ATP Inhibits Breast Cancer Migration and Bone Metastasis through Down-Regulation of CXCR4 and Purinergic Receptor P2Y11

 ,
, 
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
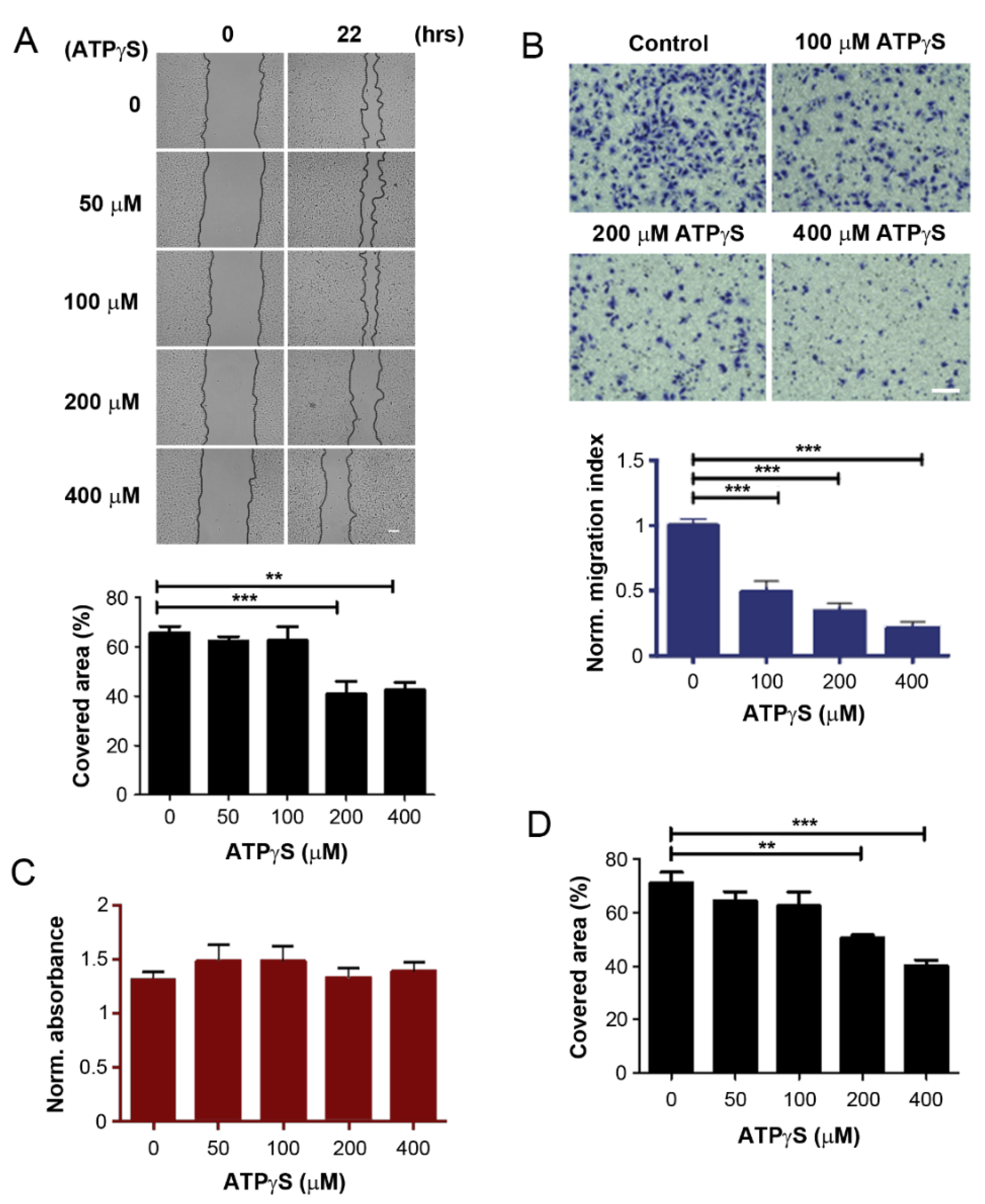
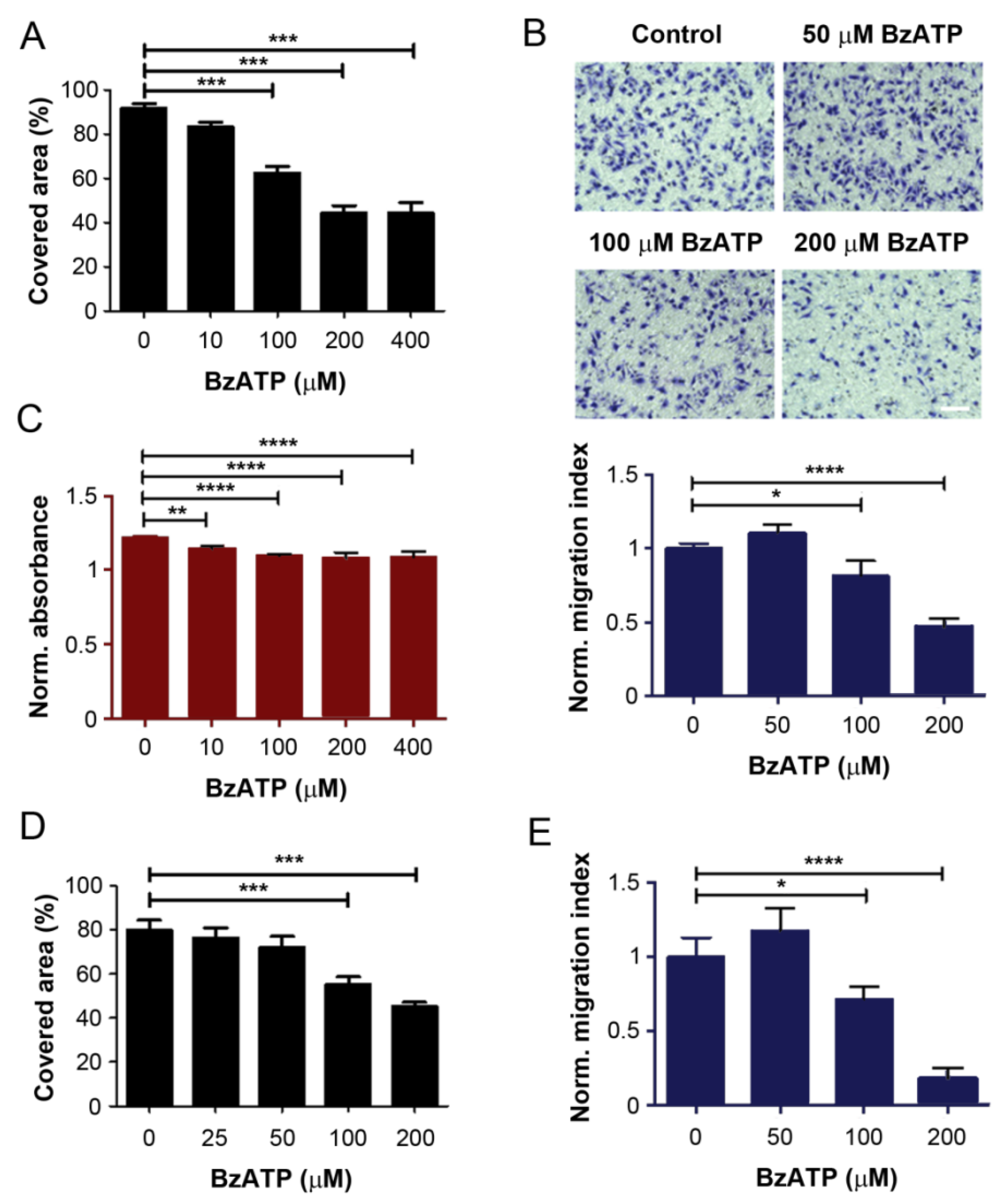
2.1. ATPγS and BzATP Inhibit Breast Cancer Cell Migration
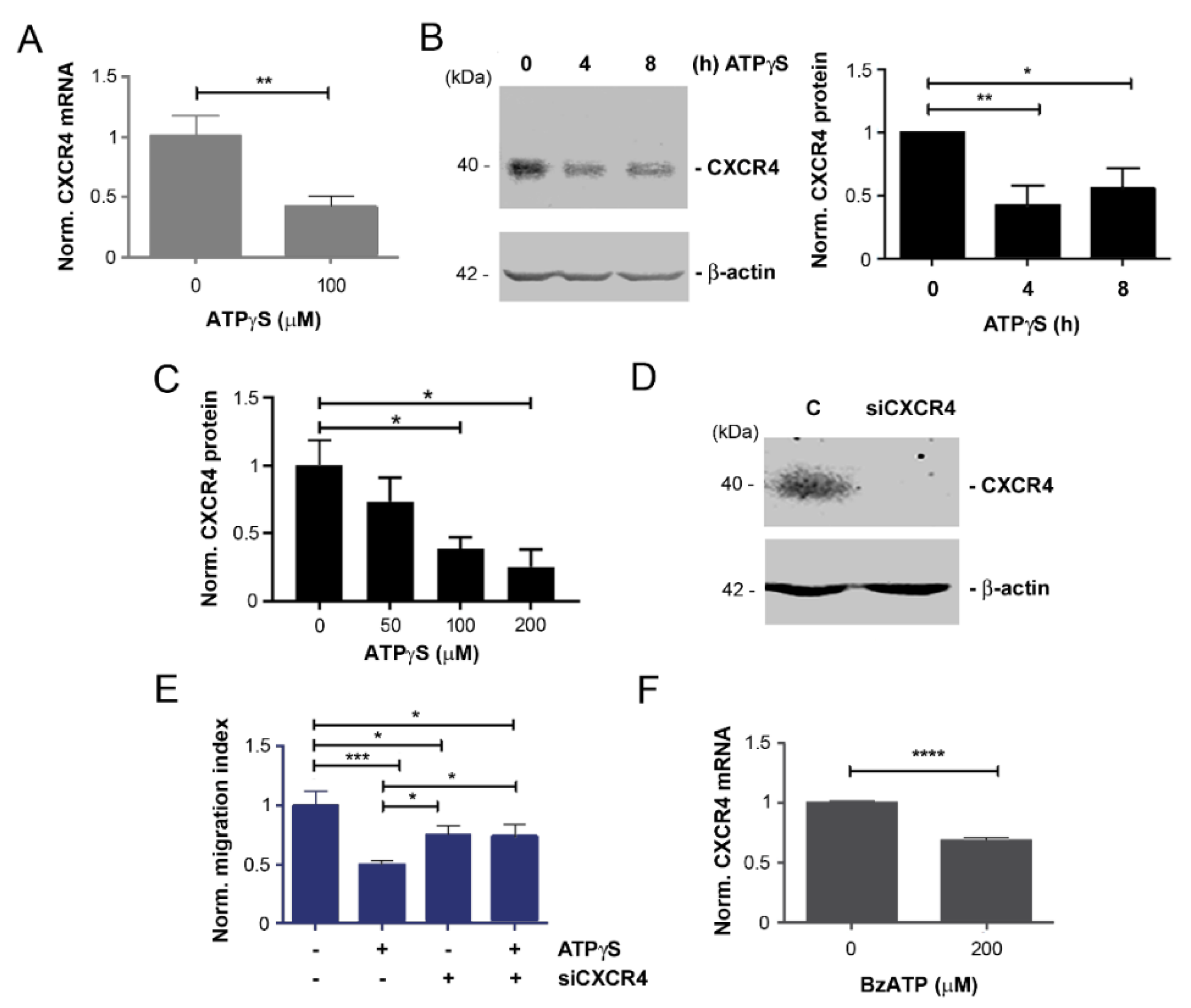
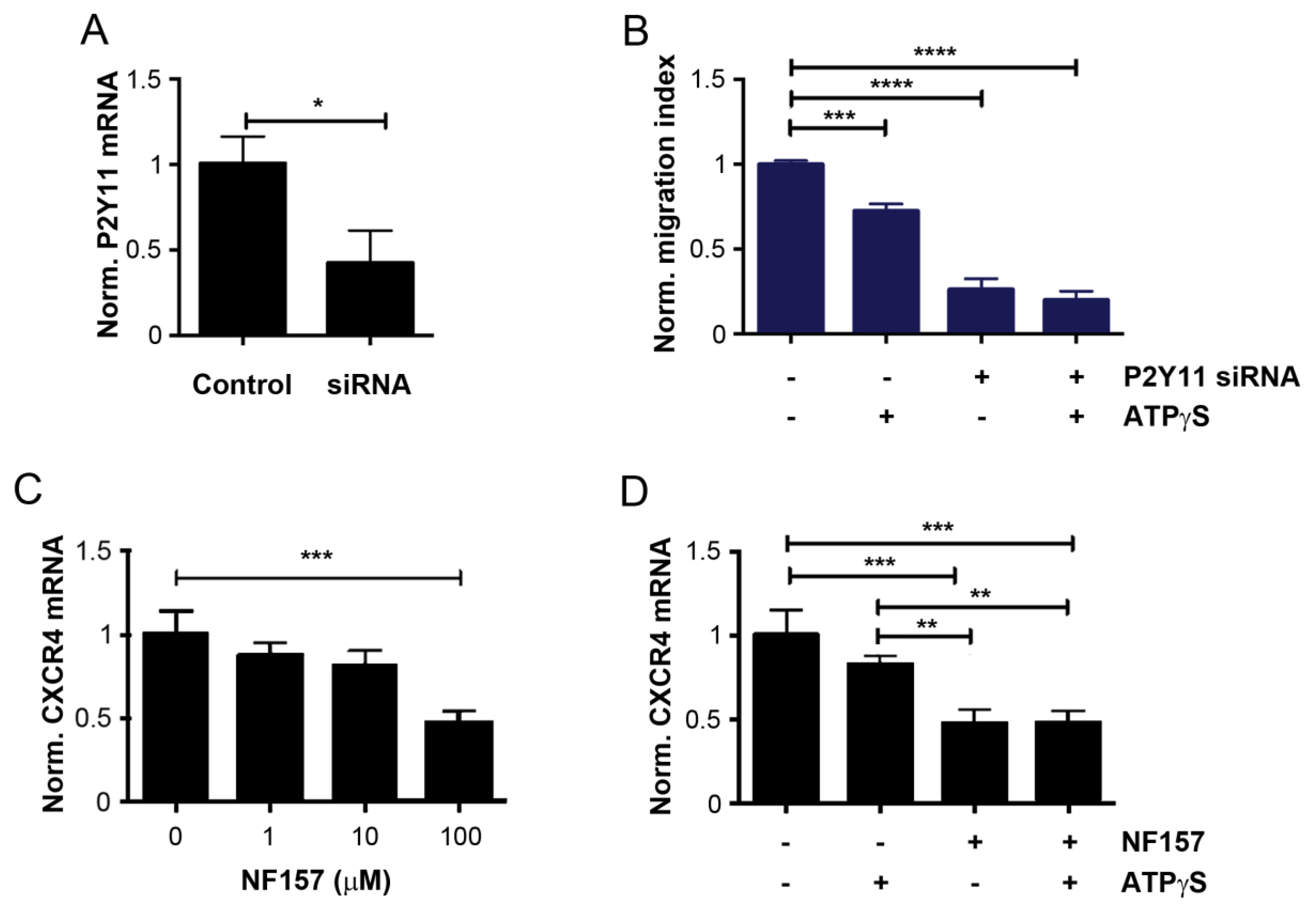
2.2. Mitigation of CXCR4 Expression by ATPγS and BzATP, and Attenuation of Inhibitory Effect of ATPγS on Cancer Cell Migration with CXCR4 Knockdown
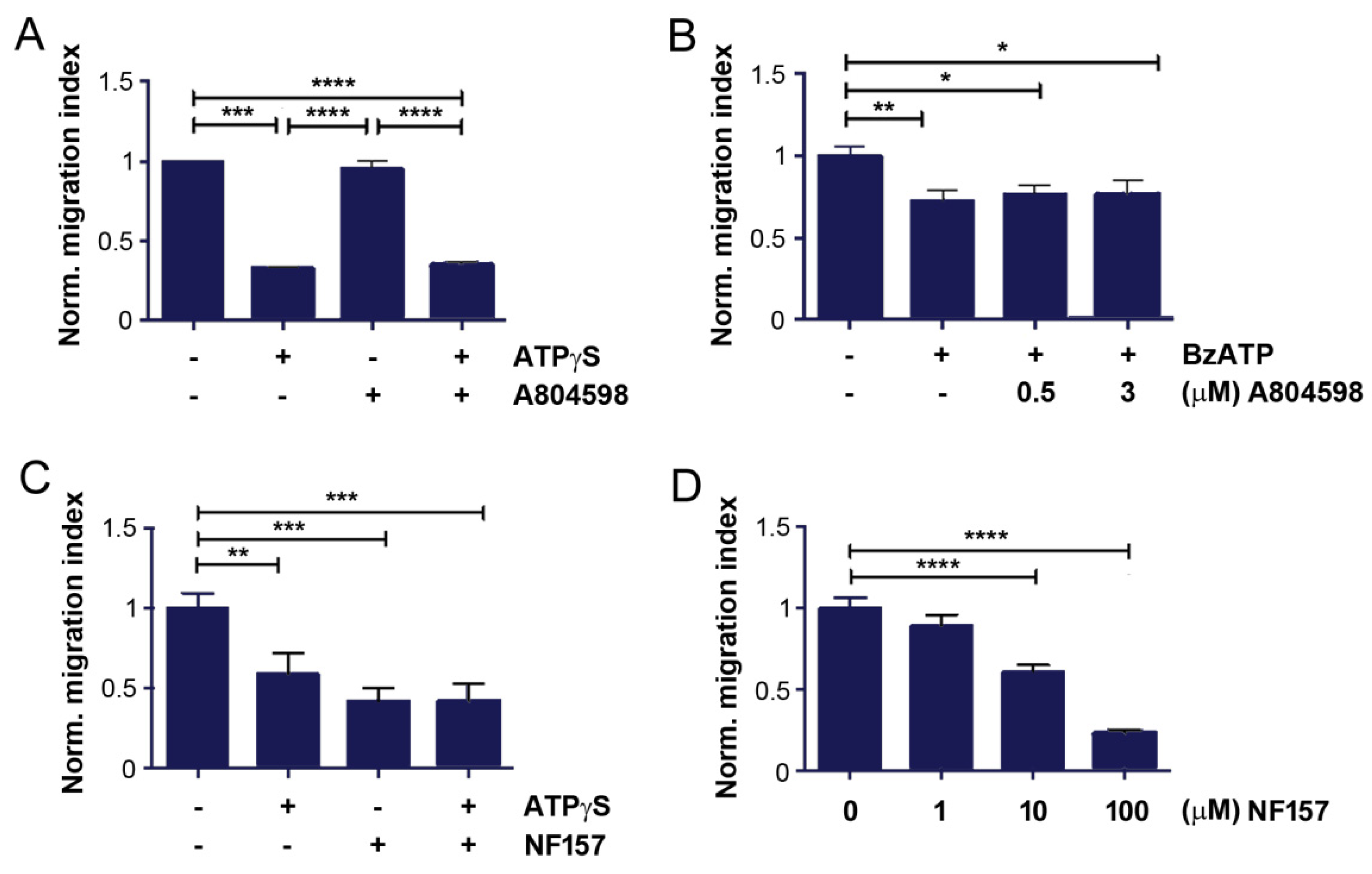
2.3. Inhibition of P2Y11 Attenuates Bone Tropic Cancer Cell Migration and CXCR4 Expression
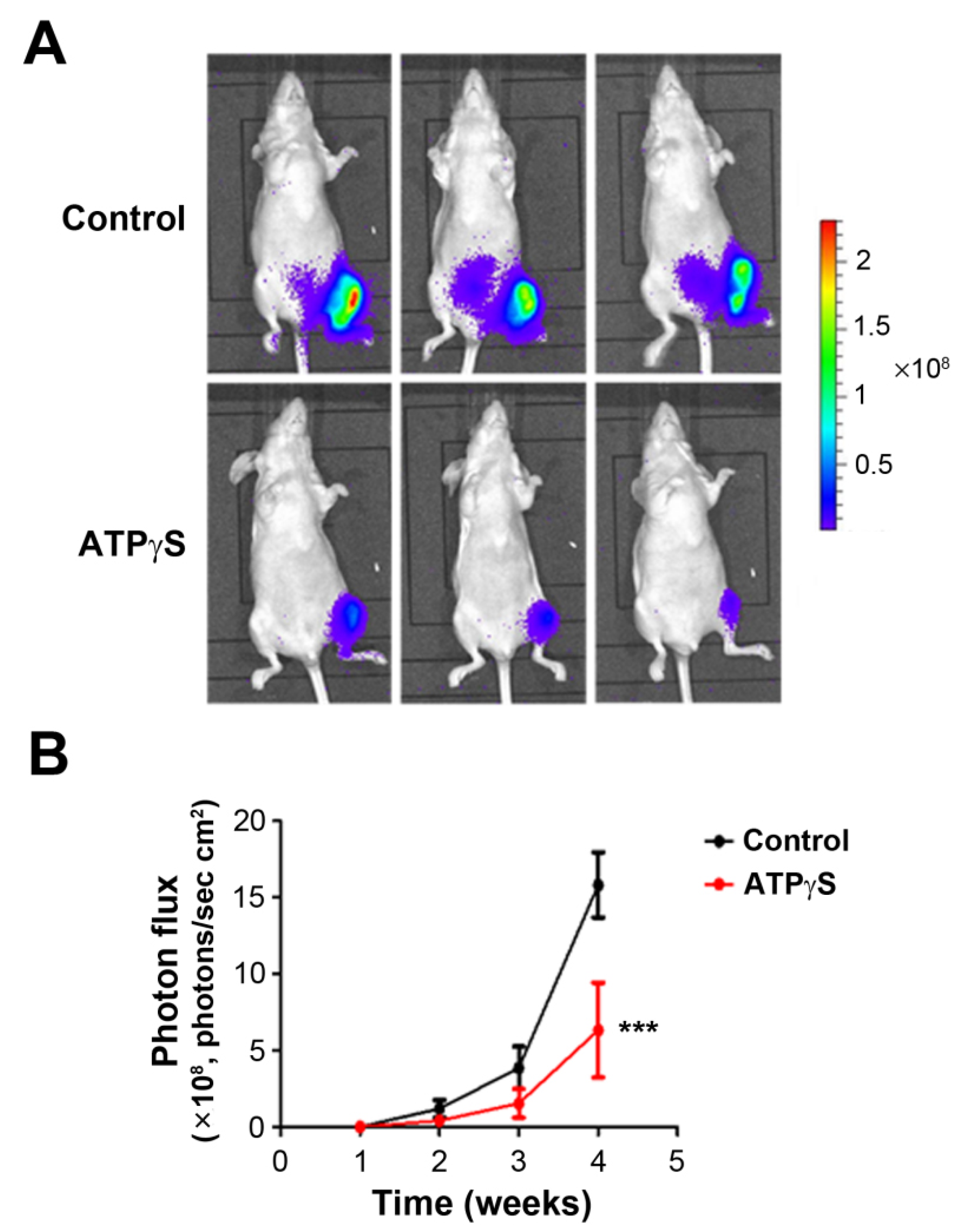
2.4. ATPγS Inhibits the Growth of Breast Cancer Cells MDA-BoM-1833 in the Tibia
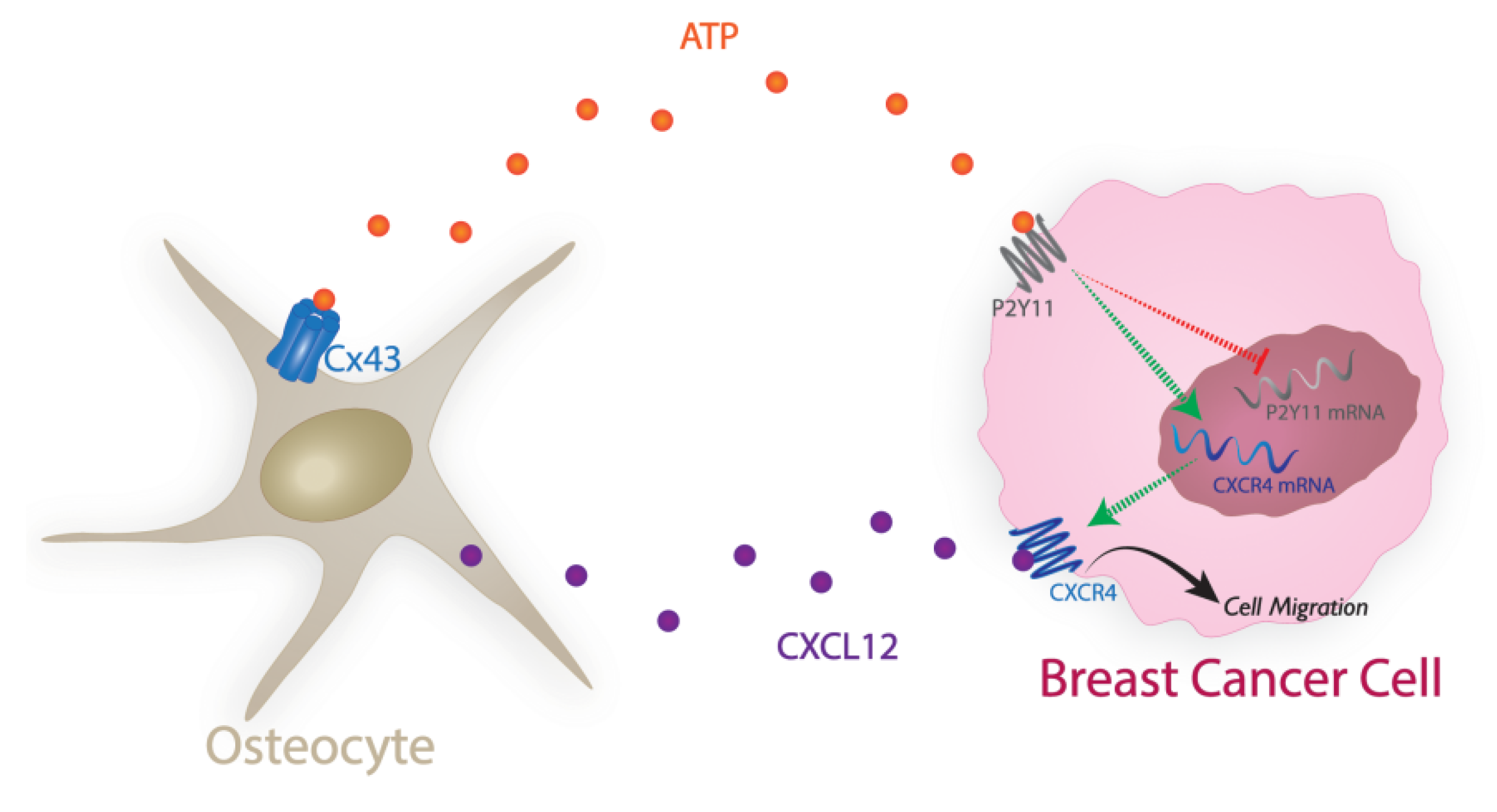
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Cultures
4.2. Wound-Healing Migration and Transwell Migration Assays
4.3. Cell Proliferation Assay
4.4. Quantitative Real Time qRT-PCR
4.5. Preparation of Crude Cell Membrane Extract and Western Blotting
4.6. Small Interfering RNA (siRNA) Transfection
4.7. Intratibial Injection and Bioluminescence Imaging
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2019. Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef]
- Roodman, G.D. Mechanism of bone metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef]
- Vacchelli, E.; Enot, D.P.; Pietrocola, F.; Zitvogel, L.; Kroemer, G. Impact of Pattern Recognition Receptors on the Prognosis of Breast Cancer Patients Undergoing Adjuvant Chemotherapy. Cancer Res. 2016, 76, 3122–3126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Virgilio, F.; Sarti, A.C.; Falzoni, S.; De Marchi, E.; Adinolfi, E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 2018, 18, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Shabbir, M.; Burnstock, G. Purinergic receptor-mediated effects of adenosine 5′-triphosphate in urological malignant diseases. Int. J. Urol. 2009, 16, 143–150. [Google Scholar] [CrossRef]
