
Emerging Role of Chimeric RNAs in Cell Plasticity and Adaptive Evolution of Cancer Cells



Abstract
:Simple Summary
Abstract
1. Introduction
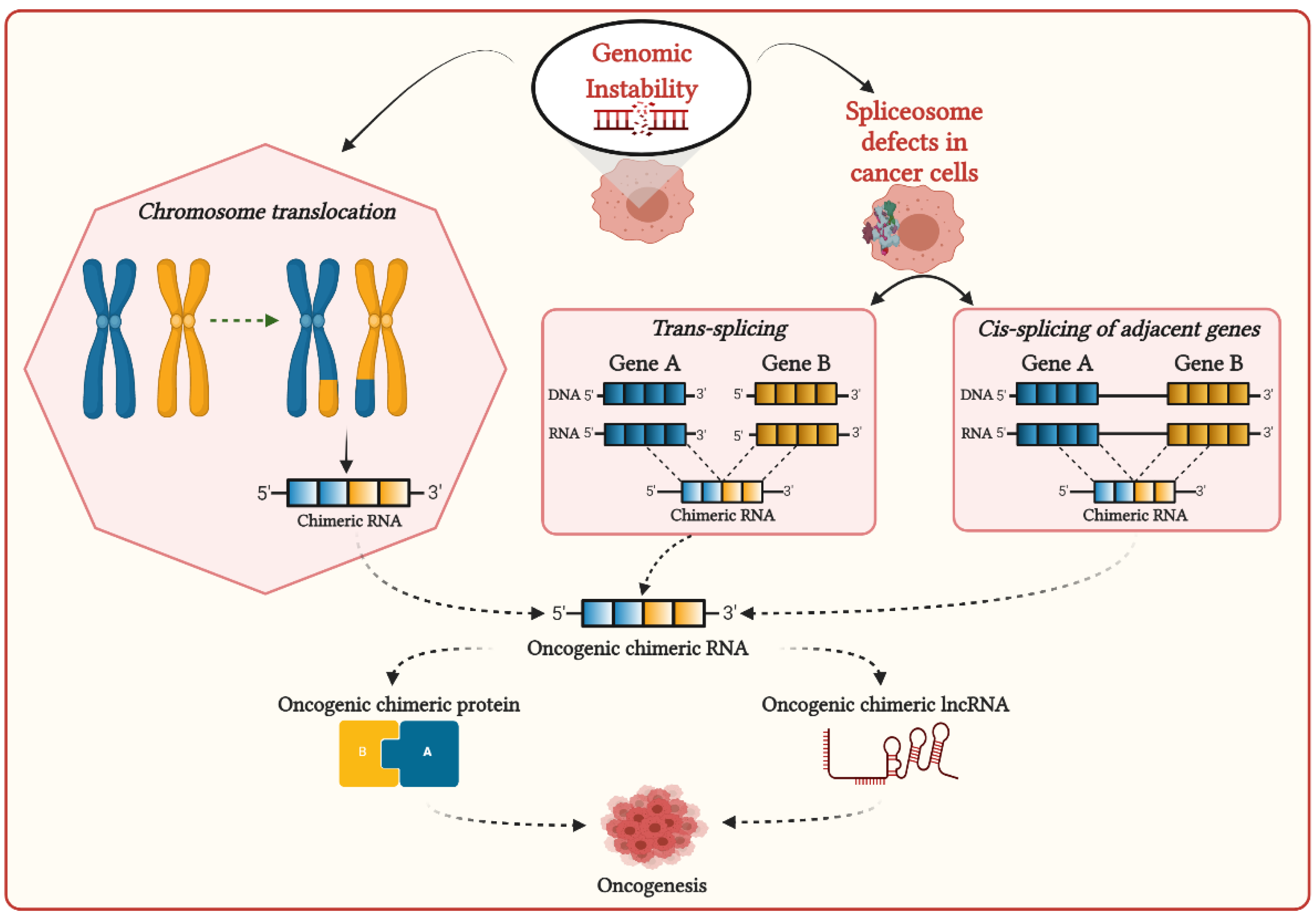
2. Mechanisms of Formation of Chimeric RNAs in Cancer Cells and Their Functional Associations with Cancer Development
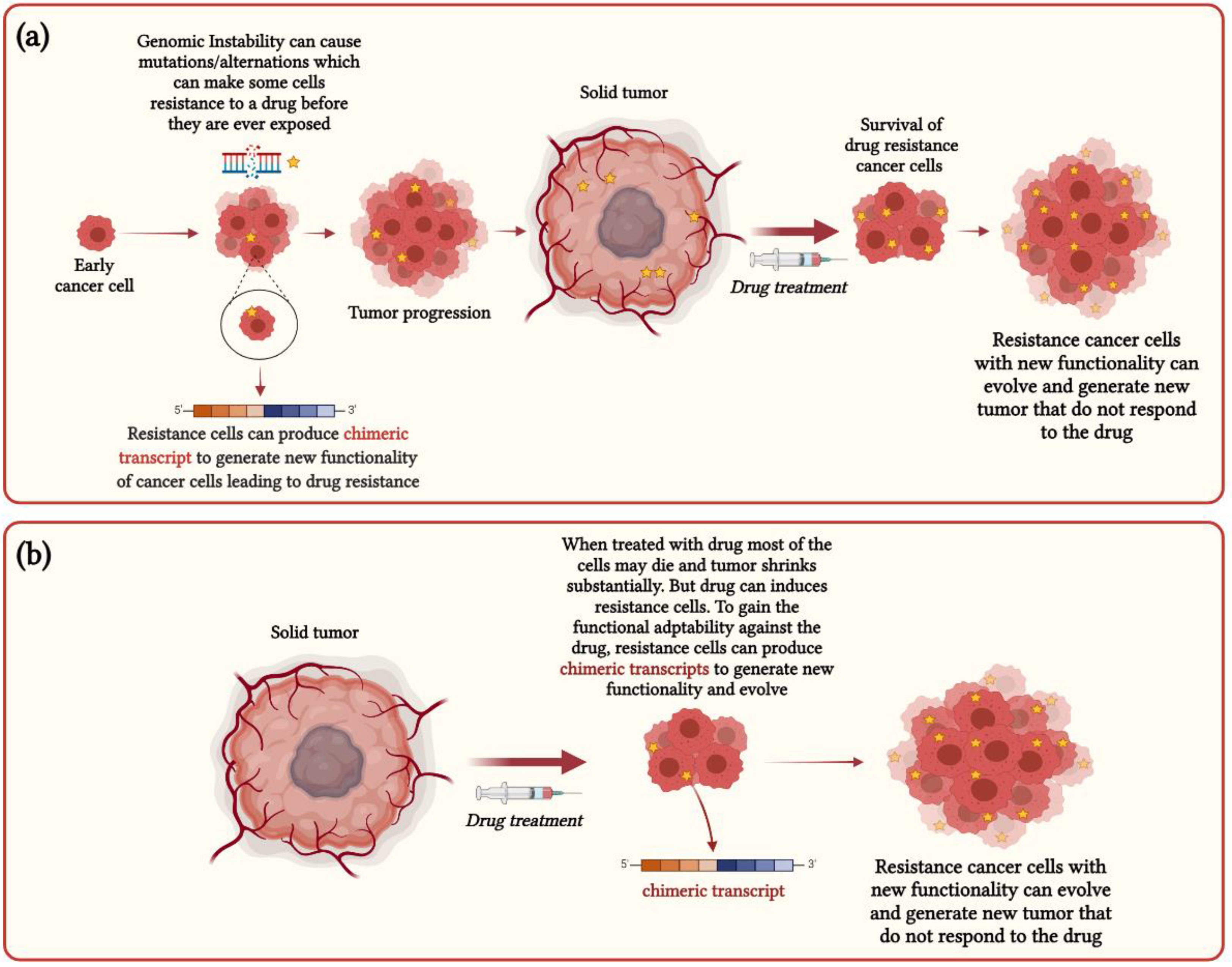
3. Functional Impact of Chimeric RNAs in Cancer Heterogeneity and Drug Resistance
4. Chimeric RNAs Are the Essential Driver for Generating Phenotypic Diversity in Cancer Cells
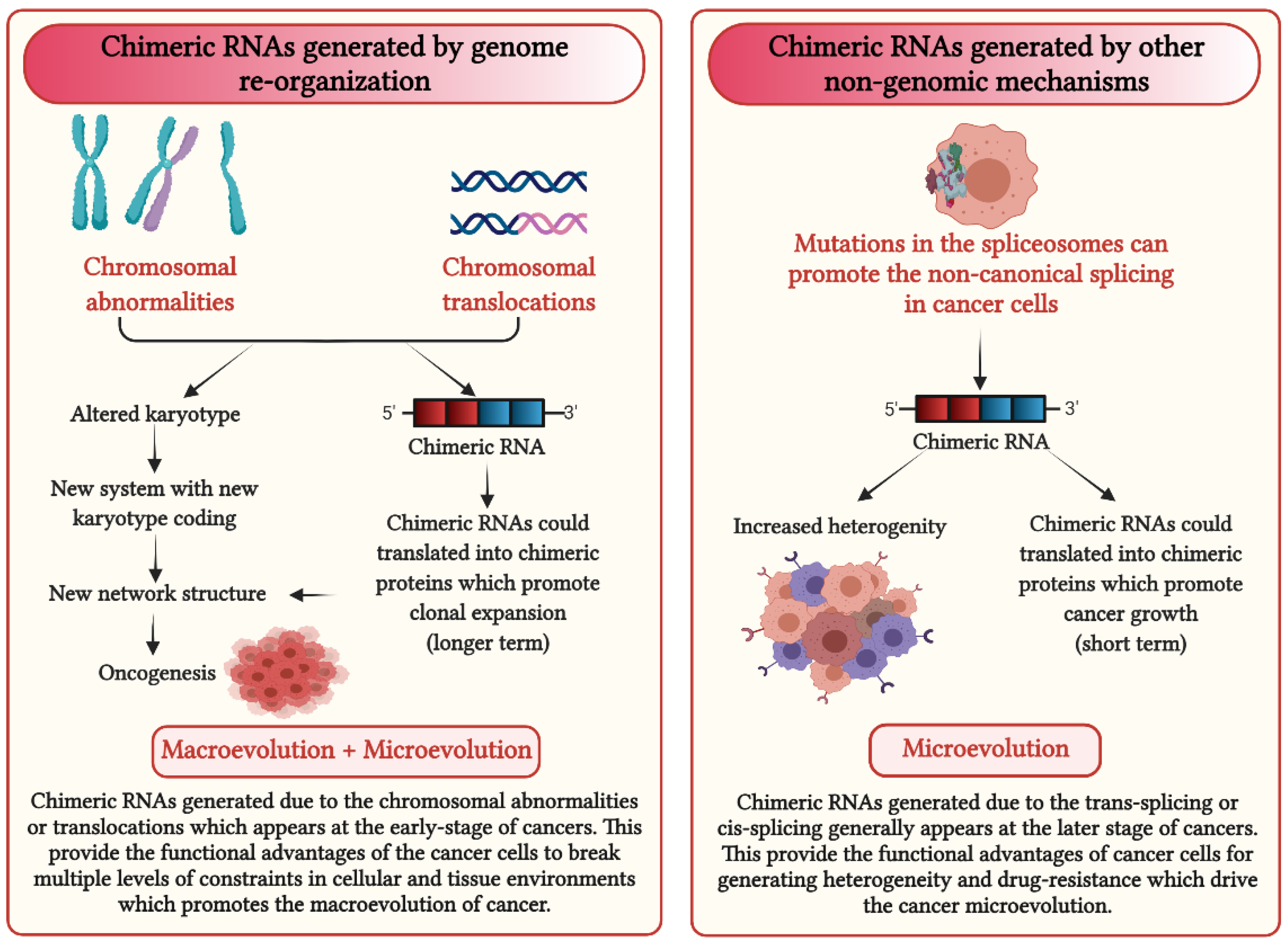
5. How Could Chimeric RNAs Lead to Cancer Evolution?
6. Conclusions and Future Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Black, J.R.M.; McGranahan, N. Genetic and non-genetic clonal diversity in cancer evolution. Nat. Rev. Cancer 2021, 21, 379–392. [Google Scholar] [CrossRef]
- Yates, L.R.; Campbell, P.J. Evolution of the cancer genome. Nat. Rev. Genet. 2012, 13, 795–806. [Google Scholar] [CrossRef] [Green Version]
- Horne, S.D.; Pollick, S.A.; Heng, H.H.Q. Evolutionary mechanism unifies the hallmarks of cancer. Int. J. Cancer 2015, 136, 2012–2021. [Google Scholar] [CrossRef] [Green Version]
- Heng, J.; Heng, H.H. Genome chaos: Creating new genomic information essential for cancer macroevolution. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef]
- Birkbak, N.J.; McGranahan, N. Cancer Genome Evolutionary Trajectories in Metastasis. Cancer Cell 2020, 37, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.H. Genome Chaos: Rethinking Genetics, Evolution, and Molecular Medicine; Academic Press Elsevier: Cambridge, MA, USA, 2019. [Google Scholar]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Marjanovic, N.D.; Weinberg, R.A.; Chaffer, C.L. Cell plasticity and heterogeneity in cancer. Clin. Chem. 2013, 59, 168–179. [Google Scholar] [CrossRef] [Green Version]
- Barber, L.J.; Davies, M.N.; Gerlinger, M. Dissecting cancer evolution at the macro-heterogeneity and micro-heterogeneity scale. Curr. Opin. Genet. Dev. 2015, 30, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Li, H.; Yang, F.; Yang, D.; Zhou, B.-B.S. Cell plasticity and genomic instability in cancer evolution. Genome Instab. Dis. 2020, 1, 301–309. [Google Scholar] [CrossRef]
- Shapiro, J.A. How chaotic is genome chaos? Cancers 2021, 13, 1358. [Google Scholar] [CrossRef]
- Heng, H.H.; Bremer, S.W.; Stevens, J.B.; Horne, S.D.; Liu, G.; Abdallah, B.Y.; Ye, K.J.; Ye, C.J. Chromosomal instability (CIN): What it is and why it is crucial to cancer evolution. Cancer Metastasis Rev. 2013, 32, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; La Choi, Y.; Kwon, M.; Park, P.J. Mechanisms and Consequences of Cancer Genome Instability: Lessons from Genome Sequencing Studies. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 283–312. [Google Scholar] [CrossRef]
- Wei, D.; Yao, Y. Genomic Instability and Cancer. J. Carcinog. Mutagen. 2014, 5, 1000165. [Google Scholar] [CrossRef]
- Li, Z.; Qin, F.; Li, H. Chimeric RNAs and their implications in cancer. Curr. Opin. Genet. Dev. 2018, 48, 36–43. [Google Scholar] [CrossRef]
