
The Role of Cancer-Associated Fibroblasts in Cancer Invasion and Metastasis


Abstract
:Simple Summary
Abstract
1. Introduction
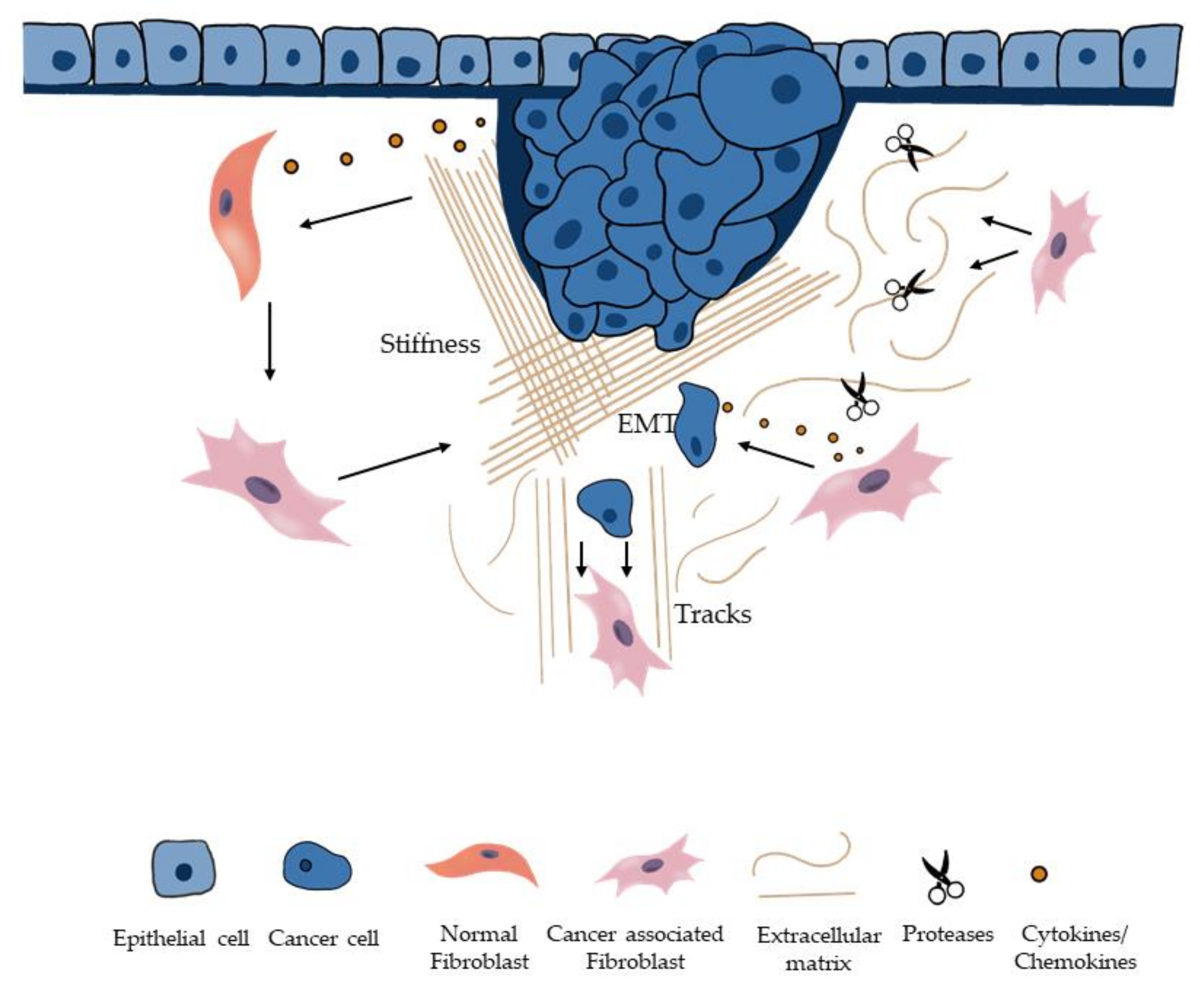
2. The Role of Fibroblasts in the Initiation of Invasion and Metastasis
2.1. CAFs and ECM Remodeling
2.2. CAFs Can Induce Stiffness of ECM
3. The Role of Fibroblasts in EMT and Migration
3.1. CAF-Secreted Factors
3.2. Stiffness in EMT
4. CAF–Tumor Communication
4.1. Communication through Exosomes
4.2. Communication through TGF-β/HGF
4.3. Communication through IL-6/IL-8
4.4. Communication through Other Factors
5. CAF-Targeted Therapies
5.1. CAF Targeting via MMP9
5.2. CAF Targeting via Hedgehog Signaling
5.3. CAF Targeting via TGF-β
5.4. CAF Reprograming
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Retsas, S. Cancer and the arts: Metastasis-as perceived through the ages. ESMO Open 2017, 2, e000226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attieh, Y.; Clark, A.G.; Grass, C.; Richon, S.; Pocard, M.; Mariani, P.; Elkhatib, N.; Betz, T.; Gurchenkov, B.; Vignjevic, D.M. Cancer-associated fibroblasts lead tumor invasion through integrin-beta3-dependent fibronectin assembly. J. Cell Biol. 2017, 216, 3509–3520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Arina, A.; Idel, C.; Hyjek, E.M.; Alegre, M.L.; Wang, Y.; Bindokas, V.P.; Weichselbaum, R.R.; Schreiber, H. Tumor-associated fibroblasts predominantly come from local and not circulating precursors. Proc. Natl. Acad. Sci. USA 2016, 113, 7551–7556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Raz, Y.; Cohen, N.; Shani, O.; Bell, R.E.; Novitskiy, S.V.; Abramovitz, L.; Levy, C.; Milyavsky, M.; Leider-Trejo, L.; Moses, H.L.; et al. Bone marrow-derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J. Exp. Med. 2018, 215, 3075–3093. [Google Scholar] [CrossRef] [Green Version]
- Bochet, L.; Lehuede, C.; Dauvillier, S.; Wang, Y.Y.; Dirat, B.; Laurent, V.; Dray, C.; Guiet, R.; Maridonneau-Parini, I.; Le Gonidec, S.; et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 2013, 73, 5657–5668. [Google Scholar] [CrossRef] [Green Version]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Friedman, G.; Levi-Galibov, O.; David, E.; Bornstein, C.; Giladi, A.; Dadiani, M.; Mayo, A.; Halperin, C.; Pevsner-Fischer, M.; Lavon, H.; et al. Cancer-associated fibroblast compositions change with breast cancer progression linking the ratio of S100A4+ and PDPN+ CAFs to clinical outcome. Nat. Cancer 2020, 1, 692–708. [Google Scholar] [CrossRef]
- Elwakeel, E.; Bruggemann, M.; Fink, A.F.; Schulz, M.H.; Schmid, T.; Savai, R.; Brune, B.; Zarnack, K.; Weigert, A. Phenotypic Plasticity of Fibroblasts during Mammary Carcinoma Development. Int. J. Mol. Sci. 2019, 20, 4438. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Courtois, E.T.; Sengupta, D.; Tan, Y.; Chen, K.H.; Goh, J.J.L.; Kong, S.L.; Chua, C.; Hon, L.K.; Tan, W.S.; et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017, 49, 708–718. [Google Scholar] [CrossRef]
- Kanzaki, R.; Pietras, K. Heterogeneity of cancer-associated fibroblasts: Opportunities for precision medicine. Cancer Sci. 2020, 111, 2708–2717. [Google Scholar] [CrossRef] [PubMed]
- Mhaidly, R.; Mechta-Grigoriou, F. Fibroblast heterogeneity in tumor micro-environment: Role in immunosuppression and new therapies. Semin. Immunol 2020, 48, 101417. [Google Scholar] [CrossRef] [PubMed]
- Nurmik, M.; Ullmann, P.; Rodriguez, F.; Haan, S.; Letellier, E. In search of definitions: Cancer-associated fibroblasts and their markers. Int. J. Cancer 2020, 146, 895–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The extracellular matrix: Tools and insights for the "omics" era. Matrix Biol. 2016, 49, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Eble, J.A.; Niland, S. The extracellular matrix in tumor progression and metastasis. Clin. Exp. Metastasis 2019, 36, 171–198. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, M.; Yu, S.; Hai, L.; Zhang, L.; Zhang, C.; Zhao, P.; Zhou, H.; Wang, S.; Yang, X. Arg kinase mediates CXCL12/CXCR4-induced invadopodia formation and invasion of glioma cells. Exp. Cell Res. 2020, 389, 111893. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Santi, A.; Kugeratski, F.G.; Zanivan, S. Cancer Associated Fibroblasts: The Architects of Stroma Remodeling. Proteomics 2018, 18, e1700167. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.L.; Wang, Z.J.; Wei, G.H.; Yang, Y.; Wang, X.W. Changes in extracellular matrix in different stages of colorectal cancer and their effects on proliferation of cancer cells. World J. Gastrointest Oncol. 2020, 12, 267–275. [Google Scholar] [CrossRef]
