
Meta-Analysis of Microdissected Breast Tumors Reveals Genes Regulated in the Stroma but Hidden in Bulk Analysis


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Search of Datasets
2.2. Data Download and Pre-Processing
2.3. Database Metadata
2.4. Meta-Analysis Pipeline
2.5. Differential Expression
2.6. Datasets’ Comparison
2.7. Collapsing p-Values
2.8. Definition of DEG Categories
2.9. Assessment of DEG Groups’ Robustness
2.10. Enrichment for Functional Categories
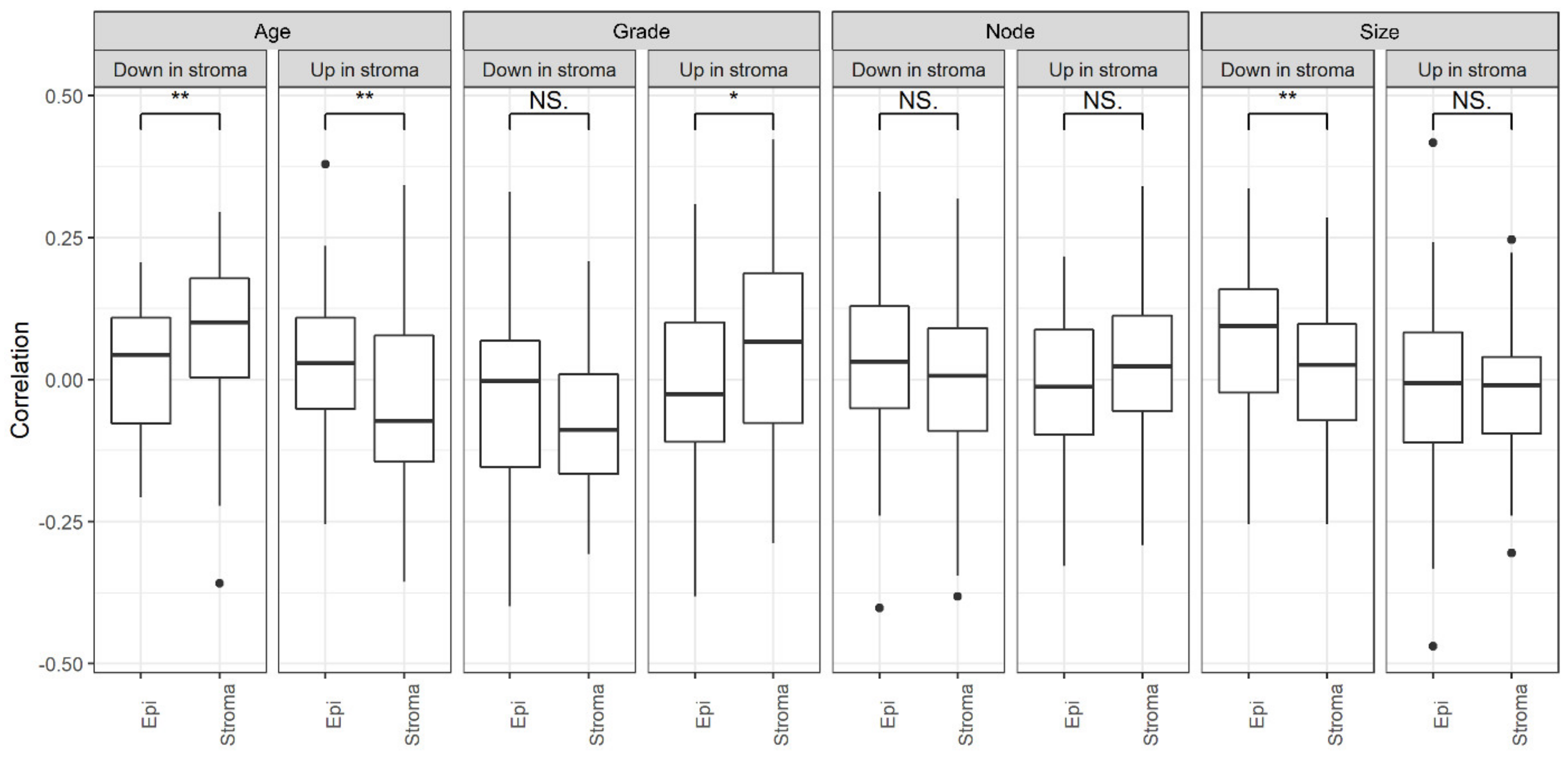
2.11. Relationship with Clinical Features
2.12. Epithelial, Stromal and Vascular Scores
2.13. Plots and Statistical Analyses
2.14. Web App
2.15. Stat3 Signatures
3. Results
3.1. Database Construction
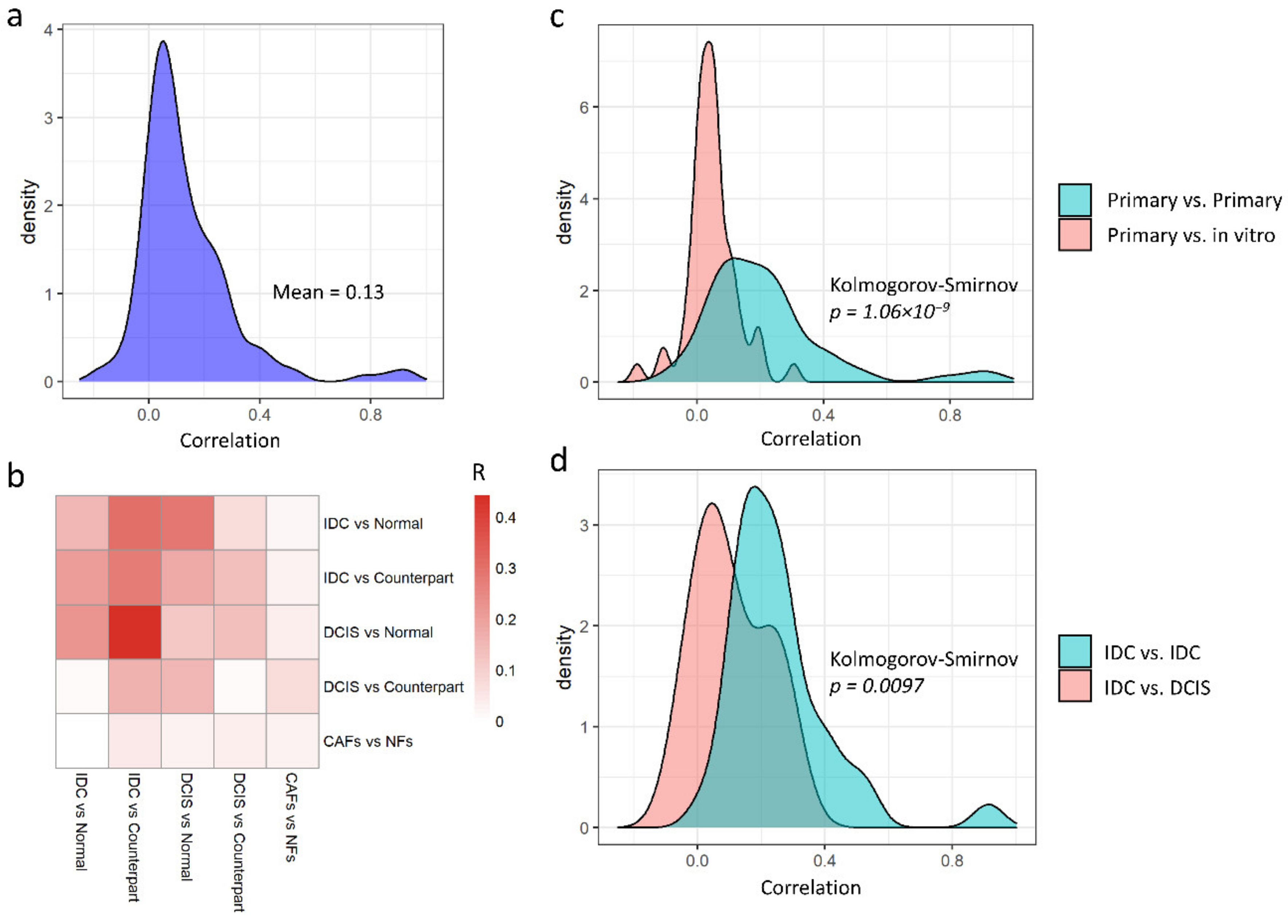
3.2. Primary and In Vitro Datasets Are Only Slightly Correlated
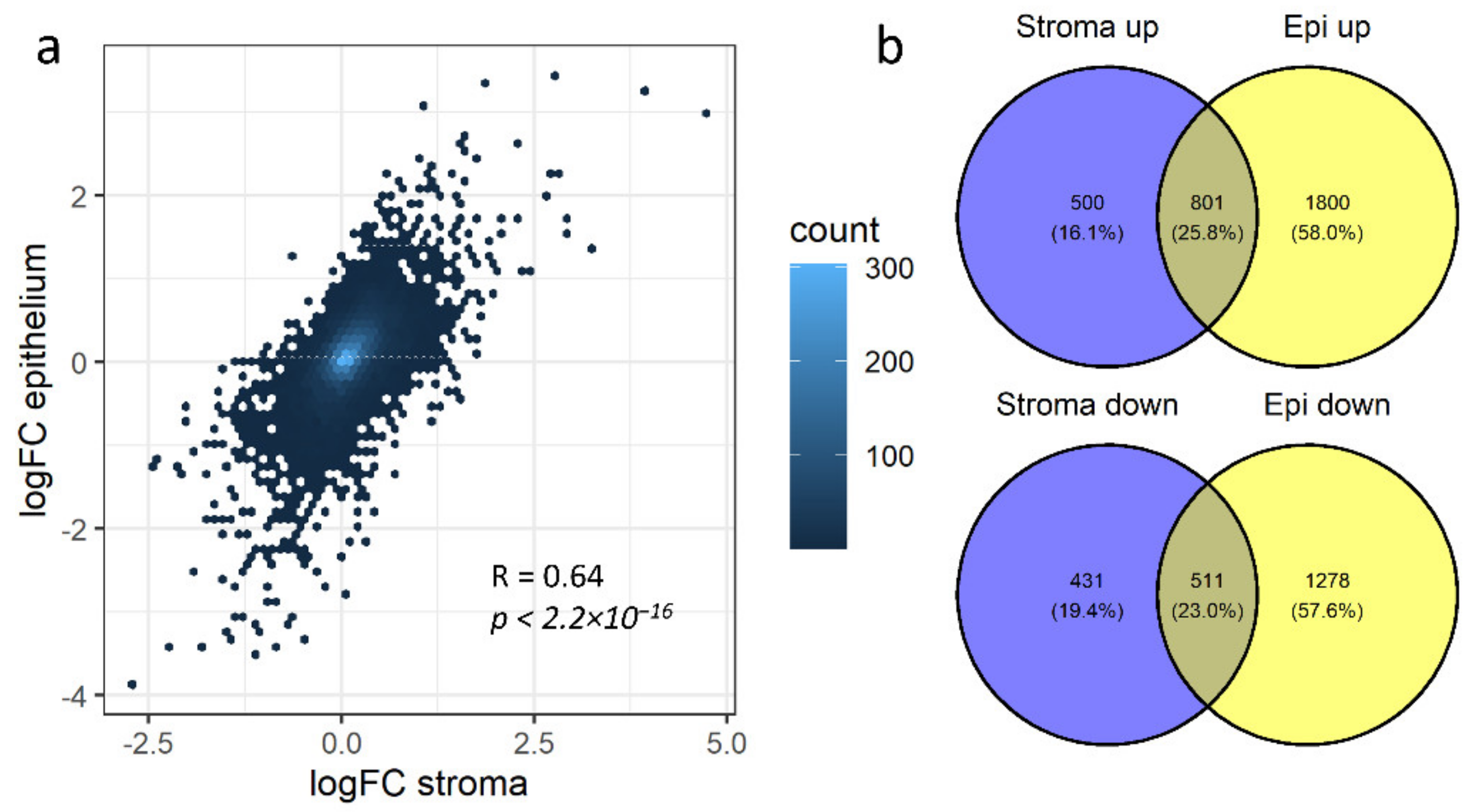
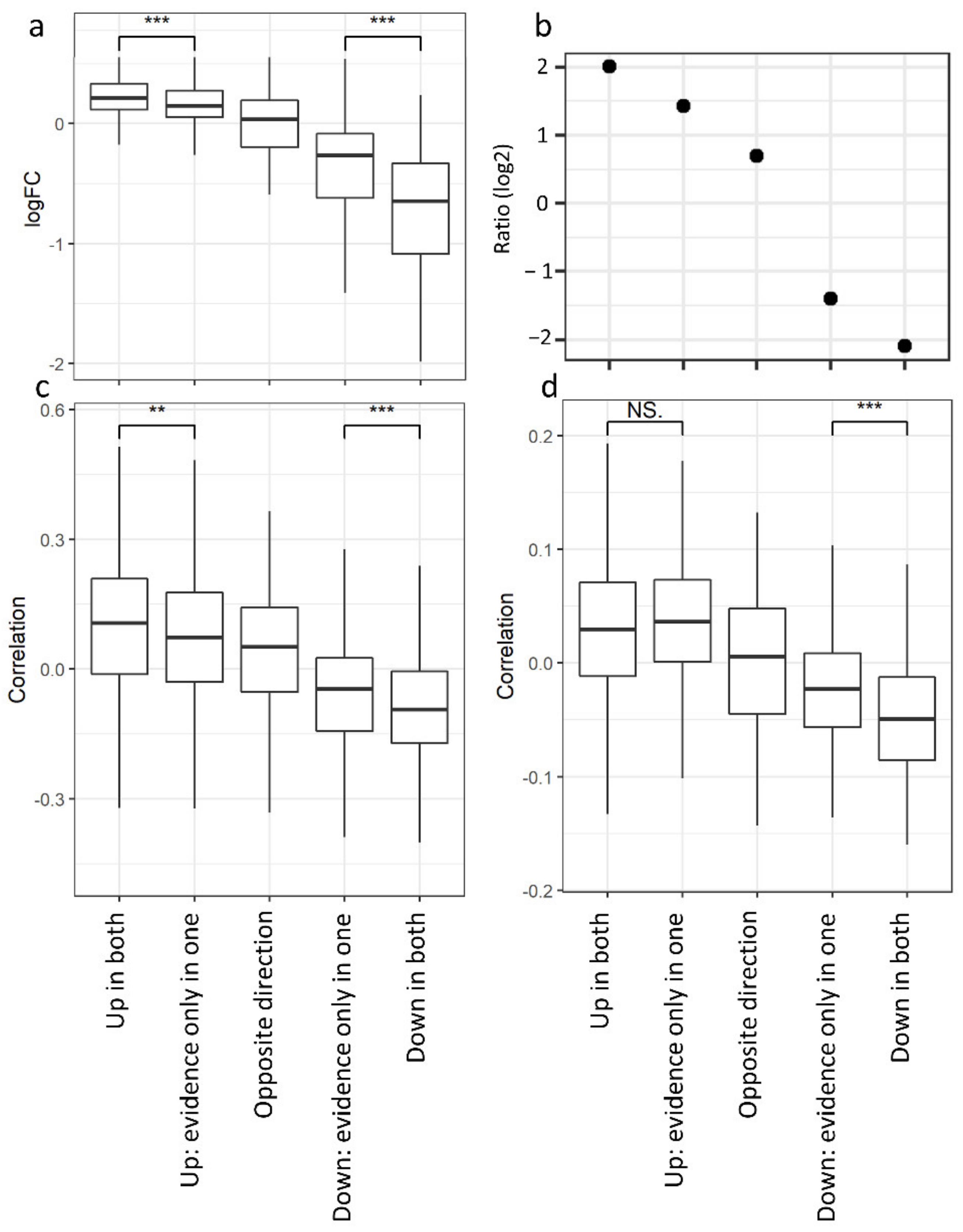
3.3. Non-Redundant Information Is Obtained by Separating Different Tissues
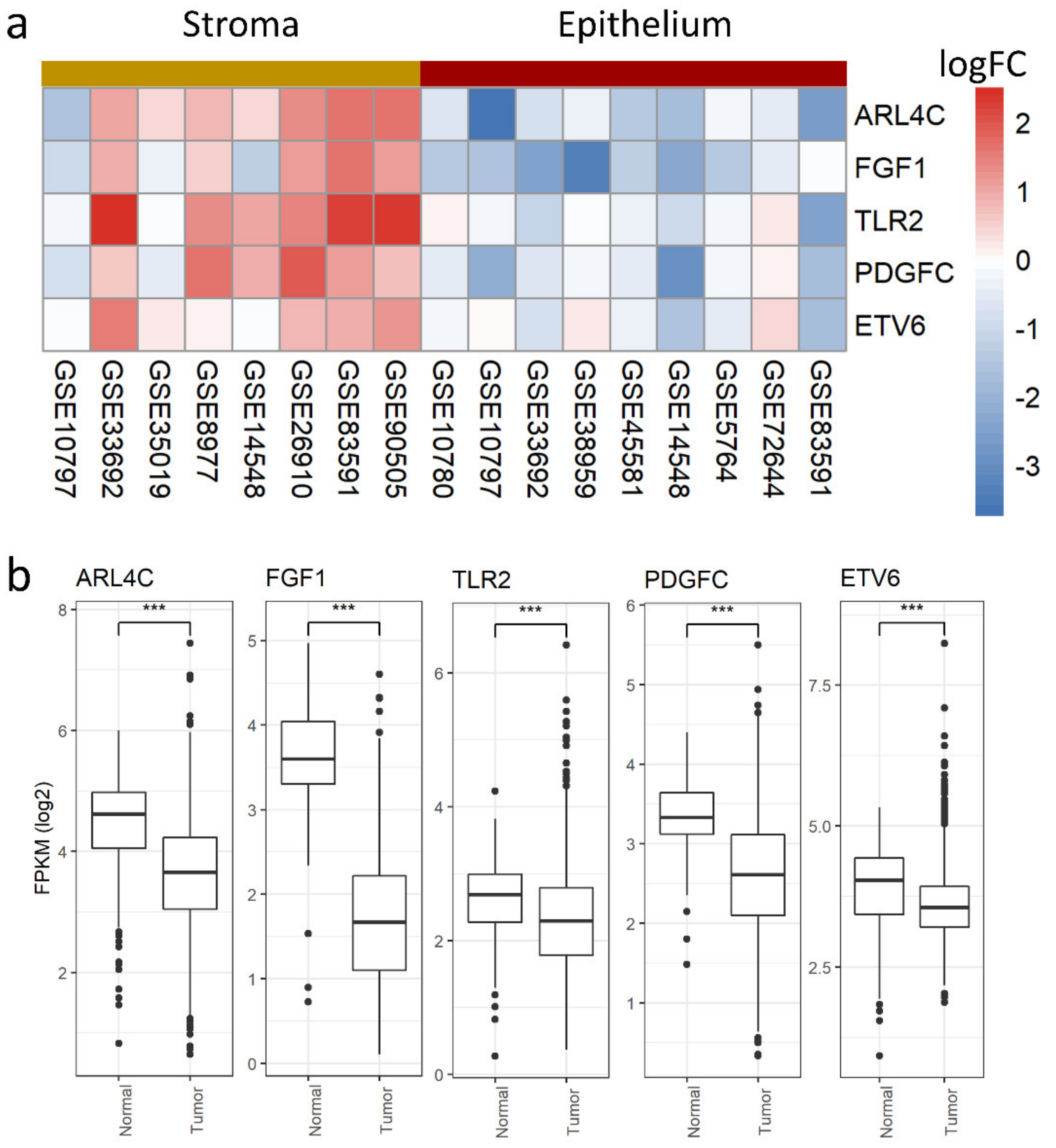
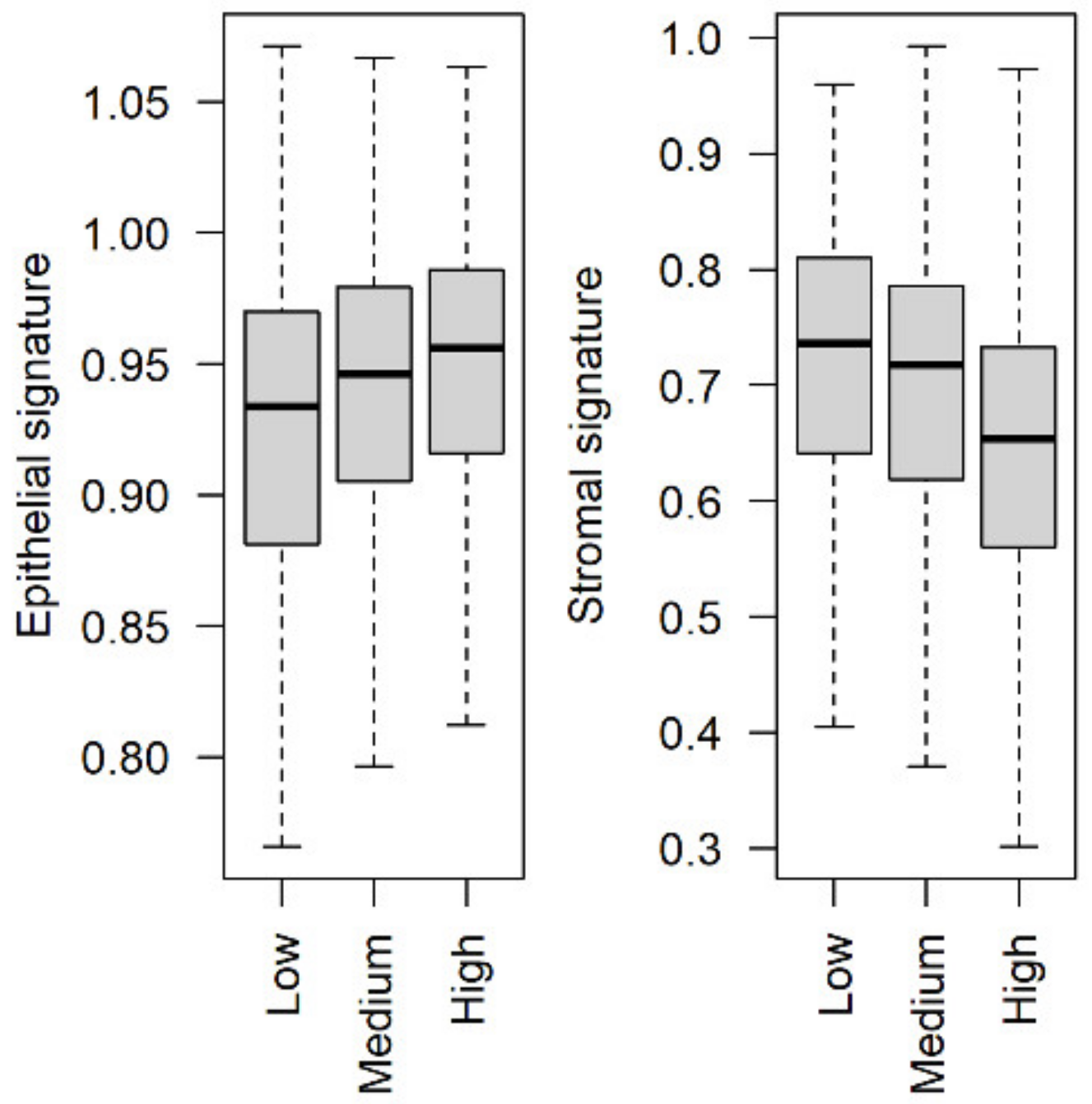
3.4. Stromal and Epithelial Markers to Impute Cell Proportions from Bulk Samples
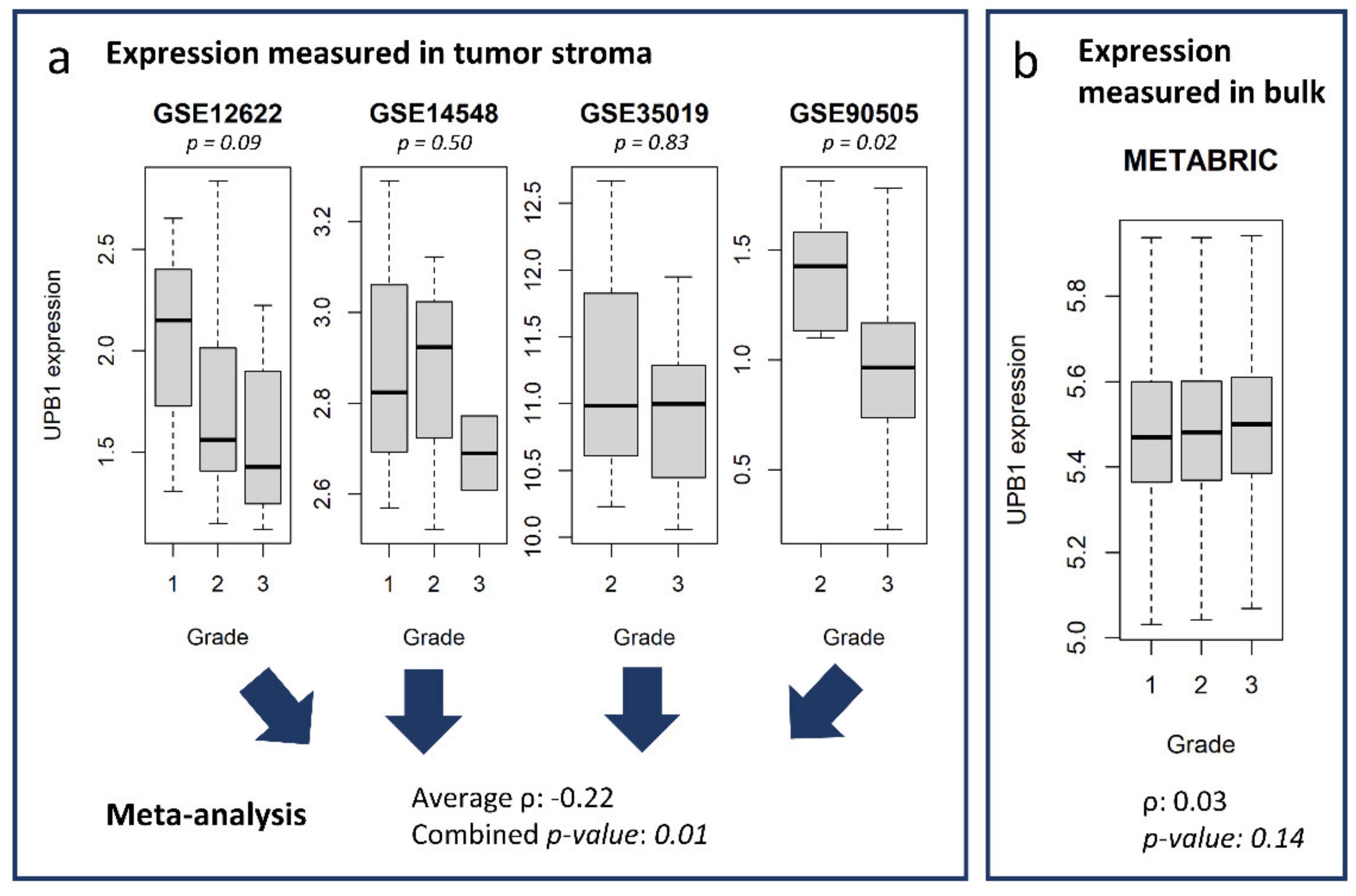
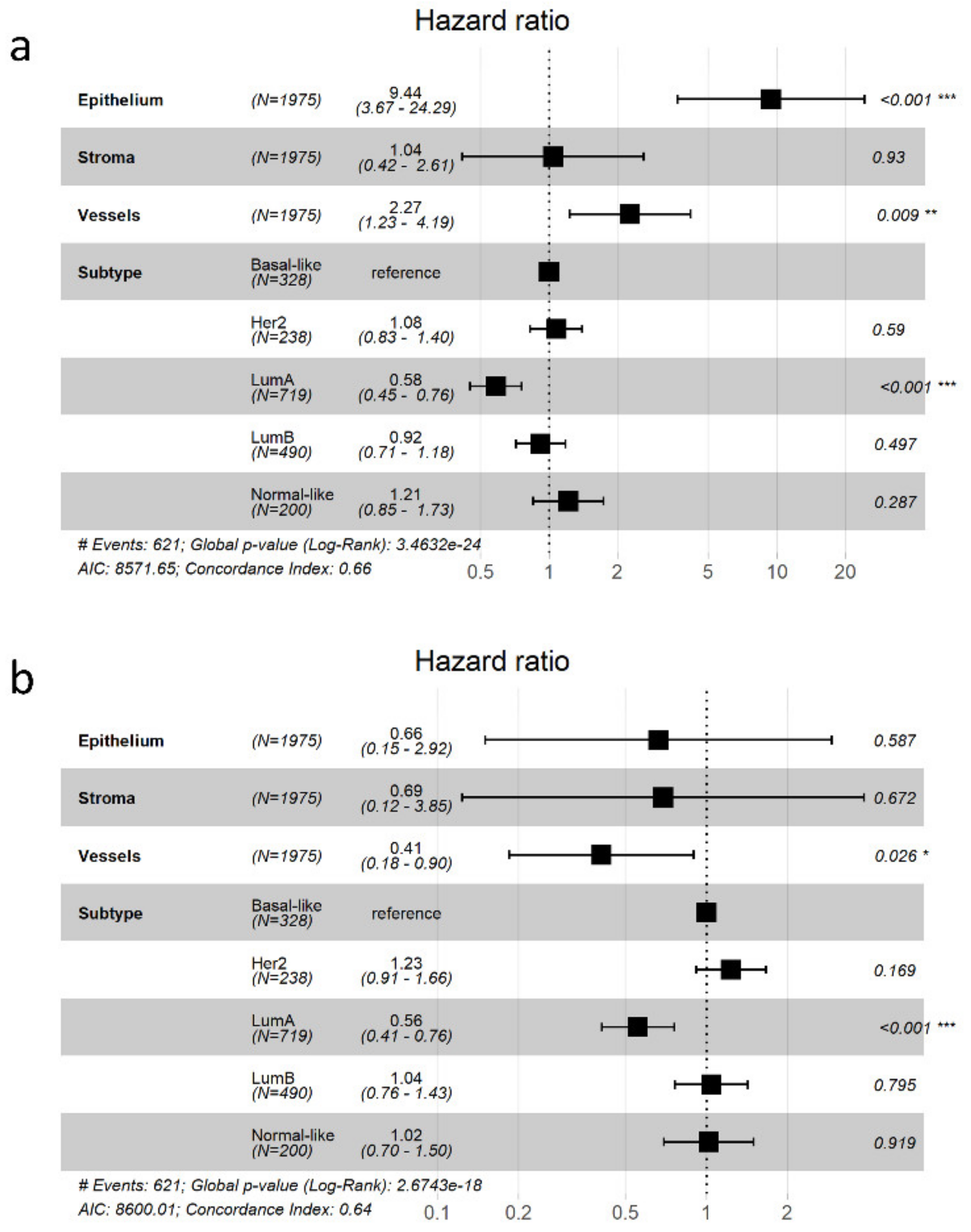
3.5. Potential Use of the Database and Web Platform
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix A.1. Details Regarding Single Annotations and Their Harmonization Compartment
Appendix A.2. Disease Status
Appendix A.3. Receptor Status (ER, PR, Her2)
Appendix A.4. TNBC Status
Appendix A.5. PAM50
Appendix A.6. Age
Appendix A.7. Grade
Appendix A.8. Size
Appendix A.9. Node
Appendix A.10. Stage
Appendix A.11. Tscore, Nscore, Mscore
Appendix A.12. Disease Free Survival
Appendix A.13. Histology
Appendix A.14. Matching
References
- Rung, J.; Brazma, A. Reuse of public genome-wide gene expression data. Nat. Rev. Genet. 2013, 14, 89–99. [Google Scholar] [CrossRef]
- Kela, I.; Ein-Dor, L.; Getz, G.; Givol, D.; Domany, E. Outcome signature genes in breast cancer: Is there a unique set? Breast Cancer Res. 2005, 7, P4.38. [Google Scholar] [CrossRef] [Green Version]
- Michiels, S.; Koscielny, S.; Hill, C. Prediction of cancer outcome with microarrays: A multiple random validation strategy. Lancet 2005, 365, 488–492. [Google Scholar] [CrossRef]
- Shen, R.; Chinnaiyan, A.M.; Ghosh, D. Pathway analysis reveals functional convergence of gene expression profiles in breast cancer. BMC Med. Genom. 2008, 1, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioannidis, J.P.A.; Ntzani, E.E.; Trikalinos, T.A.; Contopoulos-Ioannidis, D.G. Replication validity of genetic association studies. Nat. Genet. 2001, 29, 306–309. [Google Scholar] [CrossRef]
- Cui, W.; Xue, H.; Wei, L.; Jin, J.; Tian, X.; Wang, Q. High heterogeneity undermines generalization of differential expression results in RNA-Seq analysis. Hum. Genom. 2021, 15, 7. [Google Scholar] [CrossRef]
- Shedden, K.; Taylor, J.M.G.; Enkemann, S.A.; Tsao, M.-S.; Yeatman, T.J.; Gerald, W.L.; Eschrich, S.; Jurisica, I.; Giordano, T.J.; Misek, D.E.; et al. Gene expression-based survival prediction in lung adenocarcinoma: A multi-site, blinded validation study. Nat. Med. 2008, 14, 822–827. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Lappalainen, I.; Almeida-King, J.; Kumanduri, V.; Senf, A.; Spalding, J.D.; ur-Rehman, S.; Saunders, G.; Kandasamy, J.; Caccamo, M.; Leinonen, R.; et al. The European Genome-phenome Archive of human data consented for biomedical research. Nat. Genet. 2015, 47, 692–695. [Google Scholar] [CrossRef] [Green Version]
- Athar, A.; Füllgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress update—From bulk to single-cell expression data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef]
- Ramasamy, A.; Mondry, A.; Holmes, C.C.; Altman, D.G. Key issues in conducting a meta-analysis of gene expression microarray datasets. PLoS Med. 2008, 5, 1320–1332. [Google Scholar] [CrossRef]
- Taminau, J.; Lazar, C.; Meganck, S.; Nowé, A. Comparison of Merging and Meta-Analysis as Alternative Approaches for Integrative Gene Expression Analysis. ISRN Bioinform. 2014, 2014, 1–7. [Google Scholar] [CrossRef]