- Wu, H.; Li, X.; Li, H. Gene fusions and chimeric RNAs, and their implications in cancer. Genes Dis. 2019, 6, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Liang, W.W.; Foltz, S.M.; Mutharasu, G.; Jayasinghe, R.G.; Cao, S.; Liao, W.W.; Reynolds, S.M.; Wyczalkowski, M.A.; Yao, L.; et al. Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep. 2018, 23, 227–238.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.C.; Reis-Filho, J.S. Genetic heterogeneity and cancer drug resistance. Lancet Oncol. 2012, 13, e178–e185. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, S.; Jiang, J.; Lu, Y.; Nice, E.C.; Huang, C.; Zhang, J.; He, W. Emerging role of tumor cell plasticity in modifying therapeutic response. Signal. Transduct. Target. Ther. 2020, 5, 1–36. [Google Scholar] [CrossRef]
- Frenkel-Morgenstern, M.; Gorohovski, A.; Tagore, S.; Sekar, V.; Vazquez, M.; Valencia, A. ChiPPI: A novel method for mapping chimeric protein-protein interactions uncovers selection principles of protein fusion events in cancer. Nucleic Acids Res. 2017, 45, 7094–7105. [Google Scholar] [CrossRef]
- Latysheva, N.S.; Babu, M.M. Molecular Signatures of Fusion Proteins in Cancer. ACS Pharmacol. Transl. Sci. 2019, 2, 122–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, F.; Zhang, Y.; Liu, J.; Li, H. SLC45A3-ELK4 functions as a long non-coding chimeric RNA. Cancer Lett. 2017, 404, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Detroja, R.; Balamurali, D.; Matveishina, E.; Medvedeva, Y.A.; Valencia, A.; Gorohovski, A.; Frenkel-Morgenstern, M. Computational analysis of sense-antisense chimeric transcripts reveals their potential regulatory features and the landscape of expression in human cells. NAR Genom. Bioinform. 2021, 3. [Google Scholar] [CrossRef]
- Mitelman, F.; Johansson, B.; Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer 2015, 15, 371–381. [Google Scholar] [CrossRef]
- Brien, G.L.; Stegmaier, K.; Armstrong, S.A. Targeting chromatin complexes in fusion protein-driven malignancies. Nat. Rev. Cancer 2019, 19, 255–269. [Google Scholar] [CrossRef]
- Shtivelman, E.; Lifshitz, B.; Gale, R.P.; Canaani, E. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature 1985, 315, 550–554. [Google Scholar] [CrossRef]
- Collins, S.J. Retinoic acid receptors, hematopoiesis and leukemogenesis. Curr. Opin. Hematol. 2008, 15, 346–351. [Google Scholar] [CrossRef]
- Zheng, J. Oncogenic chromosomal translocations and human cancer (Review). Oncol. Rep. 2013, 30, 2011–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeed, S.; Logie, C.; Stunnenberg, H.G.; Martens, J.H.A. Genome-wide functions of PML-RARα in acute promyelocytic leukaemia. Br. J. Cancer 2011, 104, 554–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haferlach, T.; Meggendorfer, M. More than a fusion gene: The RUNX1-RUNX1T1 AML. Blood 2019, 133, 1006–1007. [Google Scholar] [CrossRef] [Green Version]
- Bailly, R.A.; Bosselut, R.; Zucman, J.; Cormier, F.; Delattre, O.; Roussel, M.; Thomas, G.; Ghysdael, J. DNA-binding and transcriptional activation properties of the EWS-FLI-1 fusion protein resulting from the t(11;22) translocation in Ewing sarcoma. Mol. Cell. Biol. 1994, 14, 3230–3241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorensen, P.H.B.; Lessnick, S.L.; Lopez-Terrada, D.; Liu, X.F.; Triche, T.J.; Denny, C.T. A second ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat. Genet. 1994, 6, 146–151. [Google Scholar] [CrossRef]