- White, N.; Burnstock, G. P2 receptors and cancer. Trends Pharmacol. Sci. 2006, 27, 211–217. [Google Scholar] [CrossRef]
- Rapaport, E. Treatment of human tumor cells with ADP or ATP yields arrest of growth in the S phase of the cell cycle. J. Cell. Physiol. 1983, 114, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.Z.; Riquelme, M.A.; Gao, X.; Ellies, L.G.; Sun, L.Z.; Jiang, J.X. Differential impact of adenosine nucleotides released by osteocytes on breast cancer growth and bone metastasis. Oncogene 2015, 34, 1831–1842. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.Z.; Riquelme, M.A.; Gu, S.; Kar, R.; Gao, X.; Sun, L.; Jiang, J.X. Osteocytic connexin hemichannels suppress breast cancer growth and bone metastasis. Oncogene 2016, 35, 5597–5607. [Google Scholar] [CrossRef] [Green Version]
- Leth-Larsen, R.; Lund, R.; Hansen, H.V.; Laenkholm, A.V.; Tarin, D.; Jensen, O.N.; Ditzel, H.J. Metastasis-related plasma membrane proteins of human breast cancer cells identified by comparative quantitative mass spectrometry. Mol. Cell. Proteom. 2009, 8, 1436–1449. [Google Scholar] [CrossRef] [Green Version]
- Di Virgilio, F.; Adinolfi, E. Extracellular purines, purinergic receptors and tumor growth. Oncogene 2017, 36, 293–303. [Google Scholar] [CrossRef]
- Choi, J.H.; Ji, Y.G.; Ko, J.J.; Cho, H.J.; Lee, D.H. Activating P2X7 Receptors Increases Proliferation of Human Pancreatic Cancer Cells via ERK1/2 and JNK. Pancreas 2018, 47, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Avanzato, D.; Genova, T.; Fiorio Pla, A.; Bernardini, M.; Bianco, S.; Bussolati, B.; Mancardi, D.; Giraudo, E.; Maione, F.; Cassoni, P.; et al. Activation of P2X7 and P2Y11 purinergic receptors inhibits migration and normalizes tumor-derived endothelial cells via cAMP signaling. Sci. Rep. 2016, 6, 32602. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Di Virgilio, F. Purinergic signalling and cancer. Purinergic Signal. 2013, 9, 491–540. [Google Scholar] [CrossRef]