- Chang, L.C.; Lin, H.M.; Sibille, E.; Tseng, G.C. Meta-analysis methods for combining multiple expression profiles: Comparisons, statistical characterization and an application guideline. BMC Bioinform. 2013, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, C.; Hu, P.; Batt, J.; Santos, C. Microarray Meta-Analysis and Cross-Platform Normalization: Integrative Genomics for Robust Biomarker Discovery. Microarrays 2015, 4, 389–406. [Google Scholar] [CrossRef]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pandey, A.; Chinnaiyan, A.M. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc. Natl. Acad. Sci. USA 2004, 101, 9309–9314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.K.; Yu, U.; Kim, S.; Yoo, O.J. Combining multiple microarray studies and modeling interstudy variation. Bioinformatics 2003, 19 (Suppl. S1), i84–i90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, F.; Breitling, R.; McEntee, C.W.; Wittner, B.S.; Nemhauser, J.L.; Chory, J. RankProd: A bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics 2006, 22, 2825–2827. [Google Scholar] [CrossRef] [Green Version]
- Cai, K.-T.; Feng, C.-X.; Zhao, J.-C.; He, R.-Q.; Ma, J.; Zhong, J.-C. Upregulated miR-203a-3p and its potential molecular mechanism in breast cancer: A study based on bioinformatics analyses and a comprehensive meta-analysis. Mol. Med. Rep. 2018, 18, 4994–5008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozovic-Spasojevic, I.; Zardavas, D.; Brohée, S.; Ameye, L.; Fumagalli, D.; Ades, F.; de Azambuja, E.; Bareche, Y.; Piccart, M.; Paesmans, M.; et al. The Prognostic Role of Androgen Receptor in Patients with Early-Stage Breast Cancer: A Meta-analysis of Clinical and Gene Expression Data. Clin. Cancer Res. 2017, 23, 2702–2712. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Wang, S.; Xiao, G.; Schiller, J.; Papadimitrakopoulou, V.; Minna, J.; Wistuba, I.I.; Xie, Y. Comprehensive evaluation of published gene expression prognostic signatures for biomarker-based lung cancer clinical studies. Ann. Oncol. 2017, 28, 733–740. [Google Scholar] [CrossRef]
- Wang, J.; Gao, P.; Song, Y.; Sun, J.; Chen, X.; Yu, H.; Wang, Y.; Wang, Z. Prognostic value of gastric cancer-associated gene signatures: Evidence based on a meta-analysis using integrated bioinformatics methods. J. Cell. Mol. Med. 2018, 22, 5743–5747. [Google Scholar] [CrossRef]
- Sandhu, V.; Labori, K.J.; Borgida, A.; Lungu, I.; Bartlett, J.; Hafezi-Bakhtiari, S.; Denroche, R.E.; Jang, G.H.; Pasternack, D.; Mbaabali, F.; et al. Meta-Analysis of 1,200 Transcriptomic Profiles Identifies a Prognostic Model for Pancreatic Ductal Adenocarcinoma. JCO Clin. Cancer Inform. 2019, 3, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yasrebi, H. Comparative study of joint analysis of microarray gene expression data in survival prediction and risk assessment of breast cancer patients. Brief. Bioinform. 2016, 17, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Tan, A.C.; Winslow, R.L.; Geman, D. Merging microarray data from separate breast cancer studies provides a robust prognostic test. BMC Bioinform. 2008, 9. [Google Scholar] [CrossRef] [Green Version]
- Long, N.P.; Jung, K.H.; Anh, N.H.; Yan, H.H.; Nghi, T.D.; Park, S.; Yoon, S.J.; Min, J.E.; Kim, H.M.; Lim, J.H.; et al. An integrative data mining and omics-based translational model for the identification and validation of oncogenic biomarkers of pancreatic cancer. Cancers 2019, 11, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Ogino, S.; Parsana, P.; Nishihara, R.; Qian, Z.; Shen, J.; Mima, K.; Masugi, Y.; Cao, Y.; Nowak, J.A.; et al. Continuity of transcriptomes among colorectal cancer subtypes based on meta-analysis. Genome Biol. 2018, 19, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Santiago, I.; Yau, C.; Heij, L.; Middleton, M.R.; Markowetz, F.; Grabsch, H.I.; Dustin, M.L.; Sivakumar, S. Immunophenotypes of pancreatic ductal adenocarcinoma: Meta-analysis of transcriptional subtypes. Int. J. Cancer 2019, 145, 1125–1137. [Google Scholar] [CrossRef] [PubMed]