- Kroll, T.G.; Sarraf, P.; Pecciarini, L.; Chen, C.J.; Mueller, E.; Spiegelman, B.M.; Fletcher, J.A. PAX8-PPARγ1 fusion in oncogene human thyroid carcinoma. Science 2000, 289, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- Cironi, L.; Petricevic, T.; Fernandes Vieira, V.; Provero, P.; Fusco, C.; Cornaz, S.; Fregni, G.; Letovanec, I.; Aguet, M.; Stamenkovic, I. The fusion protein SS18-SSX1 employs core Wnt pathway transcription factors to induce a partial Wnt signature in synovial sarcoma. Sci. Rep. 2016, 6, 22113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bruijn, D.R.H.; Allander, S.V.; Van Dijk, A.H.A.; Willemse, M.P.; Thijssen, J.; Van Groningen, J.J.M.; Meltzer, P.S.; Van Kessel, A.G. The synovial sarcoma-associated SS18-SSX2 fusion protein induces epigenetic gene (de)regulation. Cancer Res. 2006, 66, 9474–9482. [Google Scholar] [CrossRef] [Green Version]
- Persson, M.; Andrén, Y.; Mark, J.; Horlings, H.M.; Persson, F.; Stenman, G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc. Natl. Acad. Sci. USA 2009, 106, 18740–18744. [Google Scholar] [CrossRef] [Green Version]
- Wysocki, P.T.; Izumchenko, E.; Meir, J.; Ha, P.K.; Sidransky, D.; Brait, M. Adenoid cystic carcinoma: Emerging role of translocations and gene fusions. Oncotarget 2016, 7, 66239–66254. [Google Scholar] [CrossRef] [Green Version]
- Tonon, G.; Modi, S.; Wu, L.; Kubo, A.; Coxon, A.B.; Komiya, T.; O’Neil, K.; Stover, K.; El-Naggar, A.; Griffin, J.D.; et al. t(11;19)(q21;p13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling pathway. Nat. Genet. 2003, 33, 208–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Liu, J.; Gao, P.; Nakamura, M.; Cao, Y.; Shen, H.; Griffin, J.D. Transforming activity of MECT1-MAML2 fusion oncoprotein is mediated by constitutive CREB activation. EMBO J. 2005, 24, 2391–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, J.P.; Cooper, C.S. ETS gene fusions in prostate cancer. Nat. Rev. Urol. 2009, 6, 429–439. [Google Scholar] [CrossRef] [PubMed]
- White, N.M.; Feng, F.Y.; Maher, C.A. Recurrent Rearrangements in Prostate Cancer: Causes and Therapeutic Potential. Curr. Drug Targets 2013, 14, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global Survey of Phosphotyrosine Signaling Identifies Oncogenic Kinases in Lung Cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northcott, P.A.; Shih, D.J.H.; Peacock, J.; Garzia, L.; Sorana Morrissy, A.; Zichner, T.; Stútz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 2012, 487, 49–56. [Google Scholar] [CrossRef]
- Rowley, J.D. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Laxman, B.; Varambally, S.; Cao, X.; Yu, J.; Helgeson, B.E.; Cao, Q.; Prensner, J.R.; Rubin, M.A.; Shah, R.B.; et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia 2008, 10, 177-IN9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hessels, D.; Schalken, J.A. Recurrent gene fusions in prostate cancer: Their clinical implications and uses. Curr. Urol. Rep. 2013, 14, 214–222. [Google Scholar] [CrossRef]
- Haluska, F.G.; Tsujimoto, Y.; Croce, C.M. The t(8;14) chromosome translocation of the Burkitt lymphoma cell line Daudi occurred during immunoglobulin gene rearrangement and involved the heavy chain diversity region. Proc. Natl. Acad. Sci. USA 1987, 84, 6835–6839. [Google Scholar] [CrossRef] [Green Version]