- Khalid, M.; Brisson, L.; Tariq, M.; Hao, Y.; Guibon, R.; Fromont, G.; Mortadza, S.A.S.; Mousawi, F.; Manzoor, S.; Roger, S.; et al. Carcinoma-specific expression of P2Y11 receptor and its contribution in ATP-induced purinergic signalling and cell migration in human hepatocellular carcinoma cells. Oncotarget 2017, 8, 37278–37290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, E.H.; Singh, B.; Cristofanilli, M.; Gelovani, J.; Wei, C.; Vincent, L.; Cook, K.R.; Lucci, A. A CXCR4 antagonist CTCE-9908 inhibits primary tumor growth and metastasis of breast cancer. J. Surg. Res. 2009, 155, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Scala, S. Molecular Pathways: Targeting the CXCR4-CXCL12 Axis—Untapped Potential in the Tumor Microenvironment. Clin. Cancer Res. 2015, 21, 4278–4285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, L.; Magalhaes, M.A.; Coniglio, S.J.; Condeelis, J.S.; Segall, J.E. Opposing roles of CXCR4 and CXCR7 in breast cancer metastasis. Breast Cancer Res. 2011, 13, R128. [Google Scholar] [CrossRef] [Green Version]
- Bachelder, R.E.; Wendt, M.A.; Mercurio, A.M. Vascular endothelial growth factor promotes breast carcinoma invasion in an autocrine manner by regulating the chemokine receptor CXCR4. Cancer Res. 2002, 62, 7203–7206. [Google Scholar]
- Guo, F.; Wang, Y.; Liu, J.; Mok, S.C.; Xue, F.; Zhang, W. CXCL12/CXCR4: A symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene 2016, 35, 816–826. [Google Scholar] [CrossRef]
- Smith, M.C.; Luker, K.E.; Garbow, J.R.; Prior, J.L.; Jackson, E.; Piwnica-Worms, D.; Luker, G.D. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 2004, 64, 8604–8612. [Google Scholar] [CrossRef] [Green Version]