- Qadir, F.; Lalli, A.; Dar, H.H.; Hwang, S.; Aldehlawi, H.; Ma, H.; Dai, H.; Waseem, A.; Teh, M.-T. Clinical correlation of opposing molecular signatures in head and neck squamous cell carcinoma. BMC Cancer 2019, 19, 830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, T.Z.; Rouanne, M.; Tan, K.T.; Huang, R.Y.-J.; Thiery, J.-P. Molecular Subtypes of Urothelial Bladder Cancer: Results from a Meta-cohort Analysis of 2411 Tumors. Eur. Urol. 2019, 75, 423–432. [Google Scholar] [CrossRef]
- Oztemur Islakoglu, Y.; Noyan, S.; Aydos, A.; Gur Dedeoglu, B. Meta-microRNA biomarker signatures to classify breast cancer subtypes. Omics J. Integr. Biol. 2018, 22, 709–716. [Google Scholar] [CrossRef]
- Bell, R.; Barraclough, R.; Vasieva, O. Gene Expression Meta-Analysis of Potential Metastatic Breast Cancer Markers. Curr. Mol. Med. 2017, 17, 200–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; Hwang, S.G.; Kim, J.K.; Park, T.H.; Kim, Y.R.; Myeong, H.S.; Choi, J.D.; Kwon, K.; Jang, C.S.; Ro, Y.T.; et al. Identification of novel therapeutic target genes in acquired lapatinib-resistant breast cancer by integrative meta-analysis. Tumour Biol. 2016, 37, 2285–2297. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Plava, J.; Cihova, M.; Burikova, M.; Matuskova, M.; Kucerova, L.; Miklikova, S. Recent advances in understanding tumor stroma-mediated chemoresistance in breast cancer. Mol. Cancer 2019, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Lei, X.; Lei, Y.; Li, J.K.; Du, W.X.; Li, R.G.; Yang, J.; Li, J.; Li, F.; Tan, H.B. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2020, 470, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Sirota, M.; Butte, A.J. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 2015, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Schwede, M.; Waldron, L.; Mok, S.C.; Wei, W.; Basunia, A.; Merritt, M.A.; Mitsiades, C.S.; Parmigiani, G.; Harrington, D.P.; Quackenbush, J.; et al. The impact of stroma admixture on molecular subtypes and prognostic gene signatures in serous ovarian cancer. Cancer Epidemiol. Biomark. Prev. 2020, 29, 509–519. [Google Scholar] [CrossRef]
- Matikas, A.; Zerdes, I.; Lövrot, J.; Richard, F.; Sotiriou, C.; Bergh, J.; Valachis, A.; Foukakis, T. Prognostic implications of PD-L1 expression in breast cancer: Systematic review and meta-analysis of immunohistochemistry and pooled analysis of transcriptomic data. Clin. Cancer Res. 2019, 25, 5717–5726. [Google Scholar] [CrossRef] [PubMed]
- Donnem, T.; Al-Saad, S.; Al-Shibli, K.; Delghandi, M.P.; Persson, M.; Nilsen, M.N.; Busund, L.T.; Bremnes, R.M. Inverse prognostic impact of angiogenic marker expression in tumor cells versus stromal cells in non-small cell lung cancer. Clin. Cancer Res. 2007, 13, 6649–6657. [Google Scholar] [CrossRef] [Green Version]
- Sjöberg, E.; Augsten, M.; Bergh, J.; Jirström, K.; Östman, A. Expression of the chemokine CXCL14 in the tumour stroma is an independent marker of survival in breast cancer. Br. J. Cancer 2016, 114, 1117–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Eggeling, F.; Hoffmann, F. Microdissection—An Essential Prerequisite for Spatial Cancer Omics. Proteomics 2020, 20. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Chung, W.; Eum, H.H.; Lee, H.-O.; Lee, K.-M.; Lee, H.-B.; Kim, K.-T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef] [Green Version]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Steen, C.B.; Liu, C.L.; Alizadeh, A.A.; Newman, A.M. Profiling Cell Type Abundance and Expression in Bulk Tissues with CIBERSORTx. Methods Mol. Biol. 2020, 2117, 135–157. [Google Scholar] [CrossRef]
- Davis, R.T.; Blake, K.; Ma, D.; Gabra, M.B.I.; Hernandez, G.A.; Phung, A.T.; Yang, Y.; Maurer, D.; Lefebvre, A.E.Y.T.; Alshetaiwi, H.; et al. Transcriptional diversity and bioenergetic shift in human breast cancer metastasis revealed by single-cell RNA sequencing. Nat. Cell Biol. 2020, 22, 310–320. [Google Scholar] [CrossRef]
- Chen, D.T.; Nasir, A.; Culhane, A.; Venkataramu, C.; Fulp, W.; Rubio, R.; Wang, T.; Agrawal, D.; McCarthy, S.M.; Gruidl, M.; et al. Proliferative genes dominate malignancy-risk gene signature in histologically-normal breast tissue. Breast Cancer Res. Treat. 2010, 119, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Casey, T.; Bond, J.; Tighe, S.; Hunter, T.; Lintault, L.; Patel, O.; Eneman, J.; Crocker, A.; White, J.; Tessitore, J.; et al. Molecular signatures suggest a major role for stromal cells in development of invasive breast cancer. Breast Cancer Res. Treat. 2009, 114, 47–62. [Google Scholar] [CrossRef]