- Erdogan, B.; Webb, D.J. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem. Soc. Trans. 2017, 45, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Jia, Z.; Kong, J.; Zhang, F.; Fang, S.; Li, X.; Li, W.; Yang, X.; Luo, Y.; Lin, B.; et al. Carcinoma-Associated Fibroblasts Lead the Invasion of Salivary Gland Adenoid Cystic Carcinoma Cells by Creating an Invasive Track. PLoS ONE 2016, 11, e0150247. [Google Scholar] [CrossRef] [PubMed]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef]
- Neri, S.; Ishii, G.; Hashimoto, H.; Kuwata, T.; Nagai, K.; Date, H.; Ochiai, A. Podoplanin-expressing cancer-associated fibroblasts lead and enhance the local invasion of cancer cells in lung adenocarcinoma. Int. J. Cancer 2015, 137, 784–796. [Google Scholar] [CrossRef] [PubMed]
- Kalli, M.; Stylianopoulos, T. Defining the Role of Solid Stress and Matrix Stiffness in Cancer Cell Proliferation and Metastasis. Front. Oncol. 2018, 8, 55. [Google Scholar] [CrossRef]
- Voutouri, C.; Mpekris, F.; Papageorgis, P.; Odysseos, A.D.; Stylianopoulos, T. Role of constitutive behavior and tumor-host mechanical interactions in the state of stress and growth of solid tumors. PLoS ONE 2014, 9, e104717. [Google Scholar] [CrossRef] [PubMed]
- Kai, F.; Laklai, H.; Weaver, V.M. Force Matters: Biomechanical Regulation of Cell Invasion and Migration in Disease. Trends Cell Biol. 2016, 26, 486–497. [Google Scholar] [CrossRef] [Green Version]
- Acerbi, I.; Cassereau, L.; Dean, I.; Shi, Q.; Au, A.; Park, C.; Chen, Y.Y.; Liphardt, J.; Hwang, E.S.; Weaver, V.M. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr. Biol. 2015, 7, 1120–1134. [Google Scholar] [CrossRef] [Green Version]
- Bauer, J.; Emon, M.A.B.; Staudacher, J.J.; Thomas, A.L.; Zessner-Spitzenberg, J.; Mancinelli, G.; Krett, N.; Saif, M.T.; Jung, B. Increased stiffness of the tumor microenvironment in colon cancer stimulates cancer associated fibroblast-mediated prometastatic activin A signaling. Sci. Rep. 2020, 10, 50. [Google Scholar] [CrossRef]
- Gkretsi, V.; Stylianopoulos, T. Cell Adhesion and Matrix Stiffness: Coordinating Cancer Cell Invasion and Metastasis. Front. Oncol. 2018, 8, 145. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.M.; Park, I.A.; Lee, S.H.; Kim, W.H.; Bae, M.S.; Koo, H.R.; Yi, A.; Kim, S.J.; Cho, N.; Moon, W.K. Stiffness of tumours measured by shear-wave elastography correlated with subtypes of breast cancer. Eur. Radiol. 2013, 23, 2450–2458. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, H.; Sahai, E. Mechanisms and impact of altered tumour mechanics. Nat. Cell Biol. 2018, 20, 766–774. [Google Scholar] [CrossRef]
- Liu, T.; Zhou, L.; Li, D.; Andl, T.; Zhang, Y. Cancer-Associated Fibroblasts Build and Secure the Tumor Microenvironment. Front. Cell Dev. Biol. 2019, 7, 60. [Google Scholar] [CrossRef] [Green Version]
- Pankova, D.; Chen, Y.; Terajima, M.; Schliekelman, M.J.; Baird, B.N.; Fahrenholtz, M.; Sun, L.; Gill, B.J.; Vadakkan, T.J.; Kim, M.P.; et al. Cancer-Associated Fibroblasts Induce a Collagen Cross-link Switch in Tumor Stroma. Mol. Cancer Res. 2016, 14, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.; Garcia-Palmero, I.; Herrera, M.; Bartolome, R.A.; Pena, C.; Fernandez-Acenero, M.J.; Padilla, G.; Pelaez-Garcia, A.; Lopez-Lucendo, M.; Rodriguez-Merlo, R.; et al. LOXL2 Is Highly Expressed in Cancer-Associated Fibroblasts and Associates to Poor Colon Cancer Survival. Clin. Cancer Res. 2015, 21, 4892–4902. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.L.; Yang, M.H.; Tsai, M.L.; Lan, H.Y.; Su, S.H.; Chang, S.C.; Teng, H.W.; Yang, S.H.; Lan, Y.T.; Chiou, S.H.; et al. SNAIL regulates interleukin-8 expression, stem cell-like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology 2011, 141, 279–291. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, S.; Brabletz, T. The ZEB/miR-200 feedback loop--a motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef] [Green Version]
- Xiong, H.; Hong, J.; Du, W.; Lin, Y.W.; Ren, L.L.; Wang, Y.C.; Su, W.Y.; Wang, J.L.; Cui, Y.; Wang, Z.H.; et al. Roles of STAT3 and ZEB1 proteins in E-cadherin down-regulation and human colorectal cancer epithelial-mesenchymal transition. J. Biol. Chem. 2012, 287, 5819–5832. [Google Scholar] [CrossRef] [Green Version]
