
Targeting c-IAP1, c-IAP2, and Bcl-2 Eliminates Senescent Glioblastoma Cells Following Temozolomide Treatment

 , ,
, , 
 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Drug Treatment
2.2. Preparation of RNA, cDNA Synthesis, and Real Time PCR
2.3. Determination of Cell Death, Cell Cycle Progression, and Senescence
2.4. Preparation of Protein Extracts and Western Blot Analysis
2.5. Quantification and Statistical Analyses
3. Results
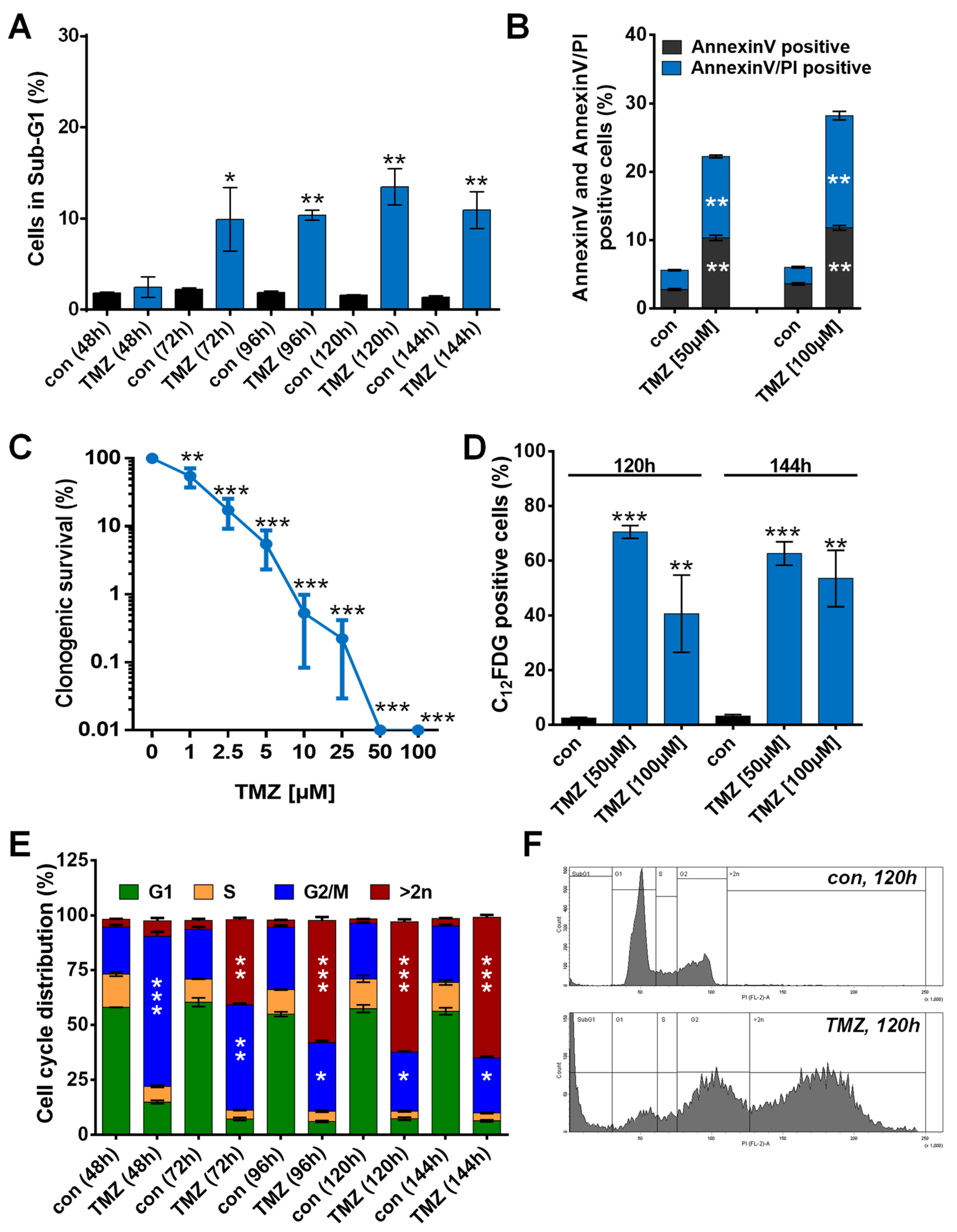
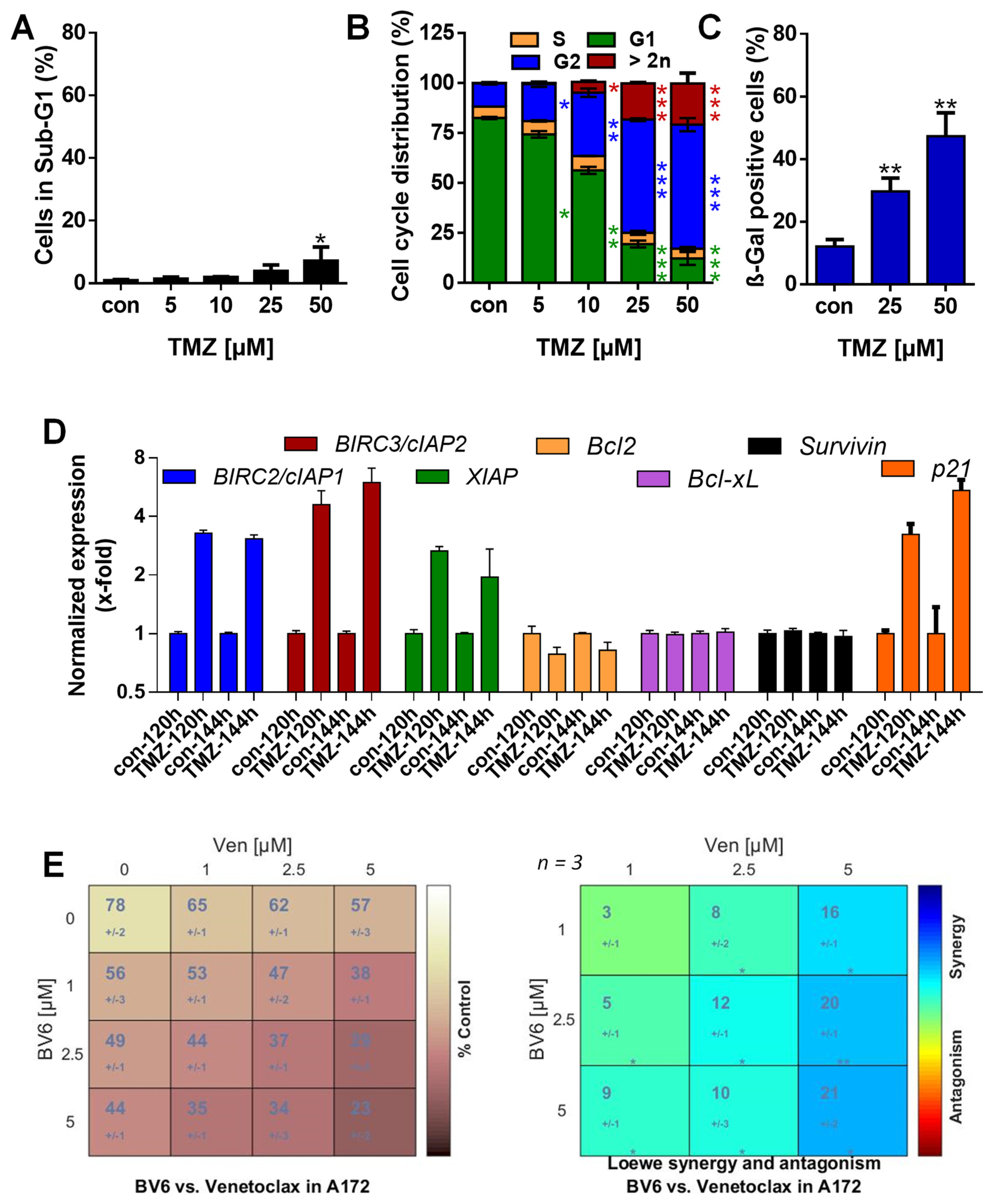
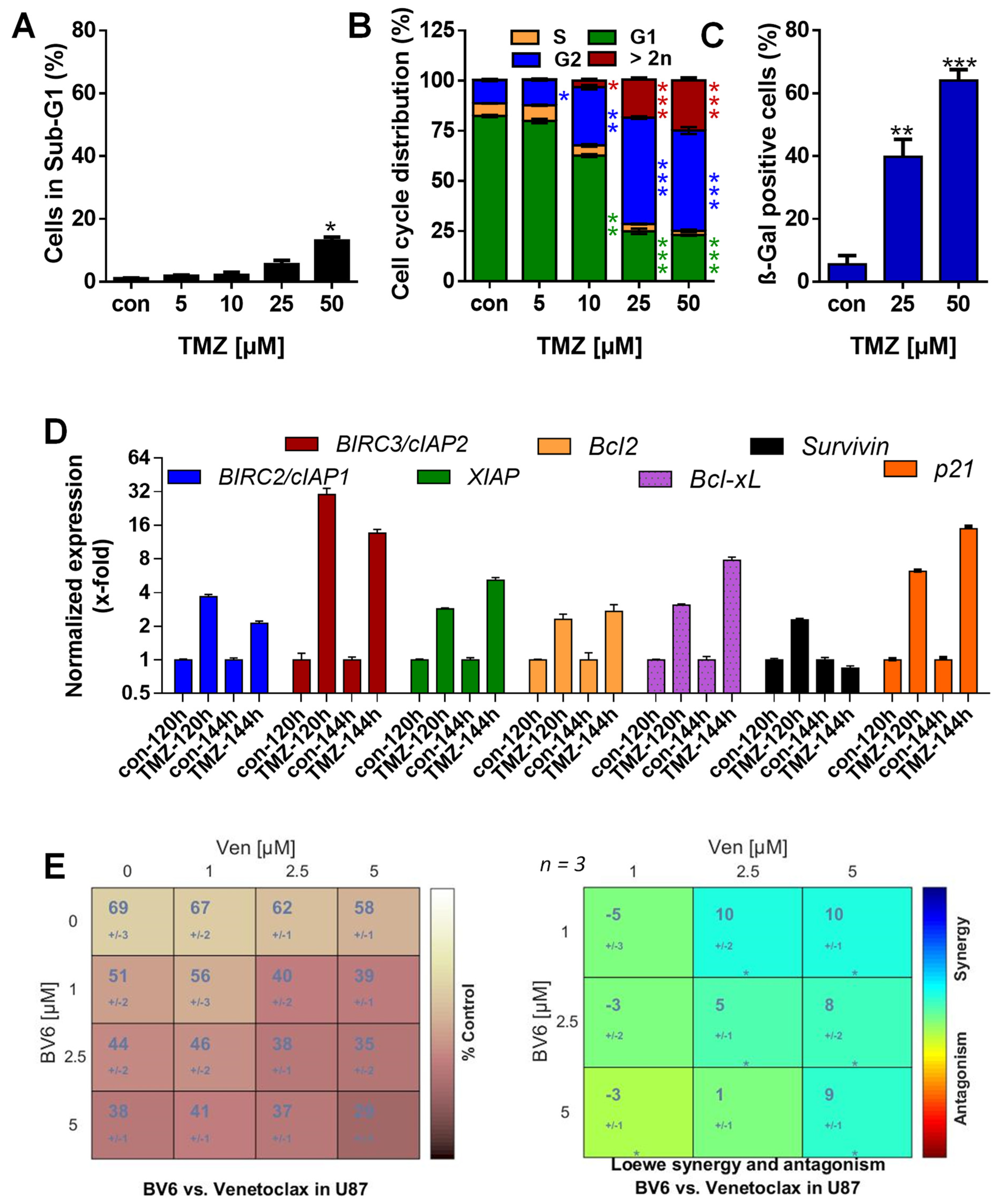
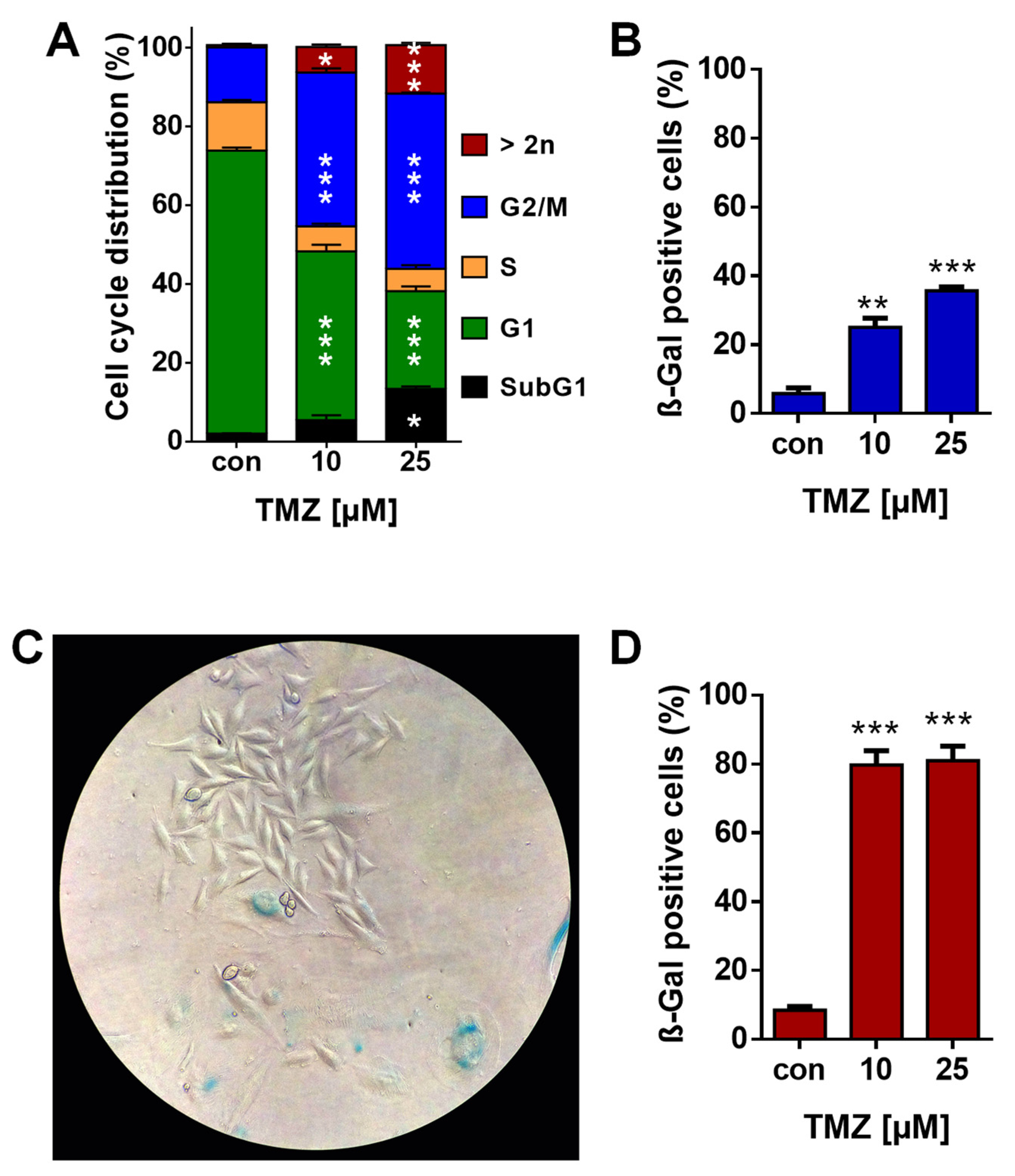
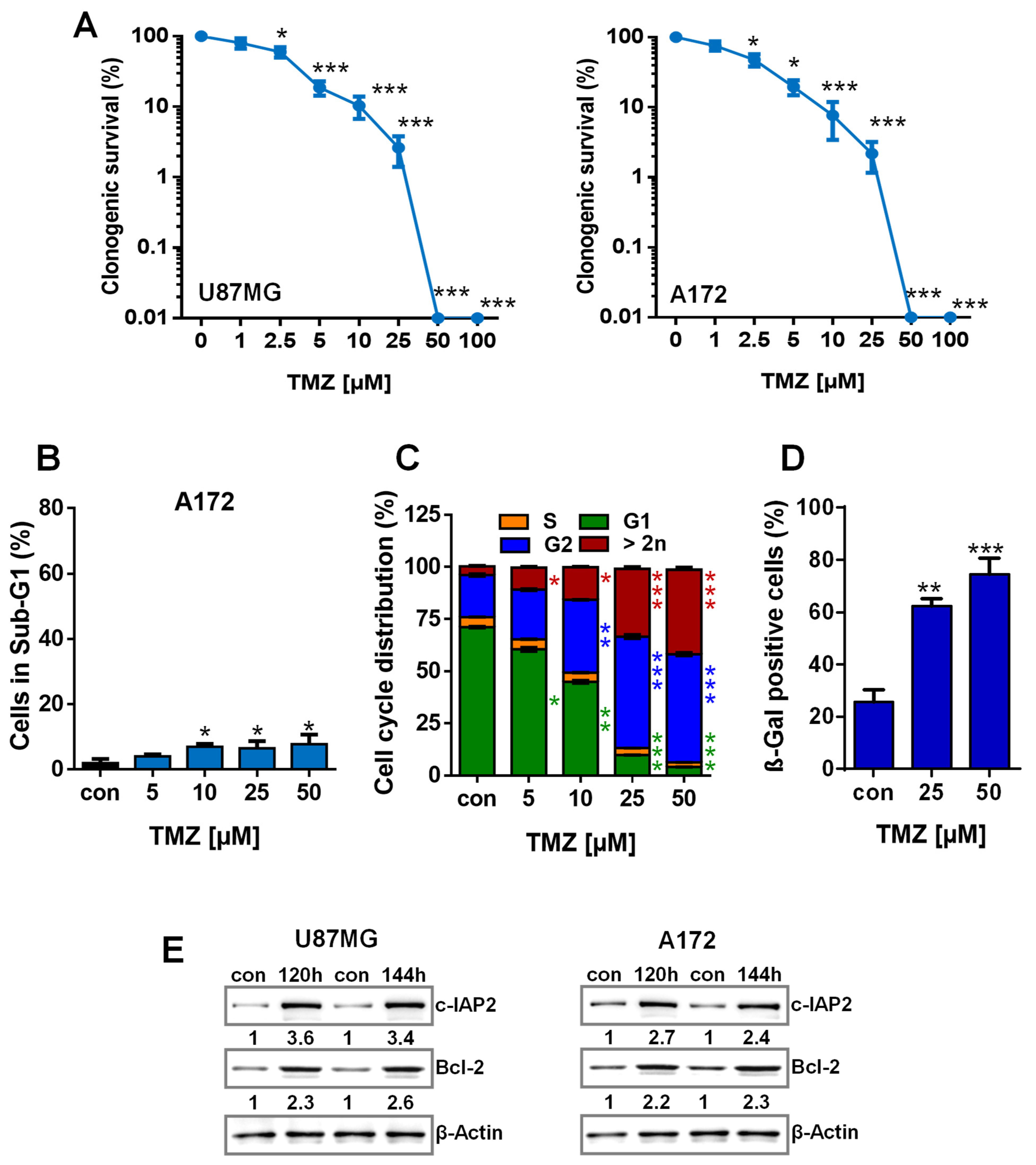
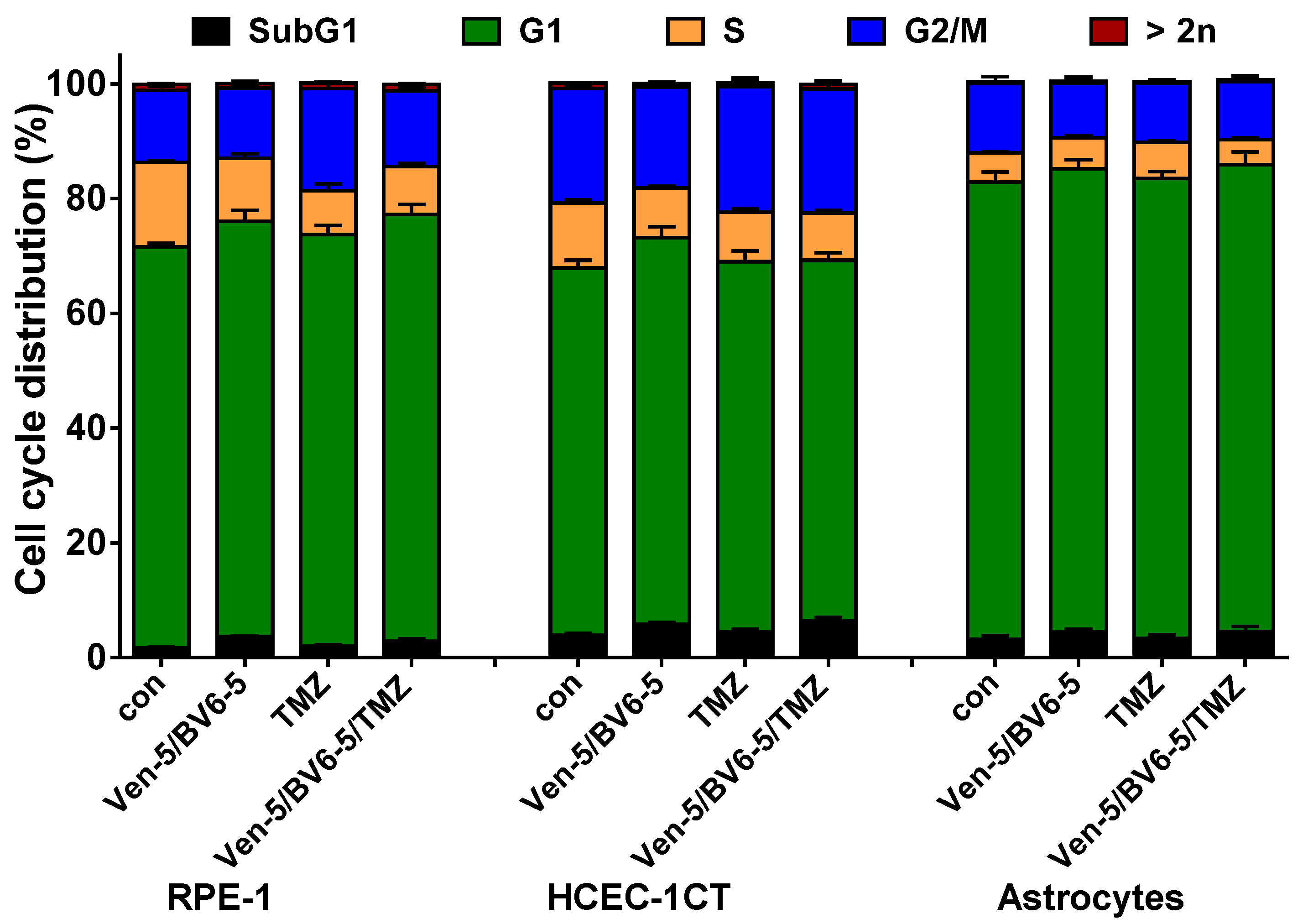
3.1. TMZ Induces Cell Death, Cell Cycle Arrest, and Senescence
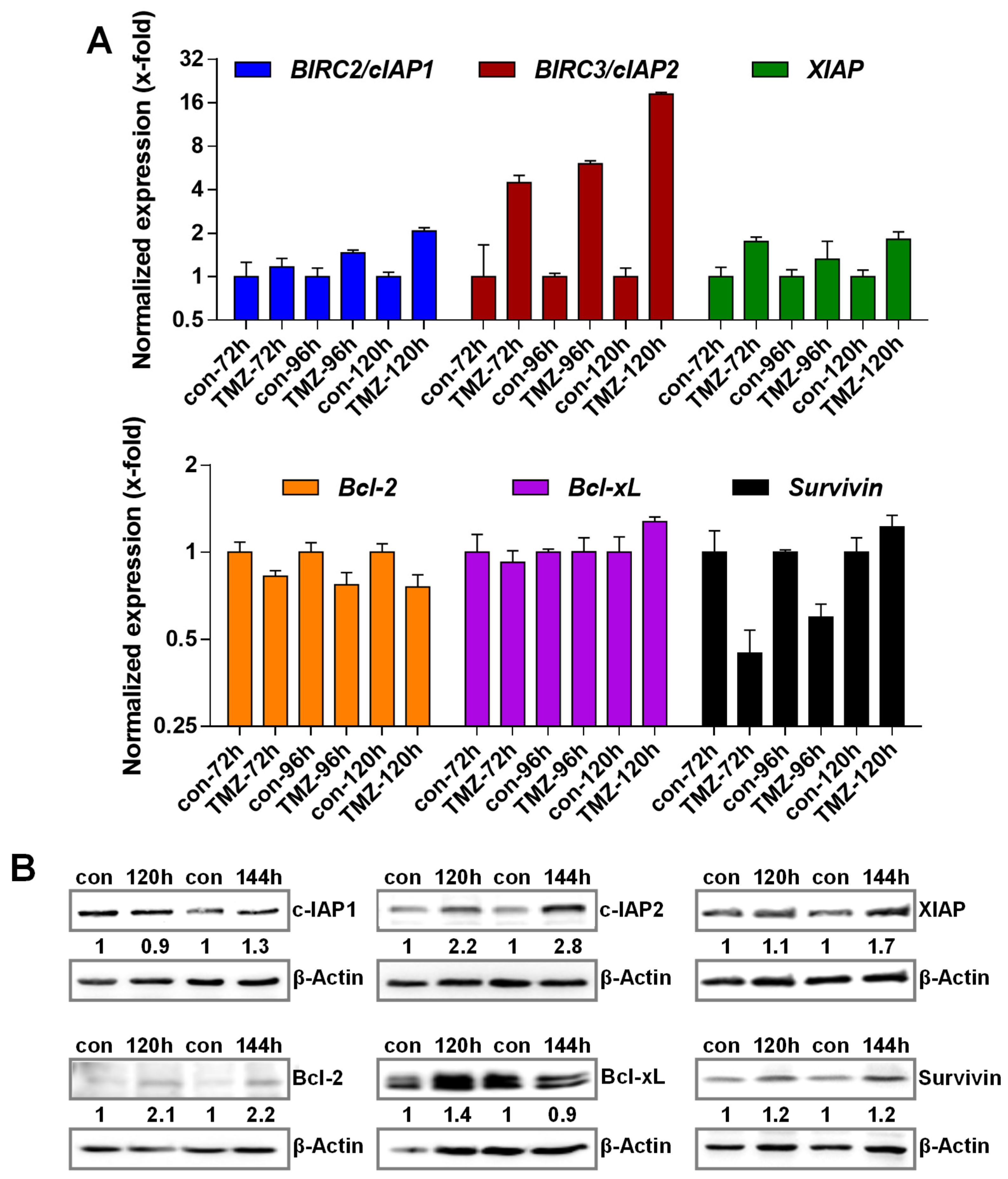
3.2. TMZ Induces Upregulation of Anti-Apoptotic Factors
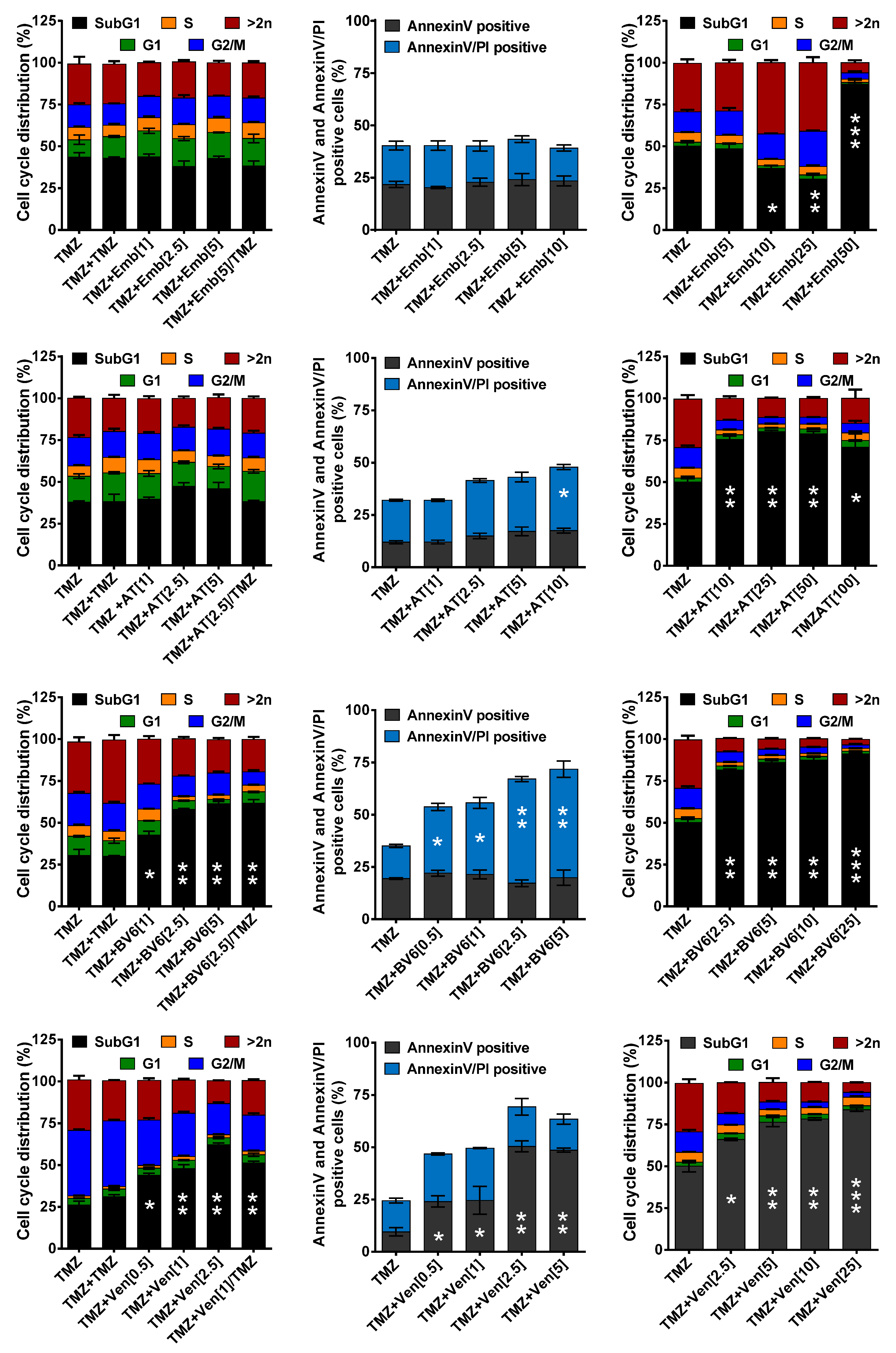
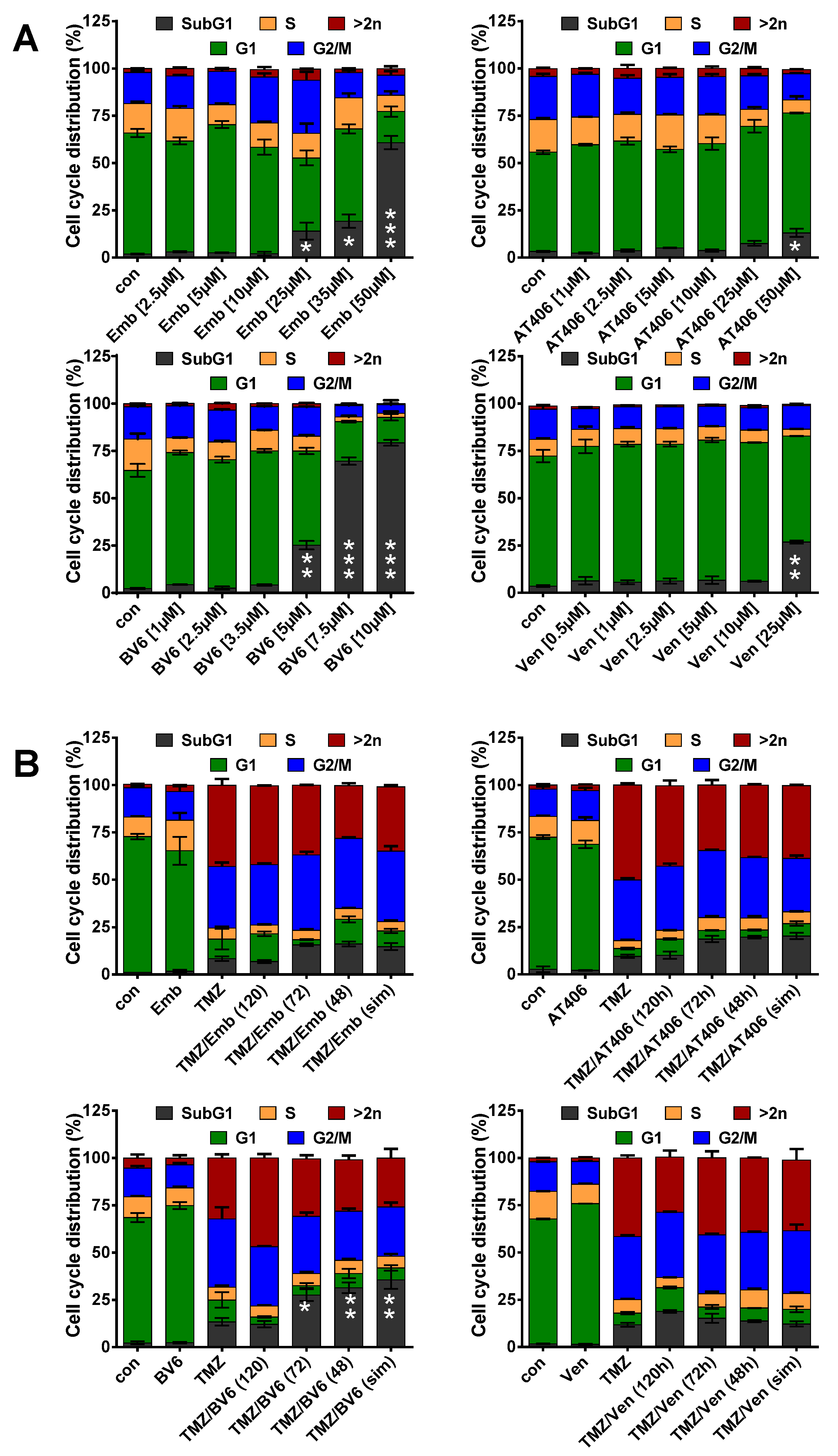
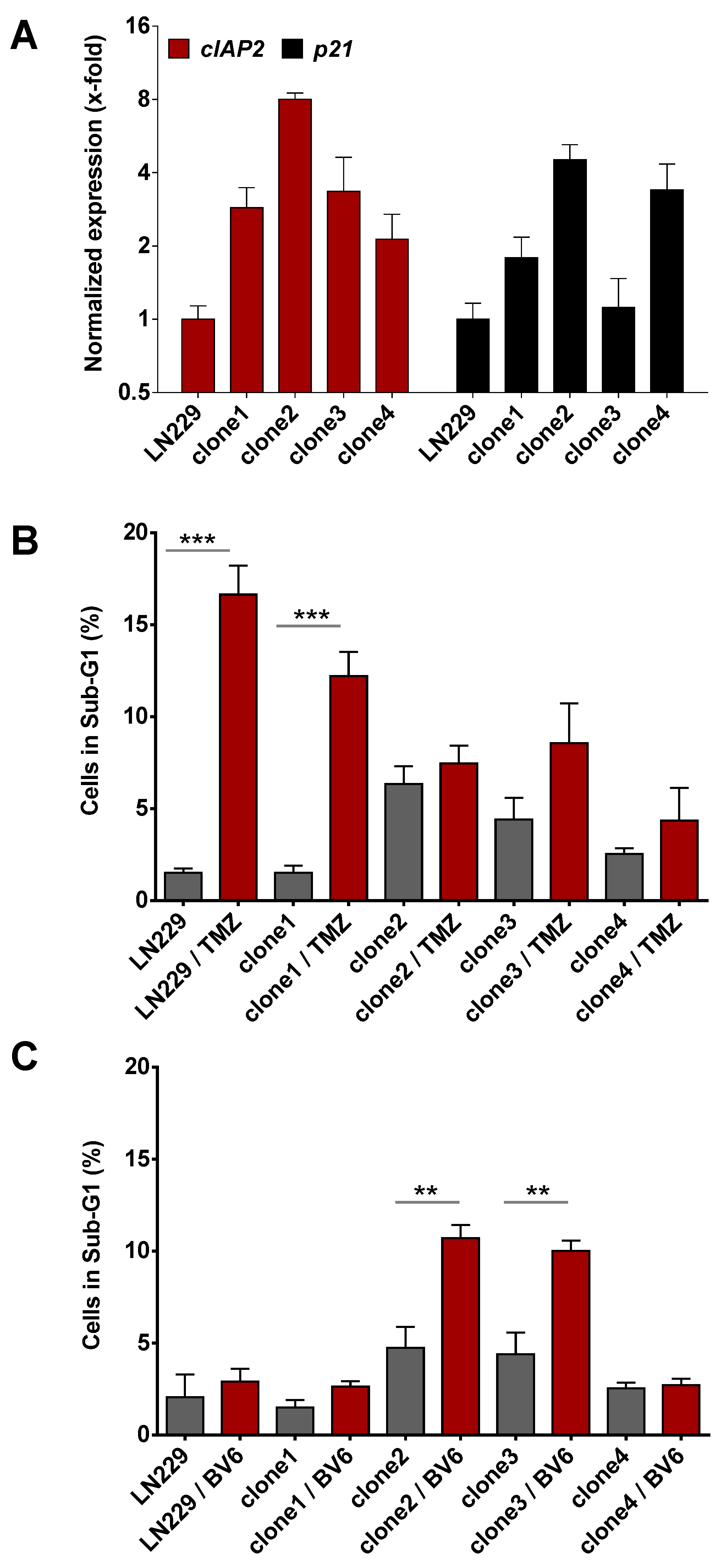
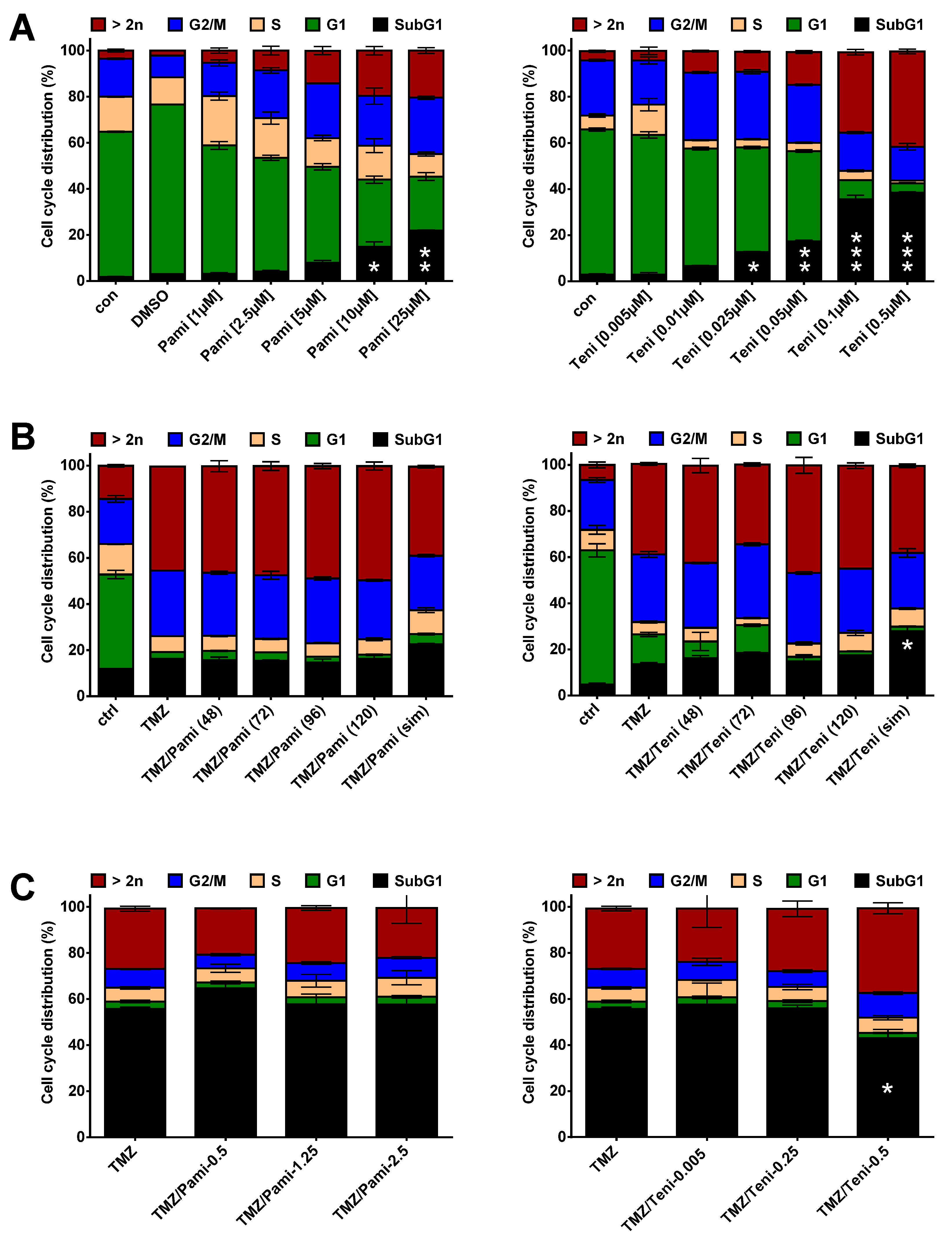
3.3. Inhibition of Anti-Apoptotic Factors at Early Time Points Impacts TMZ-Induced Cell Death
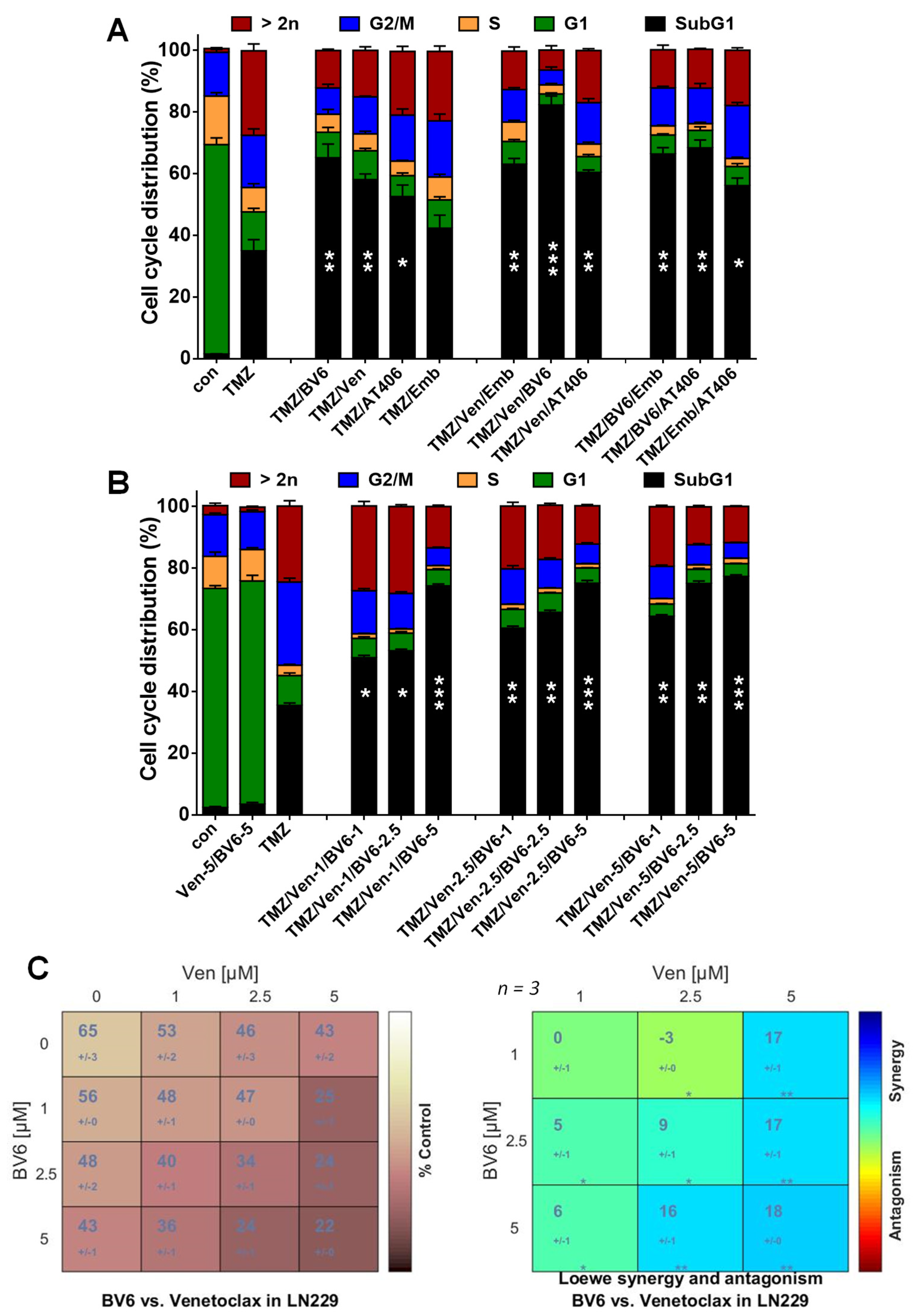
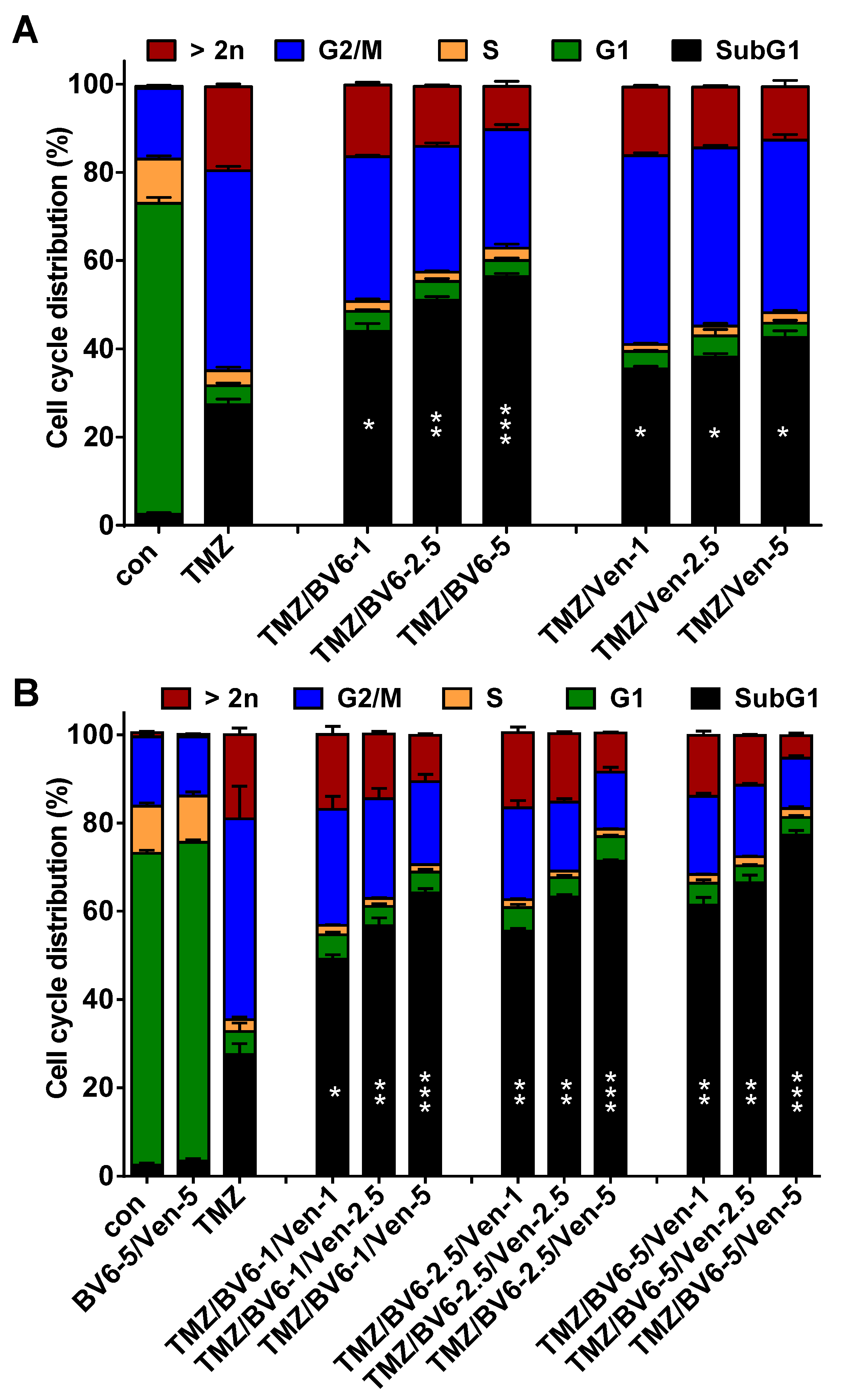
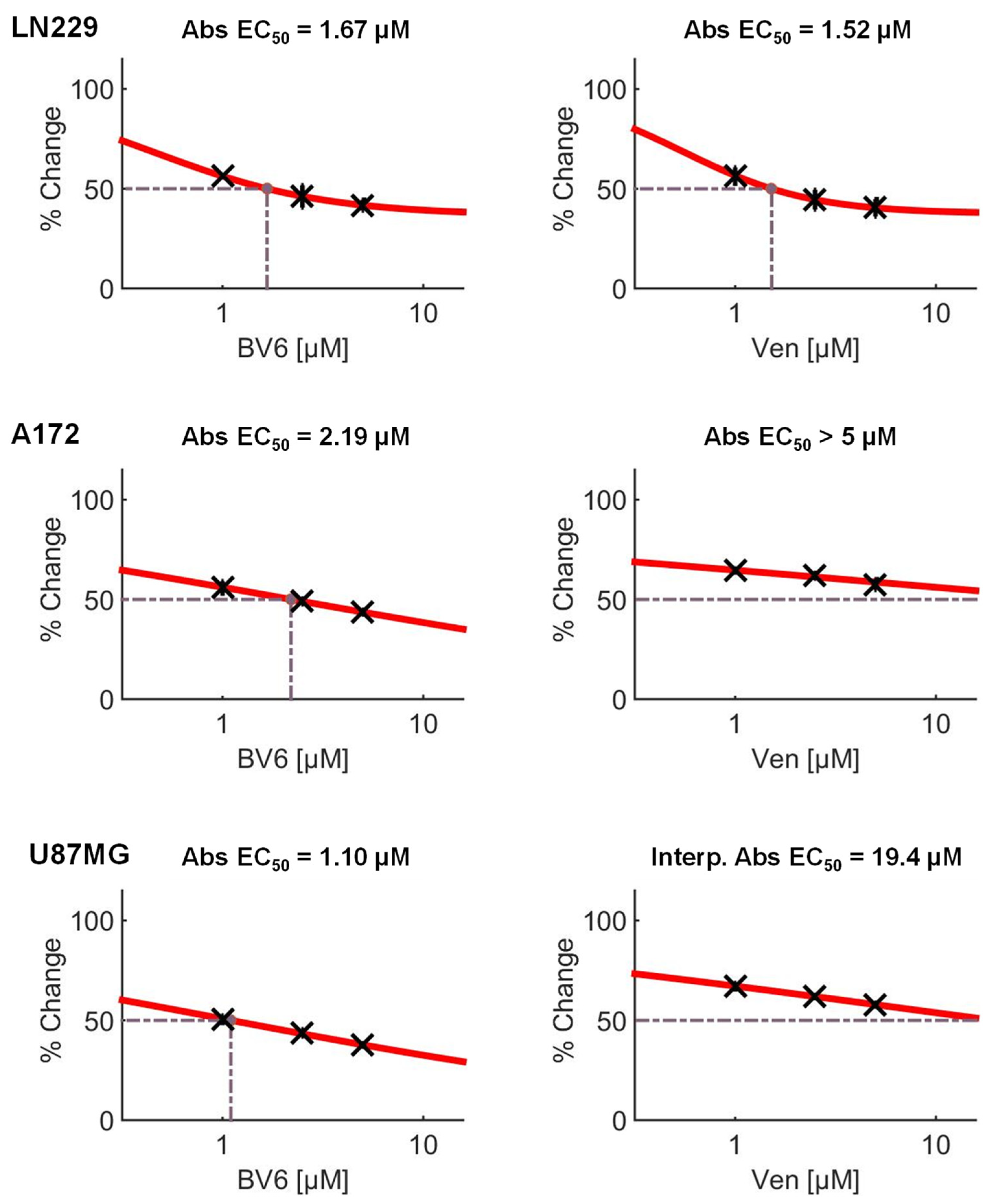
3.4. Inhibition of Anti-Apoptotic Factors Kills Senescent LN-229 Cells
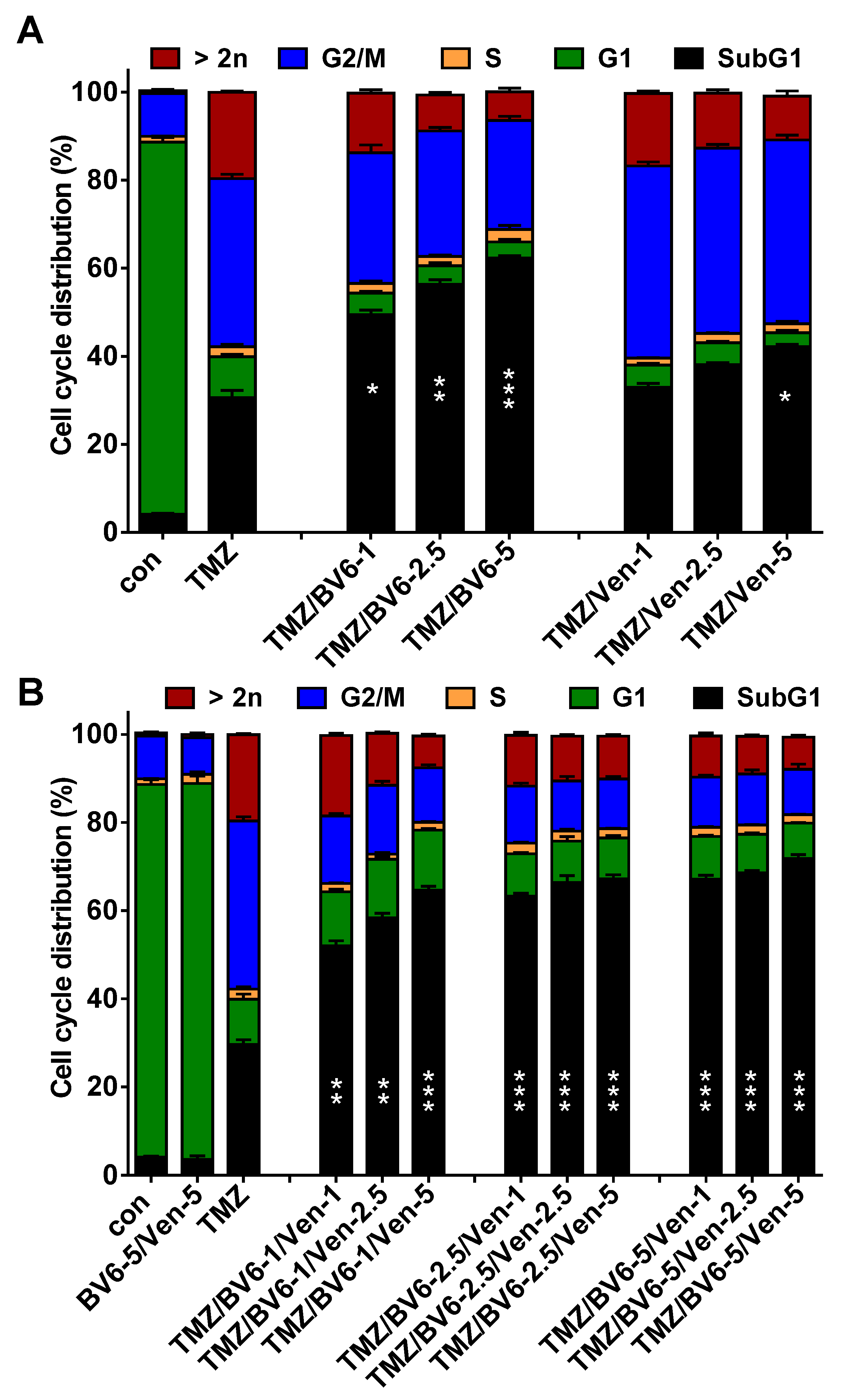
3.5. Inhibition of Anti-Apoptotic Factors Kills Senescent A172 and U87MG Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

| Gene | Primer Sequence (5′-3′) |
|---|---|
| ACTB-fw | TGGCATCCACGAAACTACC |
| ACTB-rev | GTGTTGGCGTACAGGTCTT |
| Bcl2-fw | TTCAGAGACAGCCAGGAGAAA |
| Bcl2-rev | AGTACCTGAACCGGCACCT |
| BCLxL-fw | AAGCGTAGACAAGGAGAT |
| BCLxL-rev | TAGGTGGTCATTCAGGTAA |
| BIRC2/c-IAP1-fw | TTCCCAGGTCCCTCGTATCA |
| BIRC2/c-IAP1-rev | CCGGCGGGGAAAGTTGAATA |
| BIRC3/c-IAP2-fw | TCACTCCCAGACTCTTTCCA |
| BIRC3/c-IAP2-rev | CCCCGTGTTCTACAAGTGTC |
| BIRC5/survivin-fw | ATGACTTGTGTGTGATGA |
| BIRC5/survivin-rev | GTTTGTGCTATTCTGTGAA |
| CDNK1a-fw | TACATCTTCTGCCTTAGT |
| CDNK1a-rev | TCTTAGGAACCTCTCATT |
| GAPDH-fw | CATGAGAAGTATGACAACAG |
| GAPDH-rev | ATGAGTCCTTCCACGATA |
| XIAP-fw | CCGAAGAGAAACCACATTT |
| XIAP-rev | CTGAGCCAGATCAAAGTATG |
| Protein | Molecularweight (kDa) | Antibody | RRID | Company |