- Santucci-Pereira, J.; Zeleniuch-Jacquotte, A.; Afanasyeva, Y.; Zhong, H.; Slifker, M.; Peri, S.; Ross, E.A.; López de Cicco, R.; Zhai, Y.; Nguyen, T.; et al. Genomic signature of parity in the breast of premenopausal women. Breast Cancer Res. 2019, 21, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Gregory, K.J.; Roberts, A.L.; Conlon, E.M.; Mayfield, J.A.; Hagen, M.J.; Crisi, G.M.; Bentley, B.A.; Kane, J.J.; Makari-Judson, G.; Mason, H.S.; et al. Gene expression signature of atypical breast hyperplasia and regulation by SFRP1. Breast Cancer Res. 2019, 21, 1–18. [Google Scholar] [CrossRef]
- Russo, J.; Balogh, G.A.; Russo, I.H. Full-term pregnancy induces a specific genomic signature in the human breast. Cancer Epidemiol. Biomark. Prev. 2008, 17, 51–66. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.J.; Wang, Z.; Ryan, P.D.; Isakoff, S.J.; Barmettler, A.; Fuller, A.; Muir, B.; Mohapatra, G.; Salunga, R.; Tuggle, J.T.; et al. A two-gene expression ratio predicts clinical outcome in breast cancer patients treated with tamoxifen. Cancer Cell 2004, 5, 607–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, N.; German, R.; Rao, X.; Simpson, E.; Liu, S.; Wan, J.; Liu, Y.; Sandusky, G.; Jacobsen, M.; Stoval, M.; et al. Upregulation of lipid metabolism genes in the breast prior to cancer diagnosis. npj Breast Cancer 2020, 6, 1–13. [Google Scholar] [CrossRef]
- Ma, X.-J.; Dahiya, S.; Richardson, E.; Erlander, M.; Sgroi, D.C. Gene expression profiling of the tumor microenvironment during breast cancer progression. Breast Cancer Res. 2009, 11, R7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emery, L.A.; Tripathi, A.; King, C.; Kavanah, M.; Mendez, J.; Stone, M.D.; de Las Morenas, A.; Sebastiani, P.; Rosenberg, C.L. Early dysregulation of cell adhesion and extracellular matrix pathways in breast cancer progression. Am. J. Pathol. 2009, 175, 1292–1302. [Google Scholar] [CrossRef] [Green Version]
- Graham, K.; de Las Morenas, A.; Tripathi, A.; King, C.; Kavanah, M.; Mendez, J.; Stone, M.; Slama, J.; Miller, M.; Antoine, G.; et al. Gene expression in histologically normal epithelium from breast cancer patients and from cancer-free prophylactic mastectomy patients shares a similar profile. Br. J. Cancer 2010, 102, 1284–1293. [Google Scholar] [CrossRef]
- Graham, K.; Ge, X.; de Las Morenas, A.; Tripathi, A.; Rosenberg, C.L. Gene expression profiles of estrogen receptor-positive and estrogen receptor-negative breast cancers are detectable in histologically normal breast epithelium. Clin. Cancer Res. 2011, 17, 236–246. [Google Scholar] [CrossRef] [Green Version]
- Bauer, J.A.; Chakravarthy, A.B.; Rosenbluth, J.M.; Mi, D.; Seeley, E.H.; Granja-Ingram, N.D.M.; Olivares, M.G.; Kelley, M.C.; Mayer, I.A.; Meszoely, I.M.; et al. Identification of markers of taxane sensitivity using proteomic and genomic analyses of breast tumors from patients receiving neoadjuvant paclitaxel and radiation. Clin. Cancer Res. 2010, 16, 681–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planche, A.; Bacac, M.; Provero, P.; Fusco, C.; Delorenzi, M.; Stehle, J.C.; Stamenkovic, I. Identification of prognostic molecular features in the reactive stroma of human breast and prostate cancer. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Lehmann, B.D.B.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Hosein, A.N.; Livingstone, J.; Buchanan, M.; Reid, J.F.; Hallett, M.; Basik, M. A functional in vitro model of heterotypic interactions reveals a role for interferon-positive carcinoma associated fibroblasts in breast cancer. BMC Cancer 2015, 15, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvell, D.M.E.; Kim, J.; O’Brien, J.; Tan, A.C.; Borges, V.F.; Schedin, P.; Jacobsen, B.M.; Horwitz, K.B. Genomic Signatures of Pregnancy-Associated Breast Cancer Epithelia and Stroma and their Regulation by Estrogens and Progesterone. Horm. Cancer 2013, 4, 140–153. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, E.S.; Ertel, A.; Davicioni, E.; Kline, J.; Schwartz, G.F.; Witkiewicz, A.K. Progression of ductal carcinoma in situ to invasive breast cancer is associated with gene expression programs of EMT and myoepithelia. Breast Cancer Res. Treat. 2012, 133, 1009–1024. [Google Scholar] [CrossRef]