- Francescangeli, F.; Contavalli, P.; De Angelis, M.L.; Careccia, S.; Signore, M.; Haas, T.L.; Salaris, F.; Baiocchi, M.; Boe, A.; Giuliani, A.; et al. A pre-existing population of ZEB2(+) quiescent cells with stemness and mesenchymal features dictate chemoresistance in colorectal cancer. J. Exp. Clin. Cancer Res. 2020, 39, 2. [Google Scholar] [CrossRef] [Green Version]
- Sreekumar, R.; Harris, S.; Moutasim, K.; DeMateos, R.; Patel, A.; Emo, K.; White, S.; Yagci, T.; Tulchinsky, E.; Thomas, G.; et al. Assessment of Nuclear ZEB2 as a Biomarker for Colorectal Cancer Outcome and TNM Risk Stratification. JAMA Netw. Open 2018, 1, e183115. [Google Scholar] [CrossRef] [Green Version]
- Vu, T.; Datta, P.K. Regulation of EMT in Colorectal Cancer: A Culprit in Metastasis. Cancers 2017, 9, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiori, M.E.; Di Franco, S.; Villanova, L.; Bianca, P.; Stassi, G.; De Maria, R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol. Cancer 2019, 18, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Pohl, M.; Radacz, Y.; Pawlik, N.; Schoeneck, A.; Baldus, S.E.; Munding, J.; Schmiegel, W.; Schwarte-Waldhoff, I.; Reinacher-Schick, A. SMAD4 mediates mesenchymal-epithelial reversion in SW480 colon carcinoma cells. Anticancer Res. 2010, 30, 2603–2613. [Google Scholar] [PubMed]
- Lamouille, S.; Connolly, E.; Smyth, J.W.; Akhurst, R.J.; Derynck, R. TGF-beta-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J. Cell Sci. 2012, 125, 1259–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.C.; Kim, O.H.; Lee, S.K.; Kim, S.J. IWR-1 inhibits epithelial-mesenchymal transition of colorectal cancer cells through suppressing Wnt/beta-catenin signaling as well as survivin expression. Oncotarget 2015, 6, 27146–27159. [Google Scholar] [CrossRef] [Green Version]
- Gujral, T.S.; Chan, M.; Peshkin, L.; Sorger, P.K.; Kirschner, M.W.; MacBeath, G. A noncanonical Frizzled2 pathway regulates epithelial-mesenchymal transition and metastasis. Cell 2014, 159, 844–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenos, K.J.; Miedema, D.M.; Lodestijn, S.C.; Nijman, L.E.; van den Bosch, T.; Romero Ros, X.; Lourenco, F.C.; Lecca, M.C.; van der Heijden, M.; van Neerven, S.M.; et al. Stem cell functionality is microenvironmentally defined during tumour expansion and therapy response in colon cancer. Nat. Cell Biol. 2018, 20, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Wu, X.; Tao, P.; Zhou, Q.; Li, J.; Yu, Z.; Wang, X.; Li, J.; Li, C.; Yan, M.; Zhu, Z.; et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget 2017, 8, 20741–20750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebbing, E.A.; van der Zalm, A.P.; Steins, A.; Creemers, A.; Hermsen, S.; Rentenaar, R.; Klein, M.; Waasdorp, C.; Hooijer, G.K.J.; Meijer, S.L.; et al. Stromal-derived interleukin 6 drives epithelial-to-mesenchymal transition and therapy resistance in esophageal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2019, 116, 2237–2242. [Google Scholar] [CrossRef] [Green Version]
- Goulet, C.R.; Champagne, A.; Bernard, G.; Vandal, D.; Chabaud, S.; Pouliot, F.; Bolduc, S. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of bladder cancer cells through paracrine IL-6 signalling. BMC Cancer 2019, 19, 137. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Zhang, C.; Pan, J.; Chen, L.; Qi, S.T. Interleukin6 induces an epithelialmesenchymal transition phenotype in human adamantinomatous craniopharyngioma cells and promotes tumor cell migration. Mol. Med. Rep. 2017, 15, 4123–4131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kothari, A.N.; Arffa, M.L.; Chang, V.; Blackwell, R.H.; Syn, W.K.; Zhang, J.; Mi, Z.; Kuo, P.C. Osteopontin-A Master Regulator of Epithelial-Mesenchymal Transition. J. Clin. Med. 2016, 5, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, T.H.; Yao, Y.; Yu, S.; Han, L.L.; Wang, W.J.; Guo, H.; Tian, T.; Ruan, Z.P.; Kang, X.M.; Wang, J.; et al. SDF-1/CXCR4 promotes epithelial-mesenchymal transition and progression of colorectal cancer by activation of the Wnt/beta-catenin signaling pathway. Cancer Lett. 2014, 354, 417–426. [Google Scholar] [CrossRef]
- Ping, Y.F.; Yao, X.H.; Jiang, J.Y.; Zhao, L.T.; Yu, S.C.; Jiang, T.; Lin, M.C.; Chen, J.H.; Wang, B.; Zhang, R.; et al. The chemokine CXCL12 and its receptor CXCR4 promote glioma stem cell-mediated VEGF production and tumour angiogenesis via PI3K/AKT signalling. J. Pathol. 2011, 224, 344–354. [Google Scholar] [CrossRef]
- Zhou, W.; Guo, S.; Liu, M.; Burow, M.E.; Wang, G. Targeting CXCL12/CXCR4 Axis in Tumor Immunotherapy. Curr. Med. Chem. 2019, 26, 3026–3041. [Google Scholar] [CrossRef]