- Azad, B.B.; Chatterjee, S.; Lesniak, W.G.; Lisok, A.; Pullambhatla, M.; Bhujwalla, Z.M.; Pomper, M.G.; Nimmagadda, S. A fully human CXCR4 antibody demonstrates diagnostic utility and therapeutic efficacy in solid tumor xenografts. Oncotarget 2016, 7, 12344–12358. [Google Scholar] [CrossRef] [Green Version]
- Guo, P.; You, J.O.; Yang, J.; Jia, D.; Moses, M.A.; Auguste, D.T. Inhibiting metastatic breast cancer cell migration via the synergy of targeted, pH-triggered siRNA delivery and chemokine axis blockade. Mol. Pharm. 2014, 11, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Guo, P.; McCarthy, C.; Wang, B.; Tao, Y.; Auguste, D. Peptide density targets and impedes triple negative breast cancer metastasis. Nat. Commun. 2018, 9, 2612. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordón-Cardo, C.; Guise, T.A.; Massagué, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Selicharova, I.; Sanda, M.; Mladkova, J.; Ohri, S.S.; Vashishta, A.; Fusek, M.; Jiracek, J.; Vetvicka, V. 2-DE analysis of breast cancer cell lines 1833 and 4175 with distinct metastatic organ-specific potentials: Comparison with parental cell line MDA-MB-231. Oncol. Rep. 2008, 19, 1237–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreisig, K.; Kornum, B.R. A critical look at the function of the P2Y11 receptor. Purinergic Signal. 2016, 12, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; He, H.Y.; Li, H.M.; Zheng, J.; Heng, W.J.; You, J.F.; Fang, W.G. ERK1/2 and p38 pathways are required for P2Y receptor-mediated prostate cancer invasion. Cancer Lett. 2004, 215, 239–247. [Google Scholar] [CrossRef]
- Rapaport, E.; Fishman, R.F.; Gercel, C. Growth inhibition of human tumor cells in soft-agar cultures by treatment with low levels of adenosine 5′-triphosphate. Cancer Res. 1983, 43, 4402–4406. [Google Scholar]
- White, N.; Ryten, M.; Clayton, E.; Butler, P.; Burnstock, G. P2Y purinergic receptors regulate the growth of human melanomas. Cancer Lett. 2005, 224, 81–91. [Google Scholar] [CrossRef]