- Vargas, A.C.; Reed, A.E.M.C.; Waddell, N.; Lane, A.; Reid, L.E.; Smart, C.E.; Cocciardi, S.; Da Silva, L.; Song, S.; Chenevix-Trench, G.; et al. Gene expression profiling of tumour epithelial and stromal compartments during breast cancer progression. Breast Cancer Res. Treat. 2012, 135, 153–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchou, J.; Kossenkov, A.V.; Chang, L.; Satija, C.; Herlyn, M.; Showe, L.C.; Puré, E. Human breast cancer associated fibroblasts exhibit subtype specific gene expression profiles. BMC Med. Genom. 2012, 5. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Yoshimaru, T.; Matsuo, T.; Kiyotani, K.; Miyoshi, Y.; Tanahashi, T.; Rokutan, K.; Yamaguchi, R.; Saito, A.; Imoto, S.; et al. Molecular features of triple negative breast cancer cells by genome-wide gene expression profiling analysis. Int. J. Oncol. 2013, 42, 478–506. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Stewart, S.; Nagtegaal, I.; Luo, J.; Wu, Y.; Colditz, G.; Medina, D.; Allred, D.C. Differentially expressed genes regulating the progression of ductal carcinoma in situ to invasive breast cancer. Cancer Res. 2012, 72, 4574–4586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodward, W.A.; Krishnamurthy, S.; Yamauchi, H.; El-Zein, R.; Ogura, D.; Kitadai, E.; Niwa, S.-I.; Cristofanilli, M.; Vermeulen, P.; Dirix, L.; et al. Genomic and expression analysis of microdissected inflammatory breast cancer. Breast Cancer Res. Treat. 2013, 138, 761–772. [Google Scholar] [CrossRef]
- Finak, G.; Sadekova, S.; Pepin, F.; Hallett, M.; Meterissian, S.; Halwani, F.; Khetani, K.; Souleimanova, M.; Zabolotny, B.; Omeroglu, A.; et al. Gene expression signatures of morphologically normal breast tissue identify basal-like tumors. Breast Cancer Res. 2006, 8, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Turashvili, G.; Bouchal, J.; Baumforth, K.; Wei, W.; Dziechciarkova, M.; Ehrmann, J.; Klein, J.; Fridman, E.; Skarda, J.; Srovnal, J.; et al. Novel markers for differentiation of lobular and ductal invasive breast carcinomas by laser microdissection and microarray analysis. BMC Cancer 2007, 7, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Reyngold, M.; Turcan, S.; Giri, D.; Kannan, K.; Walsh, L.A.; Viale, A.; Drobnjak, M.; Vahdat, L.T.; Lee, W.; Chan, T.A. Remodeling of the methylation landscape in breast cancer metastasis. PLoS ONE 2014, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Boersma, B.J.; Reimers, M.; Yi, M.; Ludwig, J.A.; Luke, B.T.; Stephens, R.M.; Yfantis, H.G.; Lee, D.H.; Weinstein, J.N.; Ambs, S. A stromal gene signature associated with inflammatory breast cancer. Int. J. Cancer 2008, 122, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Winslow, S.; Leandersson, K.; Edsjö, A.; Larsson, C. Prognostic stromal gene signatures in breast cancer. Breast Cancer Res. 2015, 17, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Oh, E.Y.; Christensen, S.M.; Ghanta, S.; Jeong, J.C.; Bucur, O.; Glass, B.; Montaser-Kouhsari, L.; Knoblauch, N.W.; Bertos, N.; Saleh, S.M.I.; et al. Extensive rewiring of epithelial-stromal co-expression networks in breast cancer. Genome Biol. 2015, 16. [Google Scholar] [CrossRef] [Green Version]
- Abdalla, M.; Tran-Thanh, D.; Moreno, J.; Iakovlev, V.; Nair, R.; Kanwar, N.; Abdalla, M.; Lee, J.P.Y.; Kwan, J.Y.Y.; Cawthorn, T.R.; et al. Mapping genomic and transcriptomic alterations spatially in epithelial cells adjacent to human breast carcinoma. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Medina, D.; Tsimelzon, A.; Mohsin, S.K.; Mao, S.; Wu, Y.; Allred, D.C. Alterations of gene expression in the development of early hyperplastic precursors of breast cancer. Am. J. Pathol. 2007, 171, 252–262. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of triple-negative breast cancer molecular subtypes: Implications for neoadjuvant chemotherapy selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Dowdle, J.A.; Khurshid, S.; Sullivan, N.J.; Bertos, N.; Rambani, K.; Mair, M.; Daniel, P.; Wheeler, E.; Tang, X.; et al. Discovery of Stromal Regulatory Networks that Suppress Ras-Sensitized Epithelial Cell Proliferation. Dev. Cell 2017, 41, 392–407.e6. [Google Scholar] [CrossRef] [Green Version]