- Tang, X.; Tu, G.; Yang, G.; Wang, X.; Kang, L.; Yang, L.; Zeng, H.; Wan, X.; Qiao, Y.; Cui, X.; et al. Autocrine TGF-beta1/miR-200s/miR-221/DNMT3B regulatory loop maintains CAF status to fuel breast cancer cell proliferation. Cancer Lett. 2019, 452, 79–89. [Google Scholar] [CrossRef]
- Khare, T.; Bissonnette, M.; Khare, S. CXCL12-CXCR4/CXCR7 Axis in Colorectal Cancer: Therapeutic Target in Preclinical and Clinical Studies. Int. J. Mol. Sci. 2021, 22, 7371. [Google Scholar] [CrossRef]
- Gong, J.; Lin, Y.; Zhang, H.; Liu, C.; Cheng, Z.; Yang, X.; Zhang, J.; Xiao, Y.; Sang, N.; Qian, X.; et al. Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis. 2020, 11, 267. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Farhood, B.; Mortezaee, K. Extracellular matrix (ECM) stiffness and degradation as cancer drivers. J. Cell Biochem. 2019, 120, 2782–2790. [Google Scholar] [CrossRef]
- Wei, S.C.; Fattet, L.; Tsai, J.H.; Guo, Y.; Pai, V.H.; Majeski, H.E.; Chen, A.C.; Sah, R.L.; Taylor, S.S.; Engler, A.J.; et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat. Cell Biol. 2015, 17, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zheng, Q.; Wang, Z.; Lin, X.; You, Y.; Wu, S.; Wang, Y.; Hu, C.; Xie, X.; Chen, J.; et al. Higher matrix stiffness as an independent initiator triggers epithelial-mesenchymal transition and facilitates HCC metastasis. J. Hematol. Oncol. 2019, 12, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; Del Rio Hernandez, A. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 2017, 6, e352. [Google Scholar] [CrossRef] [Green Version]
- Linares, J.; Marin-Jimenez, J.A.; Badia-Ramentol, J.; Calon, A. Determinants and Functions of CAFs Secretome During Cancer Progression and Therapy. Front. Cell Dev. Biol. 2020, 8, 621070. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Su, Y.; Zhong, S.; Cong, L.; Liu, B.; Yang, J.; Tao, Y.; He, Z.; Chen, C.; Jiang, Y. Exosomes: Key players in cancer and potential therapeutic strategy. Signal Transduct. Target. Ther. 2020, 5, 145. [Google Scholar] [CrossRef] [PubMed]
- Shelke, G.V.; Yin, Y.; Jang, S.C.; Lasser, C.; Wennmalm, S.; Hoffmann, H.J.; Li, L.; Gho, Y.S.; Nilsson, J.A.; Lotvall, J. Endosomal signalling via exosome surface TGFbeta-1. J. Extracell. Vesicles 2019, 8, 1650458. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Sohn, Y.J.; Lee, R.; Yoo, H.J.; Kang, J.Y.; Choi, N.; Na, D.; Yeon, J.H. Cancer-Associated Fibroblasts Differentiated by Exosomes Isolated from Cancer Cells Promote Cancer Cell Invasion. Int. J. Mol. Sci. 2020, 21, 8153. [Google Scholar] [CrossRef]
- Yang, X.; Li, Y.; Zou, L.; Zhu, Z. Role of Exosomes in Crosstalk Between Cancer-Associated Fibroblasts and Cancer Cells. Front. Oncol. 2019, 9, 356. [Google Scholar] [CrossRef]
- Li, W.; Zhang, X.; Wang, J.; Li, M.; Cao, C.; Tan, J.; Ma, D.; Gao, Q. TGFbeta1 in fibroblasts-derived exosomes promotes epithelial-mesenchymal transition of ovarian cancer cells. Oncotarget 2017, 8, 96035–96047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zeng, C.; Zhan, Y.; Wang, H.; Jiang, X.; Li, W. Aberrant low expression of p85alpha in stromal fibroblasts promotes breast cancer cell metastasis through exosome-mediated paracrine Wnt10b. Oncogene 2017, 36, 4692–4705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhome, R.; Goh, R.W.; Bullock, M.D.; Pillar, N.; Thirdborough, S.M.; Mellone, M.; Mirnezami, R.; Galea, D.; Veselkov, K.; Gu, Q.; et al. Exosomal microRNAs derived from colorectal cancer-associated fibroblasts: Role in driving cancer progression. Aging 2017, 9, 2666–2694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.L.; Wang, W.; Lan, X.L.; Zeng, Z.C.; Liang, Y.S.; Yan, Y.R.; Song, F.Y.; Wang, F.F.; Zhu, X.H.; Liao, W.J.; et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol. Cancer 2019, 18, 91. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, S.; Lai, Q.; Fang, Y.; Wu, C.; Liu, Y.; Li, Q.; Wang, X.; Gu, C.; Chen, J.; et al. Cancer-associated fibroblasts-derived exosomal miR-17-5p promotes colorectal cancer aggressive phenotype by initiating a RUNX3/MYC/TGF-beta1 positive feedback loop. Cancer Lett. 2020, 491, 22–35. [Google Scholar] [CrossRef]
- Matise, L.A.; Palmer, T.D.; Ashby, W.J.; Nashabi, A.; Chytil, A.; Aakre, M.; Pickup, M.W.; Gorska, A.E.; Zijlstra, A.; Moses, H.L. Lack of transforming growth factor-beta signaling promotes collective cancer cell invasion through tumor-stromal crosstalk. Breast Cancer Res. 2012, 14, R98. [Google Scholar] [CrossRef] [Green Version]