- Boerma, E.G.; Siebert, R.; Kluin, P.M.; Baudis, M. Translocations involving 8q24 in Burkitt lymphoma and other malignant lymphomas: A historical review of cytogenetics in the light of todays knowledge. Leukemia 2009, 23, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Djabali, M.; Selleri, L.; Parry, P.; Bower, M.; Young, B.D.; Evans, G.A. A trithorax–like gene is interrupted by chromosome 11q23 translocations in acute leukaemias. Nat. Genet. 1992, 2, 113–118. [Google Scholar] [CrossRef]
- Ziemin-Van Der Poel, S.; Mccabe, N.R.; Gill, H.J.; Espinosa, R.; Patel, Y.; Harden, A.; Rubinelli, P.; Smith, S.D.; Lebeau, M.M.; Rowley, I.D.; et al. Identification of a gene, MLL, that spans the breakpoint in 11q23 translocations associated with human leukemias. Proc. Natl. Acad. Sci. USA 1991, 88, 10735–10739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krivtsov, A.V.; Armstrong, S.A. MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 2007, 7, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.K.; Ma, J.; Wang, J.; Chen, X.; Gedman, A.L.; Dang, J.; Nakitandwe, J.; Holmfeldt, L.; Parker, M.; Easton, J.; et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat. Genet. 2015, 47, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, L.; Xiong, J.; DenDekker, A.; Ye, A.; Karatas, H.; Liu, L.; Wang, H.; Qin, Z.S.; Wang, S.; et al. MLL1 and MLL1 fusion proteins have distinct functions in regulating leukemic transcription program. Cell Discov. 2016, 2, 16008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parameswaran, S.; Vizeacoumar, F.S.; Kalyanasundaram Bhanumathy, K.; Qin, F.; Islam, M.F.; Toosi, B.M.; Cunningham, C.E.; Mousseau, D.D.; Uppalapati, M.C.; Stirling, P.C.; et al. Molecular characterization of an MLL1 fusion and its role in chromosomal instability. Mol. Oncol. 2019, 13, 422–440. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, C.; Pizzul, P.; Longhese, M.P.; Bonetti, D. Sensing R-Loop-Associated DNA Damage to Safeguard Genome Stability. Front. Cell Dev. Biol. 2021, 8, 1657. [Google Scholar] [CrossRef]
- Nguyen, H.D.; Leong, W.Y.; Li, W.; Reddy, P.N.G.; Sullivan, J.D.; Walter, M.J.; Zou, L.; Graubert, T.A. Spliceosome mutations induce R loop-associated sensitivity to ATR inhibition in myelodysplastic syndromes. Cancer Res. 2018, 78, 5363–5374. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Chen, J.Y.; Huang, Y.J.; Gu, Y.; Qiu, J.; Qian, H.; Shao, C.; Zhang, X.; Hu, J.; Li, H.; et al. The Augmented R-Loop Is a Unifying Mechanism for Myelodysplastic Syndromes Induced by High-Risk Splicing Factor Mutations. Mol. Cell 2018, 69, 412–425.e6. [Google Scholar] [CrossRef] [Green Version]
- Boros-Oláh, B.; Dobos, N.; Hornyák, L.; Szabó, Z.; Karányi, Z.; Halmos, G.; Roszik, J.; Székvölgyi, L. Drugging the R-loop interactome: RNA-DNA hybrid binding proteins as targets for cancer therapy. DNA Repair 2019, 84, 102642. [Google Scholar] [CrossRef]
- Tam, A.S.; Stirling, P.C. Splicing, genome stability and disease: Splice like your genome depends on it! Curr. Genet. 2019, 65, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Xie, Z.; Li, H. Intergenically Spliced Chimeric RNAs in Cancer. Trends Cancer 2016, 2, 475–484. [Google Scholar] [CrossRef] [Green Version]