- Gruosso, T.; Gigoux, M.; Manem, V.S.K.; Bertos, N.; Zuo, D.; Perlitch, I.; Saleh, S.M.I.; Zhao, H.; Souleimanova, M.; Johnson, R.M.; et al. Spatially distinct tumor immune microenvironments stratify triple-negative breast cancers. J. Clin. Investig. 2019, 129, 1785–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef]
- Saleh, S.M.I.; Bertos, N.; Gruosso, T.; Gigoux, M.; Souleimanova, M.; Zhao, H.; Omeroglu, A.; Hallett, M.T.; Park, M. Identification of interacting stromal axes in triple-negative breast cancer. Cancer Res. 2017, 77, 4673–4683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouwers, B.; Fumagalli, D.; Brohee, S.; Hatse, S.; Govaere, O.; Floris, G.; van den Eynde, K.; Bareche, Y.; Schöffski, P.; Smeets, A.; et al. The footprint of the ageing stroma in older patients with breast cancer. Breast Cancer Res. 2017, 19, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, A.; King, C.; de La Morenas, A.; Perry, V.K.; Burke, B.; Antoine, G.A.; Hirsch, E.F.; Kavanah, M.; Mendez, J.; Stone, M.; et al. Gene expression abnormalities in histologically normal breast epithelium of breast cancer patients. Int. J. Cancer 2008, 122, 1557–1566. [Google Scholar] [CrossRef]
- Jones, D.T.; Lechertier, T.; Mitter, R.; Herbert, J.M.J.; Bicknell, R.; Jones, J.L.; Li, J.L.; Buffa, F.; Harris, A.L.; Hodivala-Dilke, K. Gene Expression Analysis in Human Breast Cancer Associated Blood Vessels. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [Green Version]
- François, P.; Bertos, N.; Laferrière, J.; Sadekova, S.; Souleimanova, M.; Zhao, H.; Finak, G.; Meterissian, S.; Hallett, M.T.; Park, M. Gene-expression profiling of microdissected breast cancer microvasculature identifies distinct tumor vascular subtypes. Breast Cancer Res. 2012, 14. [Google Scholar] [CrossRef] [Green Version]
- Le, P.U.; Li, J.; Cantin, C.; Collins, C.; Nantel, A.; Tremblay, T.; Kelly, J.F.; Wang, E.; O’Connor-Mccourt, M.D.; Eg, A. Molecular Characterization of the Vasculature of Patients with Infiltrating Ductal Carcinoma Generates a Gene Signature Predictive of Breast Cancer Survival. J. Cancer Biol. Res. 2015, 3, 1068. [Google Scholar]
- Dhage, S.; Ernlund, A.; Ruggles, K.; Axelrod, D.; Berman, R.; Roses, D.; Schneider, R.J. A genomic ruler to assess oncogenic transition between breast tumor and stroma. PLoS ONE 2018, 13, e0205602. [Google Scholar] [CrossRef]
- Ershaid, N.; Sharon, Y.; Doron, H.; Raz, Y.; Shani, O.; Cohen, N.; Monteran, L.; Leider-Trejo, L.; Ben-Shmuel, A.; Yassin, M.; et al. NLRP3 inflammasome in fibroblasts links tissue damage with inflammation in breast cancer progression and metastasis. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, M.; Su, G.; Casper, C.; He, R.; Rehrauer, W.; Friedl, A. Heterogeneity of gene expression in stromal fibroblasts of human breast carcinomas and normal breast. Oncogene 2010, 29, 1732–1740. [Google Scholar] [CrossRef] [Green Version]
- Bianchini, G.; Qi, Y.; Alvarez, R.H.; Iwamoto, T.; Coutant, C.; Ibrahim, N.K.; Valero, V.; Cristofanilli, M.; Green, M.C.; Radvanyi, L.; et al. Molecular anatomy of breast cancer stroma and its prognostic value in estrogen receptor-positive and -negative cancers. J. Clin. Oncol. 2010, 28, 4316–4323. [Google Scholar] [CrossRef]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Ritchie, M.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, R.A. Statistical methods for research workers. In Breakthroughs in Statistics; Springer: Berlin/Heidelberg, Germany, 1992; pp. 66–70. [Google Scholar]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Wickham, H. ggplot2—Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2017. [Google Scholar]