|---|---|---|---|---|
| ß-Actin | 42 | sc-47778 | AB_2714189 | Santa Cruz Biotechnology |
| c-IAP1 | 70 | 7065S | AB_10890862 | Cell Signaling Technology |
| c-IAP2 | 68 | 3130S | AB_10693298 | Cell Signaling Technology |
| BCL-2 | 26 | 4223 | AB_1903909 | Cell Signaling Technology |
| BCL-xL | 26 | 2764 | AB_2228008 | Cell Signaling Technology |
| Survivin | 16 | 2808S | AB_2063948 | Cell Signaling Technology |
| XIAP | 57 | 14334S | AB_2784533 | Cell Signaling Technology |
| HRP conjugated anti-mouse | KCB002 | AB_10703407 | Rockland | |
| HRP conjugated anti-rabbit | KCB003 | AB_10702763 | Rockland |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet. Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Kaina, B.; Christmann, M. DNA repair in resistance to alkylating anticancer drugs. Int. J. Clin. Pharm. 2002, 40, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Christmann, M.; Diesler, K.; Majhen, D.; Steigerwald, C.; Berte, N.; Freund, H.; Stojanovic, N.; Kaina, B.; Osmak, M.; Ambriovic-Ristov, A.; et al. Integrin alphaVbeta3 silencing sensitizes malignant glioma cells to temozolomide by suppression of homologous recombination repair. Oncotarget 2017, 8, 27754–27771. [Google Scholar] [CrossRef] [Green Version]
- Tomicic, M.T.; Meise, R.; Aasland, D.; Berte, N.; Kitzinger, R.; Kramer, O.H.; Kaina, B.; Christmann, M. Apoptosis induced by temozolomide and nimustine in glioblastoma cells is supported by JNK/c-Jun-mediated induction of the BH3-only protein BIM. Oncotarget 2015, 6, 33755–33768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knizhnik, A.V.; Roos, W.P.; Nikolova, T.; Quiros, S.; Tomaszowski, K.H.; Christmann, M.; Kaina, B. Survival and death strategies in glioma cells: Autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS ONE 2013, 8, e55665. [Google Scholar] [CrossRef] [Green Version]
- Aasland, D.; Gotzinger, L.; Hauck, L.; Berte, N.; Meyer, J.; Effenberger, M.; Schneider, S.; Reuber, E.E.; Roos, W.P.; Tomicic, M.T.; et al. Temozolomide Induces Senescence and Repression of DNA Repair Pathways in Glioblastoma Cells via Activation of ATR-CHK1, p21, and NF-kappaB. Cancer Res. 2019, 79, 99–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayflick, L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- D’Adda di Fagagna, F. Living on a break: Cellular senescence as a DNA-damage response. Nat. Rev. Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef]