- Gao, Z.G.; Jacobson, K.A. A(2B) Adenosine Receptor and Cancer. Int. J. Mol. Sci. 2019, 20, 5139. [Google Scholar] [CrossRef] [Green Version]
- Höpfner, M.; Lemmer, K.; Jansen, A.; Hanski, C.; Riecken, E.O.; Gavish, M.; Mann, B.; Buhr, H.; Glassmeier, G.; Scherübl, H. Expression of functional P2-purinergic receptors in primary cultures of human colorectal carcinoma cells. Biochem. Biophys. Res. Commun. 1998, 251, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Beukers, M.W.; Kerkhof, C.J.; van Rhee, M.A.; Ardanuy, U.; Gurgel, C.; Widjaja, H.; Nickel, P.; AP, I.J.; Soudijn, W. Suramin analogs, divalent cations and ATP gamma S as inhibitors of ecto-ATPase. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1995, 351, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.B.; Espenel, S.; Vallard, A.; Battiston-Montagne, P.; Wozny, A.S.; Ardail, D.; Alphonse, G.; Rancoule, C.; Rodriguez-Lafrasse, C.; Magne, N. Evaluation of the Cell Invasion and Migration Process: A Comparison of the Video Microscope-based Scratch Wound Assay and the Boyden Chamber Assay. J. Vis. Exp. 2017, 129, 56337. [Google Scholar] [CrossRef] [PubMed]
- Hulkower, K.I.; Herber, R.L. Cell migration and invasion assays as tools for drug discovery. Pharmaceutics 2011, 3, 107–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Kretschmann, K.L.; Welm, A.L. Mouse models of breast cancer metastasis to bone. Cancer Metastasis Rev. 2012, 31, 579–583. [Google Scholar] [CrossRef]
- Park, J.H.; Williams, D.R.; Lee, J.H.; Lee, S.D.; Lee, J.H.; Ko, H.; Lee, G.E.; Kim, S.; Lee, J.M.; Abdelrahman, A.; et al. Potent Suppressive Effects of 1-Piperidinylimidazole Based Novel P2X7 Receptor Antagonists on Cancer Cell Migration and Invasion. J. Med. Chem. 2016, 59, 7410–7430. [Google Scholar] [CrossRef]