- Ganz, C. rintrojs: A Wrapper for the Intro.js Library. J. Open Source Softw. 2016, 1, 63. [Google Scholar] [CrossRef] [Green Version]
- Azare, J.; Leslie, K.; Al-Ahmadie, H.; Gerald, W.; Weinreb, P.H.; Violette, S.M.; Bromberg, J. Constitutively activated Stat3 induces tumorigenesis and enhances cell motility of prostate epithelial cells through integrin beta 6. Mol. Cell. Biol. 2007, 27, 4444–4453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dauer, D.J.; Ferraro, B.; Song, L.; Yu, B.; Mora, L.; Buettner, R.; Enkemann, S.; Jove, R.; Haura, E.B. Stat3 regulates genes common to both wound healing and cancer. Oncogene 2005, 24, 3397–3408. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, J.V.; Febbo, P.G.; Ramaswamy, S.; Loda, M.; Richardson, A.; Frank, D.A. Identification of a Genetic Signature of Activated Signal Transducer and Activator of Transcription 3 in Human Tumors. Cancer Res. 2005, 65, 5054–5063. [Google Scholar] [CrossRef] [Green Version]
- Tell, R.W.; Horvath, C.M. Bioinformatic analysis reveals a pattern of STAT3-associated gene expression specific to basal-like breast cancers in human tumors. Proc. Natl. Acad. Sci. USA 2014, 111, 12787–12792. [Google Scholar] [CrossRef] [Green Version]
- Sonnenblick, A.; Brohée, S.; Fumagalli, D.; Vincent, D.; Venet, D.; Ignatiadis, M.; Salgado, R.; van den Eynden, G.; Rothé, F.; Desmedt, C.; et al. Constitutive phosphorylated STAT3-associated gene signature is predictive for trastuzumab resistance in primary HER2-positive breast cancer. BMC Med. 2015, 13, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Network, C.G.A. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Klemm, F.; Bleckmann, A.; Siam, L.; Chuang, H.N.; Rietktter, E.; Behme, D.; Schulz, M.; Schaffrinski, M.; Schindler, S.; Trümper, L.; et al. β-catenin-independent WNT signaling in basal-like breast cancer and brain metastasis. Carcinogenesis 2011, 32, 434–442. [Google Scholar] [CrossRef] [Green Version]
- Monteleone, E.; Orecchia, V.; Corrieri, P.; Schiavone, D.; Avalle, L.; Moiso, E.; Savino, A.; Molineris, I.; Provero, P.; Poli, V. SP1 and STAT3 Functionally Synergize to Induce the RhoU Small GTPase and a Subclass of Non-canonical WNT Responsive Genes Correlating with Poor Prognosis in Breast Cancer. Cancers 2019, 11, 101. [Google Scholar] [CrossRef] [Green Version]
- Lopes-Coelho, F.; André, S.; Félix, A.; Serpa, J. Breast cancer metabolic cross-talk: Fibroblasts are hubs and breast cancer cells are gatherers of lipids. Mol. Cell. Endocrinol. 2018, 462, 93–106. [Google Scholar] [CrossRef]
- Yuzhalin, A.E.; Lim, S.Y.; Kutikhin, A.G.; Gordon-Weeks, A.N. Dynamic matrisome: ECM remodeling factors licensing cancer progression and metastasis. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 207–228. [Google Scholar] [CrossRef] [PubMed]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative phosphorylation as an emerging target in cancer therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boneberg, E.-M.; Legler, D.F.; Hoefer, M.M.; Ohlschlegel, C.; Steininger, H.; Füzesi, L.; Beer, G.M.; Dupont-Lampert, V.; Otto, F.; Senn, H.-J.; et al. Angiogenesis and lymphangiogenesis are downregulated in primary breast cancer. Br. J. Cancer 2009, 101, 605–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberhard, A.; Kahlert, S.; Goede, V.; Hemmerlein, B.; Plate, K.H.; Augustin, H.G. Heterogeneity of angiogenesis and blood vessel maturation in human tumors: Implications for antiangiogenic tumor therapies. Cancer Res. 2000, 60, 1388–1393. [Google Scholar]
- Al Sorkhy, M.; Fahl, Z.; Ritchie, J. Cortisol and breast cancer: A review of clinical and molecular evidence. Ann. Cancer Res. Ther. 2018, 26, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Shaul, Y.D.; Freinkman, E.; Comb, W.C.; Cantor, J.R.; Tam, W.L.; Thiru, P.; Kim, D.; Kanarek, N.; Pacold, M.E.; Chen, W.W.; et al. Dihydropyrimidine accumulation is required for the epithelial-mesenchymal transition. Cell 2014, 158, 1094–1109. [Google Scholar] [CrossRef] [Green Version]
