protein Family in Developmental and Cancer-Associated Pathways. Cell Commun. Signal. 2017, 15, 7. [Google Scholar] [CrossRef]
- Ruzinova, M.B.; Benezra, R. Id Proteins in Development, Cell Cycle and Cancer. Trends Cell Biol. 2003, 13, 410–418. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Z.; Xin, D.; Chua, C.W.; Wong, Y.C.; Leung, S.C.L.; Na, Y.; Wang, X. Prognostic Significance of Id-1 and Its Association with EGFR in Renal Cell Cancer. Histopathology 2007, 50, 484–490. [Google Scholar] [CrossRef]
- Lofstedt, T.; Jogi, A.; Sigvardsson, M.; Gradin, K.; Poellinger, L.; Pahlman, S.; Axelson, H. Induction of ID2 Expression by Hypoxia-Inducible Factor-1: A Role in Dedifferentiation of Hypoxic Neuroblastoma Cells. J. Biol. Chem. 2004, 279, 39223–39231. [Google Scholar] [CrossRef]
- Qian, C.-N.; Huang, D.; Wondergem, B.; Teh, B.T. Complexity of Tumor Vasculature in Clear Cell Renal Cell Carcinoma. Cancer 2009, 115, 2282–2289. [Google Scholar] [CrossRef]
- Courtney, K.D.; Bezwada, D.; Mashimo, T.; Pichumani, K.; Vemireddy, V.; Funk, A.M.; Wimberly, J.; McNeil, S.S.; Kapur, P.; Lotan, Y.; et al. Isotope Tracing of Human Clear Cell Renal Cell Carcinomas Demonstrates Suppressed Glucose Oxidation In Vivo. Cell Metab. 2018, 28, 793–800.e2. [Google Scholar] [CrossRef]
- Seizinger, B.R.; Rouleau, G.A.; Ozelius, L.J.; Lane, A.H.; Farmer, G.E.; Lamiell, J.M.; Haines, J.; Yuen, J.W.M.; Collins, D.; Majoor-Krakauer, D.; et al. Von Hippel–Lindau Disease Maps to the Region of Chromosome 3 Associated with Renal Cell Carcinoma. Nature 1988, 332, 268–269. [Google Scholar] [CrossRef]
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification of the von Hippel-Lindau Disease Tumor Suppressor Gene. Science 1993, 260, 1317–1320. [Google Scholar] [CrossRef] [PubMed]
- Kamura, T.; Sato, S.; Iwai, K.; Czyzyk-Krzeska, M.; Conaway, R.C.; Conaway, J.W. Activation of HIF1α Ubiquitination by a Reconstituted von Hippel-Lindau (VHL) Tumor Suppressor Complex. Proc. Natl. Acad. Sci. USA 2000, 97, 10430–10435. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1: Upstream and Downstream of Cancer Metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Simos, G.; Paraskeva, E. Hypoxia-Inducible Factors and the Regulation of Lipid Metabolism. Cells 2019, 8, 214. [Google Scholar] [CrossRef]
- Wu, G.; Wang, Q.; Xu, Y.; Li, J.; Zhang, H.; Qi, G.; Xia, Q. Targeting the Transcription Factor Receptor LXR to Treat Clear Cell Renal Cell Carcinoma: Agonist or Inverse Agonist? Cell Death Dis. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Antolín, A.A.; Mestres, J. Linking Off-Target Kinase Pharmacology to the Differential Cellular Effects Observed among PARP Inhibitors. Oncotarget 2014, 5, 3023–3028. [Google Scholar] [CrossRef] [PubMed]
- Koneru, R.; Hotte, S.J. Role of Cytokine Therapy for Renal Cell Carcinoma in the Era of Targeted Agents. Curr. Oncol. 2009, 16, S40–S44. [Google Scholar] [CrossRef]
- Gore, M.E.; Griffin, C.L.; Hancock, B.; Patel, P.M.; Pyle, L.; Aitchison, M.; James, N.; Oliver, R.T.; Mardiak, J.; Hussain, T.; et al. Interferon Alfa-2a versus Combination Therapy with Interferon Alfa-2a, Interleukin-2, and Fluorouracil in Patients with Untreated Metastatic Renal Cell Carcinoma (MRC RE04/EORTC GU 30012): An Open-Label Randomised Trial. Lancet 2010, 375, 641–648. [Google Scholar] [CrossRef]
- Li, S.; Wei, X.; He, J.; Tian, X.; Yuan, S.; Sun, L. Plasminogen Activator Inhibitor-1 in Cancer Research. Biomed. Pharmacother. 2018, 105, 83–94. [Google Scholar] [CrossRef]
- Zubac, D.P.; Wentzel-Larsen, T.; Seidal, T.; Bostad, L. Type 1 Plasminogen Activator Inhibitor (PAI-1) in Clear Cell Renal Cell Carcinoma (CCRCC) and Its Impact on Angiogenesis, Progression and Patient Survival after Radical Nephrectomy. BMC Urol. 2010, 10, 20. [Google Scholar] [CrossRef]