- Boldrini, L.; Giordano, M.; Alì, G.; Melfi, F.; Romano, G.; Lucchi, M.; Fontanini, G. P2X7 mRNA expression in non-small cell lung cancer: MicroRNA regulation and prognostic value. Oncol. Lett. 2015, 9, 449–453. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; McCormick, T.; Qi, X.; Luo, L.; Zhou, L.; Li, X.; Wang, B.C.; Gibbons, H.E.; Abdul-Karim, F.W.; Gorodeski, G.I. Activation of P2X(7)-mediated apoptosis Inhibits DMBA/TPA-induced formation of skin papillomas and cancer in mice. BMC Cancer 2009, 9, 114. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, C.; Ziegler, N.; Reiner, S.; Krasel, C.; Lohse, M.J. Agonist-selective, receptor-specific interaction of human P2Y receptors with beta-arrestin-1 and -2. J. Biol. Chem. 2008, 283, 30933–30941. [Google Scholar] [CrossRef] [Green Version]
- Ecke, D.; Hanck, T.; Tulapurkar, M.E.; Schäfer, R.; Kassack, M.; Stricker, R.; Reiser, G. Hetero-oligomerization of the P2Y11 receptor with the P2Y1 receptor controls the internalization and ligand selectivity of the P2Y11 receptor. Biochem. J. 2008, 409, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Haas, M.; Shaaban, A.; Reiser, G. Alanine-(87)-threonine polymorphism impairs signaling and internalization of the human P2Y11 receptor, when co-expressed with the P2Y1 receptor. J. Neurochem. 2014, 129, 602–613. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C. P2Y(11) Receptors: Properties, Distribution and Functions. Adv. Exp. Med. Biol. 2017, 1051, 107–122. [Google Scholar] [CrossRef] [Green Version]
- Shabbir, M.; Ryten, M.; Thompson, C.; Mikhailidis, D.; Burnstock, G. Characterization of calcium-independent purinergic receptor-mediated apoptosis in hormone-refractory prostate cancer. BJU Int. 2008, 101, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Ledderose, C.; Bromberger, S.; Slubowski, C.J.; Sueyoshi, K.; Aytan, D.; Shen, Y.; Junger, W.G. The purinergic receptor P2Y11 choreographs the polarization, mitochondrial metabolism, and migration of T lymphocytes. Sci. Signal. 2020, 13, eaba3300. [Google Scholar] [CrossRef] [PubMed]