- Calon, A.; Tauriello, D.V.; Batlle, E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin. Cancer Biol. 2014, 25, 15–22. [Google Scholar] [CrossRef]
- Hawinkels, L.J.; Paauwe, M.; Verspaget, H.W.; Wiercinska, E.; van der Zon, J.M.; van der Ploeg, K.; Koelink, P.J.; Lindeman, J.H.; Mesker, W.; ten Dijke, P.; et al. Interaction with colon cancer cells hyperactivates TGF-beta signaling in cancer-associated fibroblasts. Oncogene 2014, 33, 97–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuyuhiro, Y.; Yashiro, M.; Noda, S.; Matsuoka, J.; Hasegawa, T.; Kato, Y.; Sawada, T.; Hirakawa, K. Cancer-associated orthotopic myofibroblasts stimulates the motility of gastric carcinoma cells. Cancer Sci. 2012, 103, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xiao, C.H.; Tan, L.D.; Wang, Q.S.; Li, X.Q.; Feng, Y.M. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-beta signalling. Br. J. Cancer 2014, 110, 724–732. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, J.; Lu, Q.; Shen, B.; Huang, X.; Shen, L.; Zheng, X.; Huang, R.; Yan, J.; Guo, H. TGFbeta1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci. Rep. 2015, 5, 11924. [Google Scholar] [CrossRef] [Green Version]
- Oyanagi, J.; Kojima, N.; Sato, H.; Higashi, S.; Kikuchi, K.; Sakai, K.; Matsumoto, K.; Miyazaki, K. Inhibition of transforming growth factor-beta signaling potentiates tumor cell invasion into collagen matrix induced by fibroblast-derived hepatocyte growth factor. Exp. Cell Res. 2014, 326, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Chytil, A.; Shyr, Y.; Joly, A.; Moses, H.L. Transforming growth factor-beta signaling-deficient fibroblasts enhance hepatocyte growth factor signaling in mammary carcinoma cells to promote scattering and invasion. Mol. Cancer Res. 2008, 6, 1521–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grugan, K.D.; Miller, C.G.; Yao, Y.; Michaylira, C.Z.; Ohashi, S.; Klein-Szanto, A.J.; Diehl, J.A.; Herlyn, M.; Han, M.; Nakagawa, H.; et al. Fibroblast-secreted hepatocyte growth factor plays a functional role in esophageal squamous cell carcinoma invasion. Proc. Natl. Acad. Sci. USA 2010, 107, 11026–11031. [Google Scholar] [CrossRef] [Green Version]
- Owusu, B.Y.; Galemmo, R.; Janetka, J.; Klampfer, L. Hepatocyte Growth Factor, a Key Tumor-Promoting Factor in the Tumor Microenvironment. Cancers 2017, 9, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Su, X.; Li, Z.; Deng, L.; Liu, X.; Feng, X.; Peng, J. HGF/c-MET pathway in cancer: From molecular characterization to clinical evidence. Oncogene 2021, 40, 4625–4651. [Google Scholar] [CrossRef]
- Spina, A.; De Pasquale, V.; Cerulo, G.; Cocchiaro, P.; Della Morte, R.; Avallone, L.; Pavone, L.M. HGF/c-MET Axis in Tumor Microenvironment and Metastasis Formation. Biomedicines 2015, 3, 71–88. [Google Scholar] [CrossRef] [Green Version]
- Jedeszko, C.; Victor, B.C.; Podgorski, I.; Sloane, B.F. Fibroblast hepatocyte growth factor promotes invasion of human mammary ductal carcinoma in situ. Cancer Res. 2009, 69, 9148–9155. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Ji, J.; Jiang, J.; Cai, Q.; Wang, C.; Shi, M.; Yu, Y.; Zhu, Z.; Zhang, J. HGF-mediated crosstalk between cancer-associated fibroblasts and MET-unamplified gastric cancer cells activates coordinated tumorigenesis and metastasis. Cell Death Dis. 2018, 9, 867. [Google Scholar] [CrossRef]
- Joosten, S.P.J.; Spaargaren, M.; Clevers, H.; Pals, S.T. Hepatocyte growth factor/MET and CD44 in colorectal cancer: Partners in tumorigenesis and therapy resistance. Biochim. Biophys. Acta. Rev. Cancer 2020, 1874, 188437. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; De Sousa, E.M.F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef]
- Karakasheva, T.A.; Lin, E.W.; Tang, Q.; Qiao, E.; Waldron, T.J.; Soni, M.; Klein-Szanto, A.J.; Sahu, V.; Basu, D.; Ohashi, S.; et al. IL-6 Mediates Cross-Talk between Tumor Cells and Activated Fibroblasts in the Tumor Microenvironment. Cancer Res. 2018, 78, 4957–4970. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Cao, L.; Wang, H.; Liu, B.; Zhang, Q.; Meng, Z.; Wu, X.; Zhou, Q.; Xu, K. Cancer-associated fibroblasts enhance metastatic potential of lung cancer cells through IL-6/STAT3 signaling pathway. Oncotarget 2017, 8, 76116–76128. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Yan, M.; Wang, X.; Xu, Q.; Wang, X.; Zhu, X.; Shi, J.; Li, Z.; Zhang, J.; Chen, W. Cancer-associated Fibroblast-derived IL-6 Promotes Head and Neck Cancer Progression via the Osteopontin-NF-kappa B Signaling Pathway. Theranostics 2018, 8, 921–940. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Tian, W.; Gao, W.; Zang, R.; Wang, H.; Yang, G. Cancer-Associated Fibroblast-Derived Interleukin-8 Promotes Ovarian Cancer Cell Stemness and Malignancy Through the Notch3-Mediated Signaling. Front. Cell Dev. Biol. 2021, 9, 684505. [Google Scholar] [CrossRef]