- Grosso, A.R.; Leite, A.P.; Carvalho, S.; Matos, M.R.; Martins, F.B.; Vítor, A.C.; Desterro, J.M.P.; Carmo-Fonseca, M.; de Almeida, S.F. Pervasive transcription read-through promotes aberrant expression of oncogenes and RNA chimeras in renal carcinoma. eLife 2015, 4, e09214. [Google Scholar] [CrossRef]
- Nacu, S.; Yuan, W.; Kan, Z.; Bhatt, D.; Rivers, C.S.; Stinson, J.; Peters, B.A.; Modrusan, Z.; Jung, K.; Seshagiri, S.; et al. Deep RNA sequencing analysis of readthrough gene fusions in human prostate adenocarcinoma and reference samples. BMC Med. Genom. 2011, 4, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varley, K.E.; Gertz, J.; Roberts, B.S.; Davis, N.S.; Bowling, K.M.; Kirby, M.K.; Nesmith, A.S.; Oliver, P.G.; Grizzle, W.E.; Forero, A.; et al. Recurrent read-through fusion transcripts in breast cancer. Breast Cancer Res. Treat. 2014, 146, 287–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, C.A.; Kumar-Sinha, C.; Cao, X.; Kalyana-Sundaram, S.; Han, B.; Jing, X.; Sam, L.; Barrette, T.; Palanisamy, N.; Chinnaiyan, A.M. Transcriptome sequencing to detect gene fusions in cancer. Nature 2009, 458, 97–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rickman, D.S.; Pflueger, D.; Moss, B.; VanDoren, V.E.; Chen, C.X.; De la Taille, A.; Kuefer, R.; Tewari, A.K.; Setlur, S.R.; Demichelis, F.; et al. SLC45A3-ELK4 is a novel and frequent erythroblast transformation-specific fusion transcript in prostate cancer. Cancer Res. 2009, 69, 2734–2738. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wang, J.; Mor, G.; Sklar, J. A neoplastic gene fusion mimics trans-splicing of RNAs in normal human cells. Science 2008, 321, 1357–1361. [Google Scholar] [CrossRef]
- Gingeras, T.R. Implications of chimaeric non-co-linear transcripts. Nature 2009, 461, 206–211. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Qin, F.; Movassagh, M.; Park, H.; Golden, W.; Xie, Z.; Zhang, P.; Sklar, J.; Li, H. A chimeric RNA characteristic of rhabdomyosarcoma in normal myogenesis process. Cancer Discov. 2013, 3, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Slupianek, A.; Hoser, G.; Majsterek, I.; Bronisz, A.; Malecki, M.; Blasiak, J.; Fishel, R.; Skorski, T. Fusion Tyrosine Kinases Induce Drug Resistance by Stimulation of Homology-Dependent Recombination Repair, Prolongation of G 2/M Phase, and Protection from Apoptosis. Mol. Cell. Biol. 2002, 22, 4189–4201. [Google Scholar] [CrossRef] [Green Version]
- Bedi, A.; Barber, J.P.; Bedi, G.C.; El-Deiry, W.S.; Sidransky, D.; Vala, M.S.; Akhtar, A.J.; Hilton, J.; Jones, R.J. BCR-ABL-mediated inhibition of apoptosis with delay of G2/M transition after DNA damage: A mechanism of resistance to multiple anticancer agents. Blood 1995, 86, 1148–1158. [Google Scholar] [CrossRef] [Green Version]
- Slupianek, A.; Schmutte, C.; Tombline, G.; Nieborowska-Skorska, M.; Hoser, G.; Nowicki, M.O.; Pierce, A.J.; Fishel, R.; Skorski, T. BCR/ABL regulates mammalian RecA homologs, resulting in drug resistance. Mol. Cell 2001, 8, 795–806. [Google Scholar] [CrossRef]
- Turner, J.A.; Bemis, J.G.T.; Bagby, S.M.; Capasso, A.; Yacob, B.W.; Chimed, T.S.; Van Gulick, R.; Lee, H.; Tobin, R.; Tentler, J.J.; et al. BRAF fusions identified in melanomas have variable treatment responses and phenotypes. Oncogene 2019, 38, 1296–1308. [Google Scholar] [CrossRef]