- Muller, M. Cellular senescence: Molecular mechanisms, in vivo significance, and redox considerations. Antioxid. Redox Signal. 2009, 11, 59–98. [Google Scholar] [CrossRef] [PubMed]
- Fridman, A.L.; Tainsky, M.A. Critical pathways in cellular senescence and immortalization revealed by gene expression profiling. Oncogene 2008, 27, 5975–5987. [Google Scholar] [CrossRef] [Green Version]
- Cichowski, K.; Hahn, W.C. Unexpected pieces to the senescence puzzle. Cell 2008, 133, 958–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velarde, M.C.; Demaria, M.; Campisi, J. Senescent cells and their secretory phenotype as targets for cancer therapy. Interdiscip. Top. Gerontol. 2013, 38, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef] [Green Version]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Bavik, C.; Coleman, I.; Dean, J.P.; Knudsen, B.; Plymate, S.; Nelson, P.S. The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res. 2006, 66, 794–802. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, S.; Coppe, J.P.; Krtolica, A.; Campisi, J. Stromal-epithelial interactions in aging and cancer: Senescent fibroblasts alter epithelial cell differentiation. J. Cell Sci. 2005, 118, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Laberge, R.M.; Awad, P.; Campisi, J.; Desprez, P.Y. Epithelial-mesenchymal transition induced by senescent fibroblasts. Cancer Microenviron. 2012, 5, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Hornsby, P.J. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007, 67, 3117–3126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sage, J.; Miller, A.L.; Perez-Mancera, P.A.; Wysocki, J.M.; Jacks, T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature 2003, 424, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Beausejour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef]
- Michishita, E.; Nakabayashi, K.; Ogino, H.; Suzuki, T.; Fujii, M.; Ayusawa, D. DNA topoisomerase inhibitors induce reversible senescence in normal human fibroblasts. Biochem. Biophys. Res. Commun. 1998, 253, 667–671. [Google Scholar] [CrossRef]