- Wang, T.; Notta, F.; Navab, R.; Joseph, J.; Ibrahimov, E.; Xu, J.; Zhu, C.Q.; Borgida, A.; Gallinger, S.; Tsao, M.S. Senescent Carcinoma-Associated Fibroblasts Upregulate IL8 to Enhance Prometastatic Phenotypes. Mol. Cancer Res. 2017, 15, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Jobe, N.P.; Rosel, D.; Dvorankova, B.; Kodet, O.; Lacina, L.; Mateu, R.; Smetana, K.; Brabek, J. Simultaneous blocking of IL-6 and IL-8 is sufficient to fully inhibit CAF-induced human melanoma cell invasiveness. Histochem. Cell Biol. 2016, 146, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Tang, Z.H.; Liu, S.; Guo, S.S. Clinicopathological significance of overexpression of interleukin-6 in colorectal cancer. World J. Gastroenterol. 2017, 23, 1780–1786. [Google Scholar] [CrossRef]
- Sun, Q.; Shang, Y.; Sun, F.; Dong, X.; Niu, J.; Li, F. Interleukin-6 Promotes Epithelial-Mesenchymal Transition and Cell Invasion through Integrin beta6 Upregulation in Colorectal Cancer. Oxid. Med. Cell Longev. 2020, 2020, 8032187. [Google Scholar] [CrossRef] [PubMed]
- Angevin, E.; Tabernero, J.; Elez, E.; Cohen, S.J.; Bahleda, R.; van Laethem, J.L.; Ottensmeier, C.; Lopez-Martin, J.A.; Clive, S.; Joly, F.; et al. A phase I/II, multiple-dose, dose-escalation study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 2192–2204. [Google Scholar] [CrossRef] [Green Version]
- Brambilla, L.; Genini, D.; Laurini, E.; Merulla, J.; Perez, L.; Fermeglia, M.; Carbone, G.M.; Pricl, S.; Catapano, C.V. Hitting the right spot: Mechanism of action of OPB-31121, a novel and potent inhibitor of the Signal Transducer and Activator of Transcription 3 (STAT3). Mol. Oncol. 2015, 9, 1194–1206. [Google Scholar] [CrossRef] [Green Version]
- Fogelman, D.; Cubillo, A.; Garcia-Alfonso, P.; Miron, M.L.L.; Nemunaitis, J.; Flora, D.; Borg, C.; Mineur, L.; Vieitez, J.M.; Cohn, A.; et al. Randomized, double-blind, phase two study of ruxolitinib plus regorafenib in patients with relapsed/refractory metastatic colorectal cancer. Cancer Med. 2018, 7, 5382–5393. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; He, Z.; Ye, J.; Liu, Z.; She, X.; Gao, X.; Liang, R. Progress in Understanding the IL-6/STAT3 Pathway in Colorectal Cancer. Onco Targets Ther. 2020, 13, 13023–13032. [Google Scholar] [CrossRef]
- Lee, Y.S.; Choi, I.; Ning, Y.; Kim, N.Y.; Khatchadourian, V.; Yang, D.; Chung, H.K.; Choi, D.; LaBonte, M.J.; Ladner, R.D.; et al. Interleukin-8 and its receptor CXCR2 in the tumour microenvironment promote colon cancer growth, progression and metastasis. Br. J. Cancer 2012, 106, 1833–1841. [Google Scholar] [CrossRef]
- Najdaghi, S.; Razi, S.; Rezaei, N. An overview of the role of interleukin-8 in colorectal cancer. Cytokine 2020, 135, 155205. [Google Scholar] [CrossRef]
- Siena, S.; Sartore-Bianchi, A.; Marsoni, S.; Hurwitz, H.I.; McCall, S.J.; Penault-Llorca, F.; Srock, S.; Bardelli, A.; Trusolino, L. Targeting the human epidermal growth factor receptor 2 (HER2) oncogene in colorectal cancer. Ann. Oncol. 2018, 29, 1108–1119. [Google Scholar] [CrossRef]
- Varney, M.L.; Singh, S.; Li, A.; Mayer-Ezell, R.; Bond, R.; Singh, R.K. Small molecule antagonists for CXCR2 and CXCR1 inhibit human colon cancer liver metastases. Cancer Lett. 2011, 300, 180–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, S.; Hou, Y.; Fu, L.; Xi, L.; Yang, D.; Zhao, M.; Qin, Y.; Sun, K.; Teng, Y.; Liu, M. Cancer-associated fibroblast (CAF)-derived IL32 promotes breast cancer cell invasion and metastasis via integrin beta3-p38 MAPK signalling. Cancer Lett. 2019, 442, 320–332. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhou, Q.; Wu, X.; Xu, S.; Hu, X.; Tao, X.; Li, B.; Peng, J.; Li, D.; Shen, L.; et al. VCAM-1 secreted from cancer-associated fibroblasts enhances the growth and invasion of lung cancer cells through AKT and MAPK signaling. Cancer Lett. 2020, 473, 62–73. [Google Scholar] [CrossRef]
- Aizawa, T.; Karasawa, H.; Funayama, R.; Shirota, M.; Suzuki, T.; Maeda, S.; Suzuki, H.; Yamamura, A.; Naitoh, T.; Nakayama, K.; et al. Cancer-associated fibroblasts secrete Wnt2 to promote cancer progression in colorectal cancer. Cancer Med. 2019, 8, 6370–6382. [Google Scholar] [CrossRef] [Green Version]