- Botton, T.; Talevich, E.; Mishra, V.K.; Zhang, T.; Shain, A.H.; Berquet, C.; Gagnon, A.; Judson, R.L.; Ballotti, R.; Ribas, A.; et al. Genetic Heterogeneity of BRAF Fusion Kinases in Melanoma Affects Drug Responses. Cell Rep. 2019, 29, 573–588.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.C.; Wang, W.X.; Zhang, Q.X.; Xu, C.W.; Zhuang, W.; Du, K.Q.; Chen, G.; Lv, T.F.; Song, Y. The KIF5B-RET Fusion Gene Mutation as a Novel Mechanism of Acquired EGFR Tyrosine Kinase Inhibitor Resistance in Lung Adenocarcinoma. Clin. Lung Cancer 2019, 20, e73–e76. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Miao, Y.; Peng, Z.; Sandgren, J.; De Ståhl, T.D.; Huss, M.; Lennartsson, L.; Liu, Y.; Nistér, M.; Nilsson, S.; et al. Identification of mutations, gene expression changes and fusion transcripts by whole transcriptome RNAseq in docetaxel resistant prostate cancer cells. Springerplus 2016, 5, 1861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christie, E.L.; Pattnaik, S.; Beach, J.; Copeland, A.; Rashoo, N.; Fereday, S.; Hendley, J.; Alsop, K.; Brady, S.L.; Lamb, G.; et al. Multiple ABCB1 transcriptional fusions in drug resistant high-grade serous ovarian and breast cancer. Nat. Commun. 2019, 10, 1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, P.J.; Getz, G.; Korbel, J.O.; Stuart, J.M.; Jennings, J.L.; Stein, L.D.; Perry, M.D.; Nahal-Bose, H.K.; Ouellette, B.F.F.; Li, C.H.; et al. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, C.; Davidson, N.R.; Demircioglu, D.; Fonseca, N.A.; He, Y.; Kahles, A.; Van Lehmann, K.; Liu, F.; Shiraishi, Y.; Soulette, C.M.; et al. Genomic basis for RNA alterations in cancer. Nature 2020, 578, 129–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef]
- Moreno-Smith, M.; Lutgendorf, S.K.; Sood, A.K. Impact of stress on cancer metastasis. Futur. Oncol. 2010, 6, 1863–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreuzaler, P.; Panina, Y.; Segal, J.; Yuneva, M. Adapt and conquer: Metabolic flexibility in cancer growth, invasion and evasion. Mol. Metab. 2020, 33, 83–101. [Google Scholar] [CrossRef]
- Frenkel-Morgenstern, M.; Lacroix, V.; Ezkurdia, I.; Levin, Y.; Gabashvili, A.; Prilusky, J.; Del Pozo, A.; Tress, M.; Johnson, R.; Guigo, R.; et al. Chimeras taking shape: Potential functions of proteins encoded by chimeric RNA transcripts. Genome Res. 2012, 22, 1231–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neckles, C.; Sundara Rajan, S.; Caplen, N.J. Fusion transcripts: Unexploited vulnerabilities in cancer? Wiley Interdiscip. Rev. RNA 2020, 11, e1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniue, K.; Akimitsu, N. Fusion genes and RNAs in cancer development. Non-Coding RNA 2021, 7, 10. [Google Scholar] [CrossRef]
- Djebali, S.; Lagarde, J.; Kapranov, P.; Lacroix, V.; Borel, C.; Mudge, J.M.; Howald, C.; Foissac, S.; Ucla, C.; Chrast, J.; et al. Evidence for transcript networks composed of chimeric rnas in human cells. PLoS ONE 2012, 7, e28213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gong, M.; Yuan, H.; Park, H.G.; Frierson, H.F.; Li, H. Chimeric transcript generated by cis- splicing of adjacent genes regulates prostate cancer cell proliferation. Cancer Discov. 2012, 2, 598–607. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Lin, W.; Kannan, K.; Luo, L.; Li, J.; Chao, P.W.; Wang, Y.; Chen, Y.P.; Gu, J.; Yen, L. Aberrant chimeric RNA GOLM1-MAK10 encoding a secreted fusion protein as a molecular signature for human esophageal squamous cell carcinoma. Oncotarget 2013, 4, 2135–2143. [Google Scholar] [CrossRef] [Green Version]