- Metcalf, D. The CSFs and Cancer. Nat. Rev. Cancer 2010, 10, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Inaba, K.; Inaba, M.; Romani, N.; Aya, H.; Deguchi, M.; Ikehara, S.; Muramatsu, S.; Steinman, R.M. Generation of Large Numbers of Dendritic Cells from Mouse Bone Marrow Cultures Supplemented with Granulocyte/Macrophage Colony-Stimulating Factor. J. Exp. Med. 1992, 176, 1693–1702. [Google Scholar] [CrossRef]
- Serafini, P.; Carbley, R.; Noonan, K.A.; Tan, G.; Bronte, V.; Borrello, I. High-Dose Granulocyte-Macrophage Colony-Stimulating Factor-Producing Vaccines Impair the Immune Response through the Recruitment of Myeloid Suppressor Cells. Cancer Res. 2004, 64, 6337–6343. [Google Scholar] [CrossRef] [PubMed]
- Jinushi, M.; Nakazaki, Y.; Dougan, M.; Carrasco, D.R.; Mihm, M.; Dranoff, G. MFG-E8-Mediated Uptake of Apoptotic Cells by APCs Links the pro- and Antiinflammatory Activities of GM-CSF. J. Clin. Investig. 2007, 117, 1902–1913. [Google Scholar] [CrossRef] [PubMed]
- Filipazzi, P.; Valenti, R.; Huber, V.; Pilla, L.; Canese, P.; Iero, M.; Castelli, C.; Mariani, L.; Parmiani, G.; Rivoltini, L. Identification of a New Subset of Myeloid Suppressor Cells in Peripheral Blood of Melanoma Patients with Modulation by a Granulocyte-Macrophage Colony-Stimulation Factor-Based Antitumor Vaccine. J. Clin. Oncol. 2007, 25, 2546–2553. [Google Scholar] [CrossRef]
- Huang, X.; Hu, P.; Zhang, J. Genomic Analysis of the Prognostic Value of Colony-Stimulating Factors (CSFs) and Colony-Stimulating Factor Receptors (CSFRs) across 24 Solid Cancer Types. Ann. Transl. Med. 2020, 8, 994. [Google Scholar] [CrossRef]
- Gerharz, C.D.; Reinecke, P.; Schneider, E.M.; Schmitz, M.; Gabbert, H.E. Secretion of GM-CSF and M-CSF by Human Renal Cell Carcinomas of Different Histologic Types. Urology 2001, 58, 821–827. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Yang, S.; Tzen, C.-Y.; Lin, C.-C.; Lin, J. Renal Cell Carcinoma Producing Granulocyte Colony-Stimulating Factor. J Formos. Med. Assoc. 2006, 105, 414–417. [Google Scholar] [CrossRef]
- Yuan, J.; Luo, K.; Zhang, L.; Cheville, J.C.; Lou, Z. USP10 Regulates P53 Localization and Stability by Deubiquitinating P53. Cell 2010, 140, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ma, Y.-M.; Zheng, P.-S.; Zhang, P. GDF15 Promotes the Proliferation of Cervical Cancer Cells by Phosphorylating AKT1 and Erk1/2 through the Receptor ErbB2. J. Exp. Clin. Cancer Res. 2018, 37, 80. [Google Scholar] [CrossRef] [PubMed]
- Mimeault, M.; Batra, S.K. Divergent Molecular Mechanisms Underlying the Pleiotropic Functions of Macrophage Inhibitory Cytokine-1 in Cancer. J. Cell Physiol. 2010, 224, 626–635. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karpova, Y.; Guo, D.; Makhov, P.; Haines, A.M.; Markov, D.A.; Kolenko, V.; Tulin, A.V. Poly(ADP)-Ribosylation Inhibition: A Promising Approach for Clear Cell Renal Cell Carcinoma Therapy. Cancers 2021, 13, 4973. https://doi.org/10.3390/cancers13194973
Karpova Y, Guo D, Makhov P, Haines AM, Markov DA, Kolenko V, Tulin AV. Poly(ADP)-Ribosylation Inhibition: A Promising Approach for Clear Cell Renal Cell Carcinoma Therapy. Cancers. 2021; 13(19):4973. https://doi.org/10.3390/cancers13194973
Chicago/Turabian StyleKarpova, Yaroslava, Danping Guo, Peter Makhov, Adam M. Haines, Dmitriy A. Markov, Vladimir Kolenko, and Alexei V. Tulin. 2021. "Poly(ADP)-Ribosylation Inhibition: A Promising Approach for Clear Cell Renal Cell Carcinoma Therapy" Cancers 13, no. 19: 4973. https://doi.org/10.3390/cancers13194973