- Chamberland, J.P.; Antonow, L.T.; Dias Santos, M.; Ritter, B. NECAP2 controls clathrin coat recruitment to early endosomes for fast endocytic recycling. J. Cell Sci. 2016, 129, 2625–2637. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Weigert, A.; Reu, S.; Guenther, S.; Mansouri, S.; Bassaly, B.; Gattenlöhner, S.; Grimminger, F.; Pullamsetti, S.S.; Seeger, W.; et al. Spatial density and distribution of tumor-associated macrophages predict survival in non-small cell lung carcinoma. Cancer Res. 2020, 80, 4414–4425. [Google Scholar] [CrossRef] [PubMed]
- Medrek, C.; Pontén, F.; Jirström, K.; Leandersson, K. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer 2012, 12, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jackute, J.; Zemaitis, M.; Pranys, D.; Sitkauskiene, B.; Miliauskas, S.; Vaitkiene, S.; Sakalauskas, R. Distribution of M1 and M2 macrophages in tumor islets and stroma in relation to prognosis of non-small cell lung cancer. BMC Immunol. 2018, 19, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.-H.; Qin, L.; Li, X. Role of STAT3 signaling pathway in breast cancer. Cell Commun. Signal. 2020, 18, 33. [Google Scholar] [CrossRef] [Green Version]
- Pathria, P.; Gotthardt, D.; Prchal-Murphy, M.; Putz, E.-M.; Holcmann, M.; Schlederer, M.; Grabner, B.; Crncec, I.; Svinka, J.; Musteanu, M.; et al. Myeloid STAT3 promotes formation of colitis-associated colorectal cancer in mice. Oncoimmunology 2015, 4, e998529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savino, A.; Provero, P.; Poli, V. Differential co-expression analyses allow the identification of critical signalling pathways altered during tumour transformation and progression. Int. J. Mol. Sci. 2020, 21, 9461. [Google Scholar] [CrossRef] [PubMed]
- Nitta, H.; Kelly, B.; Allred, C.; Jewell, S.; Banks, P.; Dennis, E.; Grogan, T.M. The assessment of HER2 status in breast cancer: The past, the present, and the future. Pathol. Int. 2016, 66, 313–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haibe-Kains, B.; Desmedt, C.; Loi, S.; Culhane, A.; Bontempi, G.; Quackenbush, J.; Sotiriou, C. A Three-Gene Model to Robustly Identify Breast Cancer Molecular Subtypes. J. Natl. Cancer Inst. 2012, 104, 311–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hortobagyi, G.N.; Edge, S.B.; Giuliano, A. New and Important Changes in the TNM Staging System for Breast Cancer. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 457–467. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | # of Samples | # of Datasets |
|---|---|---|
| System | ||
| Primary tumor | 2048 | 43 |
| In vitro | 96 | 5 |
| Compartment | ||
| Epithelium | 1230 | 32 |
| Stroma | 664 | 21 |
| Vessels | 64 | 4 |
| Adipose | 16 | 1 |
| Disease status | ||
| Invasive BC | 990 | 31 |
| Tumor (other) | 296 | 11 |
| Normal counterpart | 370 | 17 |
| Normal | 326 | 16 |
| ER status | ||
| Positive | 502 | 20 |
| Negative | 419 | 23 |
| PR status | ||
| Positive | 306 | 16 |
| Negative | 435 | 21 |
| Her2 status | ||
| Positive | 302 | 18 |
| Negative | 661 | 24 |
| LN positivity | ||
| Positive | 309 | 15 |
| Negative | 228 | 17 |
| PAM50 subtype | ||
| Basal-like | 120 | 16 |
| Her2+ | 71 | 13 |
| LumA | 194 | 16 |
| LumB | 140 | 16 |
| Normal-like | 35 | 10 |
| Age | |
|---|---|
| Median (range) | 55 (27–94) |
| # annotated samples | 756 |
| # of datasets | 18 |
| Size (in mm) | |
| Median (range) | 24 (4–161) |
| # annotated samples | 378 |
| # of datasets | 9 |
| Grade | # of samples |
| I | 50 |
| II | 189 |
| III | 312 |
| # of datasets | 12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savino, A.; De Marzo, N.; Provero, P.; Poli, V. Meta-Analysis of Microdissected Breast Tumors Reveals Genes Regulated in the Stroma but Hidden in Bulk Analysis. Cancers 2021, 13, 3371. https://doi.org/10.3390/cancers13133371
Savino A, De Marzo N, Provero P, Poli V. Meta-Analysis of Microdissected Breast Tumors Reveals Genes Regulated in the Stroma but Hidden in Bulk Analysis. Cancers. 2021; 13(13):3371. https://doi.org/10.3390/cancers13133371
Chicago/Turabian StyleSavino, Aurora, Niccolò De Marzo, Paolo Provero, and Valeria Poli. 2021. "Meta-Analysis of Microdissected Breast Tumors Reveals Genes Regulated in the Stroma but Hidden in Bulk Analysis" Cancers 13, no. 13: 3371. https://doi.org/10.3390/cancers13133371