- Chitikova, Z.V.; Gordeev, S.A.; Bykova, T.V.; Zubova, S.G.; Pospelov, V.A.; Pospelova, T.V. Sustained activation of DNA damage response in irradiated apoptosis-resistant cells induces reversible senescence associated with mTOR downregulation and expression of stem cell markers. Cell Cycle 2014, 13, 1424–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Schmitt, C.A. The dynamic nature of senescence in cancer. Nat. Cell Biol. 2019, 21, 94–101. [Google Scholar] [CrossRef]
- Yu, Y.; Schleich, K.; Yue, B.; Ji, S.; Lohneis, P.; Kemper, K.; Silvis, M.R.; Qutob, N.; van Rooijen, E.; Werner-Klein, M.; et al. Targeting the Senescence-Overriding Cooperative Activity of Structurally Unrelated H3K9 Demethylases in Melanoma. Cancer Cell 2018, 33, 322–336.e328. [Google Scholar] [CrossRef] [Green Version]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Dabritz, J.H.M.; Zhao, Z.; Yu, Y.; Dorr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- Leikam, C.; Hufnagel, A.L.; Otto, C.; Murphy, D.J.; Muhling, B.; Kneitz, S.; Nanda, I.; Schmid, M.; Wagner, T.U.; Haferkamp, S.; et al. In vitro evidence for senescent multinucleated melanocytes as a source for tumor-initiating cells. Cell Death Dis. 2015, 6, e1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomicic, M.T.; Christmann, M. Targeting anticancer drug-induced senescence in glioblastoma therapy. Oncotarget 2018, 9, 37466–37467. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The Clinical Potential of Senolytic Drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Roig, A.I.; Eskiocak, U.; Hight, S.K.; Kim, S.B.; Delgado, O.; Souza, R.F.; Spechler, S.J.; Wright, W.E.; Shay, J.W. Immortalized epithelial cells derived from human colon biopsies express stem cell markers and differentiate in vitro. Gastroenterology 2010, 138, 1012–1021.e5. [Google Scholar] [CrossRef]
- Tomicic, M.T.; Krämer, F.; Nguyen, A.; Schwarzenbach, C.; Christmann, M. Oxaliplatin-Induced Senescence in Colorectal Cancer Cells Depends on p14ARF-Mediated Sustained p53 Activation. Cancers 2021, 13, 2019. [Google Scholar] [CrossRef]