- Kramer, N.; Schmollerl, J.; Unger, C.; Nivarthi, H.; Rudisch, A.; Unterleuthner, D.; Scherzer, M.; Riedl, A.; Artaker, M.; Crncec, I.; et al. Autocrine WNT2 signaling in fibroblasts promotes colorectal cancer progression. Oncogene 2017, 36, 5460–5472. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.; Iglesias, M.; Cespedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.; et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Qi, F.; Zhao, F.; Li, G.; Shao, S.; Zhang, X.; Yuan, L.; Feng, Y. Cancer-associated fibroblasts enhance tumor-associated macrophages enrichment and suppress NK cells function in colorectal cancer. Cell Death Dis. 2019, 10, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comito, G.; Giannoni, E.; Segura, C.P.; Barcellos-de-Souza, P.; Raspollini, M.R.; Baroni, G.; Lanciotti, M.; Serni, S.; Chiarugi, P. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene 2014, 33, 2423–2431. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Sakakura, K.; Kudo, T.; Toyoda, M.; Kaira, K.; Oyama, T.; Chikamatsu, K. Cancer-associated fibroblasts promote an immunosuppressive microenvironment through the induction and accumulation of protumoral macrophages. Oncotarget 2017, 8, 8633–8647. [Google Scholar] [CrossRef] [Green Version]
- Ziani, L.; Safta-Saadoun, T.B.; Gourbeix, J.; Cavalcanti, A.; Robert, C.; Favre, G.; Chouaib, S.; Thiery, J. Melanoma-associated fibroblasts decrease tumor cell susceptibility to NK cell-mediated killing through matrix-metalloproteinases secretion. Oncotarget 2017, 8, 19780–19794. [Google Scholar] [CrossRef] [Green Version]
- Ziani, L.; Chouaib, S.; Thiery, J. Alteration of the Antitumor Immune Response by Cancer-Associated Fibroblasts. Front. Immunol. 2018, 9, 414. [Google Scholar] [CrossRef]
- Marshall, D.C.; Lyman, S.K.; McCauley, S.; Kovalenko, M.; Spangler, R.; Liu, C.; Lee, M.; O’Sullivan, C.; Barry-Hamilton, V.; Ghermazien, H.; et al. Selective Allosteric Inhibition of MMP9 Is Efficacious in Preclinical Models of Ulcerative Colitis and Colorectal Cancer. PLoS ONE 2015, 10, e0127063. [Google Scholar] [CrossRef] [Green Version]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [Green Version]
- Riaz, S.K.; Ke, Y.; Wang, F.; Kayani, M.A.; Malik, M.F.A. Influence of SHH/GLI1 axis on EMT mediated migration and invasion of breast cancer cells. Sci. Rep. 2019, 9, 6620. [Google Scholar] [CrossRef]
- Mazzocca, A.; Fransvea, E.; Dituri, F.; Lupo, L.; Antonaci, S.; Giannelli, G. Down-regulation of connective tissue growth factor by inhibition of transforming growth factor beta blocks the tumor-stroma cross-talk and tumor progression in hepatocellular carcinoma. Hepatology 2010, 51, 523–534. [Google Scholar] [CrossRef]
- Zhang, Q.; Hou, X.; Evans, B.J.; VanBlaricom, J.L.; Weroha, S.J.; Cliby, W.A. LY2157299 Monohydrate, a TGF-betaR1 Inhibitor, Suppresses Tumor Growth and Ascites Development in Ovarian Cancer. Cancers 2018, 10, 260. [Google Scholar] [CrossRef] [Green Version]
- Mardhian, D.F.; Storm, G.; Bansal, R.; Prakash, J. Nano-targeted relaxin impairs fibrosis and tumor growth in pancreatic cancer and improves the efficacy of gemcitabine in vivo. J. Control. Release 2018, 290, 1–10. [Google Scholar] [CrossRef]
- Yao, Y.; Guo, Q.; Cao, Y.; Qiu, Y.; Tan, R.; Yu, Z.; Zhou, Y.; Lu, N. Artemisinin derivatives inactivate cancer-associated fibroblasts through suppressing TGF-beta signaling in breast cancer. J. Exp. Clin. Cancer Res. 2018, 37, 282. [Google Scholar] [CrossRef]
- Froeling, F.E.; Feig, C.; Chelala, C.; Dobson, R.; Mein, C.E.; Tuveson, D.A.; Clevers, H.; Hart, I.R.; Kocher, H.M. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-beta-catenin signaling to slow tumor progression. Gastroenterology 2011, 141, 1486–1497, 1497.e1–1497.e14. [Google Scholar] [CrossRef]
- Han, X.; Li, Y.; Xu, Y.; Zhao, X.; Zhang, Y.; Yang, X.; Wang, Y.; Zhao, R.; Anderson, G.J.; Zhao, Y.; et al. Reversal of pancreatic desmoplasia by re-educating stellate cells with a tumour microenvironment-activated nanosystem. Nat. Commun. 2018, 9, 3390. [Google Scholar] [CrossRef] [Green Version]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Juric, V.; O’Sullivan, C.; Stefanutti, E.; Kovalenko, M.; Greenstein, A.; Barry-Hamilton, V.; Mikaelian, I.; Degenhardt, J.; Yue, P.; Smith, V.; et al. MMP-9 inhibition promotes anti-tumor immunity through disruption of biochemical and physical barriers to T-cell trafficking to tumors. PLoS ONE 2018, 13, e0207255. [Google Scholar] [CrossRef] [Green Version]