- Rao, C.V.; Wolf, D.M.; Arkin, A.P. Control, exploitation and tolerance of intracellular noise. Nature 2002, 420, 231–237. [Google Scholar] [CrossRef]
- Acar, M.; Mettetal, J.T.; Van Oudenaarden, A. Stochastic switching as a survival strategy in fluctuating environments. Nat. Genet. 2008, 40, 471–475. [Google Scholar] [CrossRef]
- Richard, M.; Yvert, G. How does evolution tune biological noise? Front. Genet. 2014, 5, 374. [Google Scholar] [CrossRef] [Green Version]
- Espinosa-Soto, C.; Martin, O.C.; Wagner, A. Phenotypic plasticity can facilitate adaptive evolution in gene regulatory circuits. BMC Evol. Biol. 2011, 11, 5. [Google Scholar] [CrossRef] [Green Version]
- Fraser, D.; Kærn, M. A chance at survival: Gene expression noise and phenotypic diversification strategies. Mol. Microbiol. 2009, 71, 1333–1340. [Google Scholar] [CrossRef]
- Heng, J.; Heng, H.H. Karyotype coding: The creation and maintenance of system information for complexity and biodiversity. Biosystems 2021, 208, 104476. [Google Scholar] [CrossRef]
- Heng, H.H.Q.; Bremer, S.W.; Stevens, J.B.; Ye, K.J.; Liu, G.; Ye, C.J. Genetic and epigenetic heterogeneity in cancer: A genome-centric perspective. J. Cell. Physiol. 2009, 220, 538–547. [Google Scholar] [CrossRef]
- Brock, A.; Krause, S.; Ingber, D.E. Control of cancer formation by intrinsic genetic noise and microenvironmental cues. Nat. Rev. Cancer 2015, 15, 499–509. [Google Scholar] [CrossRef]
- Swanton, C.; Beck, S. Epigenetic Noise Fuels Cancer Evolution. Cancer Cell 2014, 26, 775–776. [Google Scholar] [CrossRef] [Green Version]
- Gerstung, M.; Jolly, C.; Leshchiner, I.; Dentro, S.C.; Gonzalez, S.; Rosebrock, D.; Mitchell, T.J.; Rubanova, Y.; Anur, P.; Yu, K.; et al. The evolutionary history of 2,658 cancers. Nature 2020, 578, 122–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahir, N.; Sun, R.; Gallahan, D.; Gatenby, R.A.; Curtis, C. Characterizing the ecological and evolutionary dynamics of cancer. Nat. Genet. 2020, 52, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Bozic, I.; Reiter, J.G.; Allen, B.; Antal, T.; Chatterjee, K.; Shah, P.; Moon, Y.S.; Yaqubie, A.; Kelly, N.; Le, D.T.; et al. Evolutionary dynamics of cancer in response to targeted combination therapy. eLife 2013, 2, e00747. [Google Scholar] [CrossRef]
- Liu, G.; Stevens, J.B.; Horne, S.D.; Abdallah, B.Y.; Ye, K.J.; Bremer, S.W.; Ye, C.J.; Chen, D.J.; Heng, H.H. Genome chaos: Survival strategy during crisis. Cell Cycle 2014, 13, 528–537. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Chimeric RNA | Gene 1 | Gene 2 | Associated Cancers | Potential Function | Chromosomal Aberrations |
|---|---|---|---|---|---|
| BCR-ABL | BCR | ABL | Chronic myelogenous leukemia (CML) | BCR-ABL fusion protein alters constitutively active ABL1 kinase and activates a variety of signaling pathways that promote CML development [30] | Translocation t(9;22) |
| PML-RARα | PML | RARα | Acute promyelocytic leukemia (APL) | PML-RARα fusion protein interplays with retinoic X receptors (RXR) and promotes the deregulation of epigenetic modifications [31,32,33] | Translocation t(15;17) |