- Christmann, M.; Kaina, B. Nuclear translocation of mismatch repair proteins MSH2 and MSH6 as a response of cells to alkylating agents. J. Biol. Chem. 2000, 275, 36256–36262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Veroli, G.Y.; Fornari, C.; Wang, D.; Mollard, S.; Bramhall, J.L.; Richards, F.M.; Jodrell, D.I. Combenefit: An interactive platform for the analysis and visualization of drug combinations. Bioinformatics 2016, 32, 2866–2868. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Kaina, B. Are There Thresholds in Glioblastoma Cell Death Responses Triggered by Temozolomide? Int. J. Mol. Sci. 2019, 20, 1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratenwerth, B.; Geisen, S.M.; He, Y.; Beltzig, L.; Sturla, S.J.; Kaina, B. Molecular dosimetry of temozolomide: Quantification of critical lesions, correlation to cell death responses and threshold doses. Mol. Cancer Ther. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Eich, M.; Roos, W.P.; Nikolova, T.; Kaina, B. Contribution of ATM and ATR to the resistance of glioblastoma and malignant melanoma cells to the methylating anticancer drug temozolomide. Mol. Cancer Ther. 2013, 12, 2529–2540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiros, S.; Roos, W.P.; Kaina, B. Processing of O6-methylguanine into DNA double-strand breaks requires two rounds of replication whereas apoptosis is also induced in subsequent cell cycles. Cell Cycle 2010, 9, 168–178. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Qin, L.; Feng, R.; Hu, G.; Sun, H.; He, Y.; Zhang, R. Emerging senolytic agents derived from natural products. Mech. Ageing Dev. 2019, 181, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Reich, T.R.; Schwarzenbach, C.; Vilar, J.B.; Unger, S.; Muhlhausler, F.; Nikolova, T.; Poplawski, A.; Baymaz, H.I.; Beli, P.; Christmann, M.; et al. Localization matters: Nuclear-trapped Survivin sensitizes glioblastoma cells to temozolomide by elevating cellular senescence and impairing homologous recombination. Cell. Mol. Life Sci. CMLS 2021, 78, 5587–5604. [Google Scholar] [CrossRef]
- Kuffer, C.; Kuznetsova, A.Y.; Storchova, Z. Abnormal mitosis triggers p53-dependent cell cycle arrest in human tetraploid cells. Chromosoma 2013, 122, 305–318. [Google Scholar] [CrossRef]