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef]
- Butler, G.S.; Overall, C.M. Updated biological roles for matrix metalloproteinases and new "intracellular" substrates revealed by degradomics. Biochemistry 2009, 48, 10830–10845. [Google Scholar] [CrossRef]
- Fields, G.B. The Rebirth of Matrix Metalloproteinase Inhibitors: Moving Beyond the Dogma. Cells 2019, 8, 984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadler-Olsen, E.; Winberg, J.O.; Uhlin-Hansen, L. Matrix metalloproteinases in cancer: Their value as diagnostic and prognostic markers and therapeutic targets. Tumour Biol. 2013, 34, 2041–2051. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.T.; Zhao, W.D.; He, W.; Wei, W. Hedgehog signaling pathway mediates invasion and metastasis of hepatocellular carcinoma via ERK pathway. Acta. Pharmacol. Sin. 2012, 33, 691–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele, N.G.; Biffi, G.; Kemp, S.B.; Zhang, Y.; Drouillard, D.; Syu, L.; Hao, Y.; Oni, T.E.; Brosnan, E.; Elyada, E.; et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 2023–2037. [Google Scholar] [CrossRef]
- Valenti, G.; Quinn, H.M.; Heynen, G.; Lan, L.; Holland, J.D.; Vogel, R.; Wulf-Goldenberg, A.; Birchmeier, W. Cancer Stem Cells Regulate Cancer-Associated Fibroblasts via Activation of Hedgehog Signaling in Mammary Gland Tumors. Cancer Res. 2017, 77, 2134–2147. [Google Scholar] [CrossRef] [Green Version]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Szczepny, A.; Rogers, S.; Jayasekara, W.S.N.; Park, K.; McCloy, R.A.; Cochrane, C.R.; Ganju, V.; Cooper, W.A.; Sage, J.; Peacock, C.D.; et al. The role of canonical and non-canonical Hedgehog signaling in tumor progression in a mouse model of small cell lung cancer. Oncogene 2017, 36, 5544–5550. [Google Scholar] [CrossRef] [PubMed]
- Gu, D.; Schlotman, K.E.; Xie, J. Deciphering the role of hedgehog signaling in pancreatic cancer. J. Biomed. Res. 2016, 30, 353–360. [Google Scholar] [CrossRef] [Green Version]
- Rajurkar, M.; De Jesus-Monge, W.E.; Driscoll, D.R.; Appleman, V.A.; Huang, H.; Cotton, J.L.; Klimstra, D.S.; Zhu, L.J.; Simin, K.; Xu, L.; et al. The activity of Gli transcription factors is essential for Kras-induced pancreatic tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, E1038–E1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Young, C.D.; Zhou, H.; Wang, X. Transforming Growth Factor-beta Signaling in Fibrotic Diseases and Cancer-Associated Fibroblasts. Biomolecules 2020, 10, 1666. [Google Scholar] [CrossRef]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]


{kind=link}
| Pathway | Compound | Targets | Cancer | Mechanism | Ref. |
|---|---|---|---|---|---|
| MMP9 | GS-5745 | MMP9 | Colorectal | Inhibition of tumor growth and metastasis | [123] |
| Hedgehog | Smo | Pancreatic | Decrease in fibroblast accumulation and easier drug delivery | [124] | |
| Shh/Gli pathway | Breast | Reduction in cell migration and invasion | [125] | ||
| TGF-β | LY2109761 | TGF-β receptor1 | Hepatic | Reduction in the stromal compartment | [126] |
| Galunisertib | Ovarian | Prevention of CAF activation and reduction in proliferation and invasion | [127] | ||
| Realaxin-2 | Smad2 pathway | Pancreatic | Decrease in tumor growth and increase in sensitivity of gemcitabine | [128] | |
| Artesiminin | TGF-β | Breast | Inactivation of CAFs and inhibition of cancer growth and metastasis | [129] | |
| Reprograming | ATRA | HSP47 | Pancreatic | Reprograming of PSCs, reduction in Wnt signaling, and modulation of proliferation, apoptosis, and invasion | [130] |
| Nano-system (ATRA+ anti HSP47) | Reprograming of PSCs, reduction in ECM production, and increase in chemotherapeutic effects | [131] | |||
| VDR ligand | Induction of quiescent PSCs from activated PSCs | [132] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asif, P.J.; Longobardi, C.; Hahne, M.; Medema, J.P. The Role of Cancer-Associated Fibroblasts in Cancer Invasion and Metastasis. Cancers 2021, 13, 4720. https://doi.org/10.3390/cancers13184720
Asif PJ, Longobardi C, Hahne M, Medema JP. The Role of Cancer-Associated Fibroblasts in Cancer Invasion and Metastasis. Cancers. 2021; 13(18):4720. https://doi.org/10.3390/cancers13184720
Chicago/Turabian StyleAsif, Paris Jabeen, Ciro Longobardi, Michael Hahne, and Jan Paul Medema. 2021. "The Role of Cancer-Associated Fibroblasts in Cancer Invasion and Metastasis" Cancers 13, no. 18: 4720. https://doi.org/10.3390/cancers13184720