- Communi, D.; Suarez-Huerta, N.; Dussossoy, D.; Savi, P.; Boeynaems, J.M. Cotranscription and intergenic splicing of human P2Y11 and SSF1 genes. J. Biol. Chem. 2001, 276, 16561–16566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, T.P.; Shen, H.; Liu, L.X.; Shu, Y.Q. The impact of chemokine receptor CXCR4 on breast cancer prognosis: A meta-analysis. Cancer Epidemiol. 2013, 37, 725–731. [Google Scholar] [CrossRef]
- Dewan, M.Z.; Ahmed, S.; Iwasaki, Y.; Ohba, K.; Toi, M.; Yamamoto, N. Stromal cell-derived factor-1 and CXCR4 receptor interaction in tumor growth and metastasis of breast cancer. Biomed. Pharmacother. 2006, 60, 273–276. [Google Scholar] [CrossRef]
- Wu, J.; Wu, X.; Liang, W.; Chen, C.; Zheng, L.; An, H. Clinicopathological and prognostic significance of chemokine receptor CXCR4 overexpression in patients with esophageal cancer: A meta-analysis. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 3709–3715. [Google Scholar] [CrossRef]
- Jiang, Q.; Sun, Y.; Liu, X. CXCR4 as a prognostic biomarker in gastrointestinal cancer: A meta-analysis. Biomark. Biochem. Indic. Expo. Response Susceptibility Chem. 2019, 24, 510–516. [Google Scholar] [CrossRef]
- Lv, S.; Yang, Y.; Kwon, S.; Han, M.; Zhao, F.; Kang, H.; Dai, C.; Wang, R. The association of CXCR4 expression with prognosis and clinicopathological indicators in colorectal carcinoma patients: A meta-analysis. Histopathology 2014, 64, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, S. CXCR4/CXCL12 in non-small-cell lung cancer metastasis to the brain. Int. J. Mol. Sci. 2013, 14, 1713–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.; Marian, D.; Weich, E.; Engl, T.; Wedel, S.; Relja, B.; Jonas, D.; Blaheta, R.A. CXCR4 chemokine receptor engagement modifies integrin dependent adhesion of renal carcinoma cells. Exp. Cell Res. 2007, 313, 4051–4065. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Ordoñez, A.; Seoane, S.; Cabezas, P.; Eiro, N.; Sendon-Lago, J.; Macia, M.; Garcia-Caballero, T.; Gonzalez, L.O.; Sanchez, L.; Vizoso, F.; et al. Breast cancer metastasis to liver and lung is facilitated by Pit-1-CXCL12-CXCR4 axis. Oncogene 2018, 37, 1430–1444. [Google Scholar] [CrossRef] [PubMed]
- Lapteva, N.; Yang, A.G.; Sanders, D.E.; Strube, R.W.; Chen, S.Y. CXCR4 knockdown by small interfering RNA abrogates breast tumor growth in vivo. Cancer Gene Ther. 2005, 12, 84–89. [Google Scholar] [CrossRef]
- Yang, M.; Zeng, C.; Li, P.; Qian, L.; Ding, B.; Huang, L.; Li, G.; Jiang, H.; Gong, N.; Wu, W. Impact of CXCR4 and CXCR7 knockout by CRISPR/Cas9 on the function of triple-negative breast cancer cells. OncoTargets Ther. 2019, 12, 3849–3858. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Stamatoyannopoulos, G.; Song, C.Z. Down-regulation of CXCR4 by inducible small interfering RNA inhibits breast cancer cell invasion in vitro. Cancer Res. 2003, 63, 4801–4804. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Riquelme, M.A.; Tian, Y.; Zhao, D.; Acosta, F.M.; Gu, S.; Jiang, J.X. ATP Inhibits Breast Cancer Migration and Bone Metastasis through Down-Regulation of CXCR4 and Purinergic Receptor P2Y11. Cancers 2021, 13, 4293. https://doi.org/10.3390/cancers13174293
Liu X, Riquelme MA, Tian Y, Zhao D, Acosta FM, Gu S, Jiang JX. ATP Inhibits Breast Cancer Migration and Bone Metastasis through Down-Regulation of CXCR4 and Purinergic Receptor P2Y11. Cancers. 2021; 13(17):4293. https://doi.org/10.3390/cancers13174293
Chicago/Turabian StyleLiu, Xiaowen, Manuel A. Riquelme, Yi Tian, Dezhi Zhao, Francisca M. Acosta, Sumin Gu, and Jean X. Jiang. 2021. "ATP Inhibits Breast Cancer Migration and Bone Metastasis through Down-Regulation of CXCR4 and Purinergic Receptor P2Y11" Cancers 13, no. 17: 4293. https://doi.org/10.3390/cancers13174293