- Walen, K.H. Genetic stability of senescence reverted cells: Genome reduction division of polyploidy cells, aneuploidy and neoplasia. Cell Cycle 2008, 7, 1623–1629. [Google Scholar] [CrossRef]
- Mosieniak, G.; Sikora, E. Polyploidy: The link between senescence and cancer. Curr. Pharm. Des. 2010, 16, 734–740. [Google Scholar] [CrossRef] [Green Version]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: From discovery to translation. J. Intern. Med. 2020, 288, 518–536. [Google Scholar] [CrossRef]
- Wang, E. Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res. 1995, 55, 2284–2292. [Google Scholar] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Shahbandi, A.; Rao, S.G.; Anderson, A.Y.; Frey, W.D.; Olayiwola, J.O.; Ungerleider, N.A.; Jackson, J.G. BH3 mimetics selectively eliminate chemotherapy-induced senescent cells and improve response in TP53 wild-type breast cancer. Cell Death Differ. 2020, 27, 3097–3116. [Google Scholar] [CrossRef] [PubMed]
- Malaquin, N.; Vancayseele, A.; Gilbert, S.; Antenor-Habazac, L.; Olivier, M.A.; Ait Ali Brahem, Z.; Saad, F.; Delouya, G.; Rodier, F. DNA Damage- But Not Enzalutamide-Induced Senescence in Prostate Cancer Promotes Senolytic Bcl-xL Inhibitor Sensitivity. Cells 2020, 9, 1593. [Google Scholar] [CrossRef] [PubMed]
- Marschall, V.; Fulda, S. Smac mimetic-induced upregulation of interferon-beta sensitizes glioblastoma to temozolomide-induced cell death. Cell Death Dis. 2015, 6, e1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Berglund, A.; Kenchappa, R.S.; Forsyth, P.A.; Mule, J.J.; Etame, A.B. BIRC3 is a novel driver of therapeutic resistance in Glioblastoma. Sci. Rep. 2016, 6, 21710. [Google Scholar] [CrossRef] [Green Version]
- Tomicic, M.T. Replica to the Opinion Letter regarding the article “Sensitization of colorectal cancer cells to irinotecan by the Survivin inhibitor LLP3 depends on XAF1 proficiency in the context of mutated p53” (Arch Toxicol https://doi.org/10.1007/s00204-018-240-x). Arch. Toxicol. 2018, 92, 3243–3244. [Google Scholar] [CrossRef]
- Steigerwald, C.; Rasenberger, B.; Christmann, M.; Tomicic, M.T. Sensitization of colorectal cancer cells to irinotecan by the Survivin inhibitor LLP3 depends on XAF1 proficiency in the context of mutated p53. Arch. Toxicol. 2018, 92, 2645–2648. [Google Scholar] [CrossRef]