| RUNX1–RUNX1T1 | RUNX1 | RUNX1T1 | Acute promyelocytic leukemia (APL) | RUNX1–RUNX1T1 fusion protein interacts with other proteins to repress transcription and induce leukemogenesis in myeloid progenitor cells [34] | Translocation t(8;21) |
| EWS–FLI1 | EWS | FLI1 | Ewing’s sarcoma (EWS) | EWS–FLI1 fusion transcription factors upregulate genes associated with the cell cycle, invasion, and proliferation pathways [35] | Translocation t(11;22) |
| EWS–ERG | EWS | ERG | Ewing’s sarcoma (EWS) | EWS–ERG fusion transcription factors upregulate genes associated with the cell cycle, invasion, and proliferation pathways [36] | Translocation t(21;22) |
| PAX8-PPARγ1 | PAX8 | PPARγ1 | Thyroid follicular carcinomas | PAX8-PPARγ1 fusion protein can act as a dominant-negative inhibitor of wild-type PPARγ and can activate or repress PAX8-responsive genes [37] | Translocation t(2;3)(q13;p25) |
| SS18–SSX1 | SS18 | SSX1 | Synovial sarcoma | SS18-SSX1 fusion protein employs core Wnt pathway transcription factors to induce Wnt target gene expression in synovial sarcoma [38] | Translocation t(X;18) |
| SS18–SSX2 | SS18 | SSX2 | Synovial sarcoma | SS18-SSX2 fusion protein induces epigenetic gene deregulation and promotes the development of synovial sarcoma [39] | Translocation t(X;18) |
| MYB-NFIB | MYB | NFIB | Adenoid cystic carcinomas (ACC) | MYB-NFIB fusion proteins promote the upregulation of MYB, which can drive the development of adenoid cystic carcinoma (ACC) [40,41] | Translocation t(6;9)(q22–23;p23–24) |
| MECT1-MAML2 | MECT1 | MAML2 | Mucoepidermoid carcinoma | MECT1-MAML2 fusion protein undermine two signaling pathways, CREB and Notch, that could be potentially important in cancer development [42,43] | Translocation t(11;19)(q14–21;p12–13) |
| TMPRSS2-ERG | TMPRSS2 | ERG | Prostate cancer | Expression of TMPRSS2-ERG chimeric transcripts induces overexpression of the transcription factor ERG, which promotes invasion in human prostate cancer development [44,45] | Del(q22) and Translocation t(7;21)(1,26–28) |
| EML4–ALK | EML4 | ALK | Non-small-cell lung cancer (NSCLC) | Generation of this EML4–ALK chimeric RNA leads cancer transformation by activating downstream reactions in the ALK signaling pathway [46] | Inversion of chromosome 2 (inv2) (p21:p23) |
| PVT1–MYC | PVT1 | MYC | Medulloblastoma | Chromothripsis in medulloblastoma promotes the recurrent translocations, which enable the fusion of lncRNA PVT1 to MYC gene as a consequence of the continuous oncogenic effect via MYC amplification [47] | Chromothripsis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukherjee, S.; Heng, H.H.; Frenkel-Morgenstern, M. Emerging Role of Chimeric RNAs in Cell Plasticity and Adaptive Evolution of Cancer Cells. Cancers 2021, 13, 4328. https://doi.org/10.3390/cancers13174328
Mukherjee S, Heng HH, Frenkel-Morgenstern M. Emerging Role of Chimeric RNAs in Cell Plasticity and Adaptive Evolution of Cancer Cells. Cancers. 2021; 13(17):4328. https://doi.org/10.3390/cancers13174328
Chicago/Turabian StyleMukherjee, Sumit, Henry H. Heng, and Milana Frenkel-Morgenstern. 2021. "Emerging Role of Chimeric RNAs in Cell Plasticity and Adaptive Evolution of Cancer Cells" Cancers 13, no. 17: 4328. https://doi.org/10.3390/cancers13174328