- Tomicic, M.T.; Steigerwald, C.; Rasenberger, B.; Brozovic, A.; Christmann, M. Functional mismatch repair and inactive p53 drive sensitization of colorectal cancer cells to irinotecan via the IAP antagonist BV6. Arch. Toxicol. 2019, 93, 2265–2277. [Google Scholar] [CrossRef] [PubMed]
- Stepanenko, A.A.; Chekhonin, V.P. On the Critical Issues in Temozolomide Research in Glioblastoma: Clinically Relevant Concentrations and MGMT-independent Resistance. Biomedicines 2019, 7, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, L.A.; Eckardt, J.R.; Baker, S.D.; Eckhardt, S.G.; Dugan, M.; Forral, K.; Reidenberg, P.; Statkevich, P.; Weiss, G.R.; Rinaldi, D.A.; et al. Phase I and pharmacokinetic study of temozolomide on a daily-for-5-days schedule in patients with advanced solid malignancies. J. Clin. Oncol. 1999, 17, 2604–2613. [Google Scholar] [CrossRef]
- Rosso, L.; Brock, C.S.; Gallo, J.M.; Saleem, A.; Price, P.M.; Turkheimer, F.E.; Aboagye, E.O. A N. model for prediction of drug distribution in tumor and normal tissues: Pharmacokinetics of temozolomide in glioma patients. Cancer Res. 2009, 69, 120–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostermann, S.; Csajka, C.; Buclin, T.; Leyvraz, S.; Lejeune, F.; Decosterd, L.A.; Stupp, R. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 3728–3736. [Google Scholar] [CrossRef] [Green Version]
- Portnow, J.; Badie, B.; Chen, M.; Liu, A.; Blanchard, S.; Synold, T.W. The neuropharmacokinetics of temozolomide in patients with resectable brain tumors: Potential implications for the current approach to chemoradiation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 7092–7098. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.; Weingart, J.; Nduom, E.K.; Harfi, T.T.; George, R.T.; McAreavey, D.; Ye, X.; Anders, N.M.; Peer, C.; Figg, W.D.; et al. The effect of an adenosine A2A agonist on intra-tumoral concentrations of temozolomide in patients with recurrent glioblastoma. Fluids Barriers CNS 2018, 15, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermisson, M.; Klumpp, A.; Wick, W.; Wischhusen, J.; Nagel, G.; Roos, W.; Kaina, B.; Weller, M. O6-methylguanine DNA methyltransferase and p53 status predict temozolomide sensitivity in human malignant glioma cells. J. Neurochem. 2006, 96, 766–776. [Google Scholar] [CrossRef]
- Strobel, H.; Baisch, T.; Fitzel, R.; Schilberg, K.; Siegelin, M.D.; Karpel-Massler, G.; Debatin, K.M.; Westhoff, M.A. Temozolomide and Other Alkylating Agents in Glioblastoma Therapy. Biomedicines 2019, 7, 69. [Google Scholar] [CrossRef] [Green Version]
- D’Alessandris, Q.G.; Biffoni, M.; Martini, M.; Runci, D.; Buccarelli, M.; Cenci, T.; Signore, M.; Stancato, L.; Olivi, A.; De Maria, R.; et al. The clinical value of patient-derived glioblastoma tumorspheres in predicting treatment response. Neuro Oncol. 2017, 19, 1097–1108. [Google Scholar] [CrossRef] [Green Version]
- Villalva, C.; Cortes, U.; Wager, M.; Tourani, J.M.; Rivet, P.; Marquant, C.; Martin, S.; Turhan, A.G.; Karayan-Tapon, L. O6-Methylguanine-methyltransferase (MGMT) promoter methylation status in glioma stem-like cells is correlated to temozolomide sensitivity under differentiation-promoting conditions. Int. J. Mol. Sci. 2012, 13, 6983–6994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, M.; McCully, C.; Godwin, K.; Balis, F.M. Plasma and cerebrospinal fluid pharmacokinetics of intravenous temozolomide in non-human primates. J. Neuro Oncol. 2003, 61, 203–207. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwarzenbach, C.; Tatsch, L.; Brandstetter Vilar, J.; Rasenberger, B.; Beltzig, L.; Kaina, B.; Tomicic, M.T.; Christmann, M. Targeting c-IAP1, c-IAP2, and Bcl-2 Eliminates Senescent Glioblastoma Cells Following Temozolomide Treatment. Cancers 2021, 13, 3585. https://doi.org/10.3390/cancers13143585
Schwarzenbach C, Tatsch L, Brandstetter Vilar J, Rasenberger B, Beltzig L, Kaina B, Tomicic MT, Christmann M. Targeting c-IAP1, c-IAP2, and Bcl-2 Eliminates Senescent Glioblastoma Cells Following Temozolomide Treatment. Cancers. 2021; 13(14):3585. https://doi.org/10.3390/cancers13143585
Chicago/Turabian StyleSchwarzenbach, Christian, Larissa Tatsch, Juliana Brandstetter Vilar, Birgit Rasenberger, Lea Beltzig, Bernd Kaina, Maja T. Tomicic, and Markus Christmann. 2021. "Targeting c-IAP1, c-IAP2, and Bcl-2 Eliminates Senescent Glioblastoma Cells Following Temozolomide Treatment" Cancers 13, no. 14: 3585. https://doi.org/10.3390/cancers13143585