
Evaluation of Darolutamide (ODM201) Efficiency on Androgen Receptor Mutants Reported to Date in Prostate Cancer Patients

 , , ,
, , ,  , , ,
, , ,
Abstract
:Simple Summary
Abstract
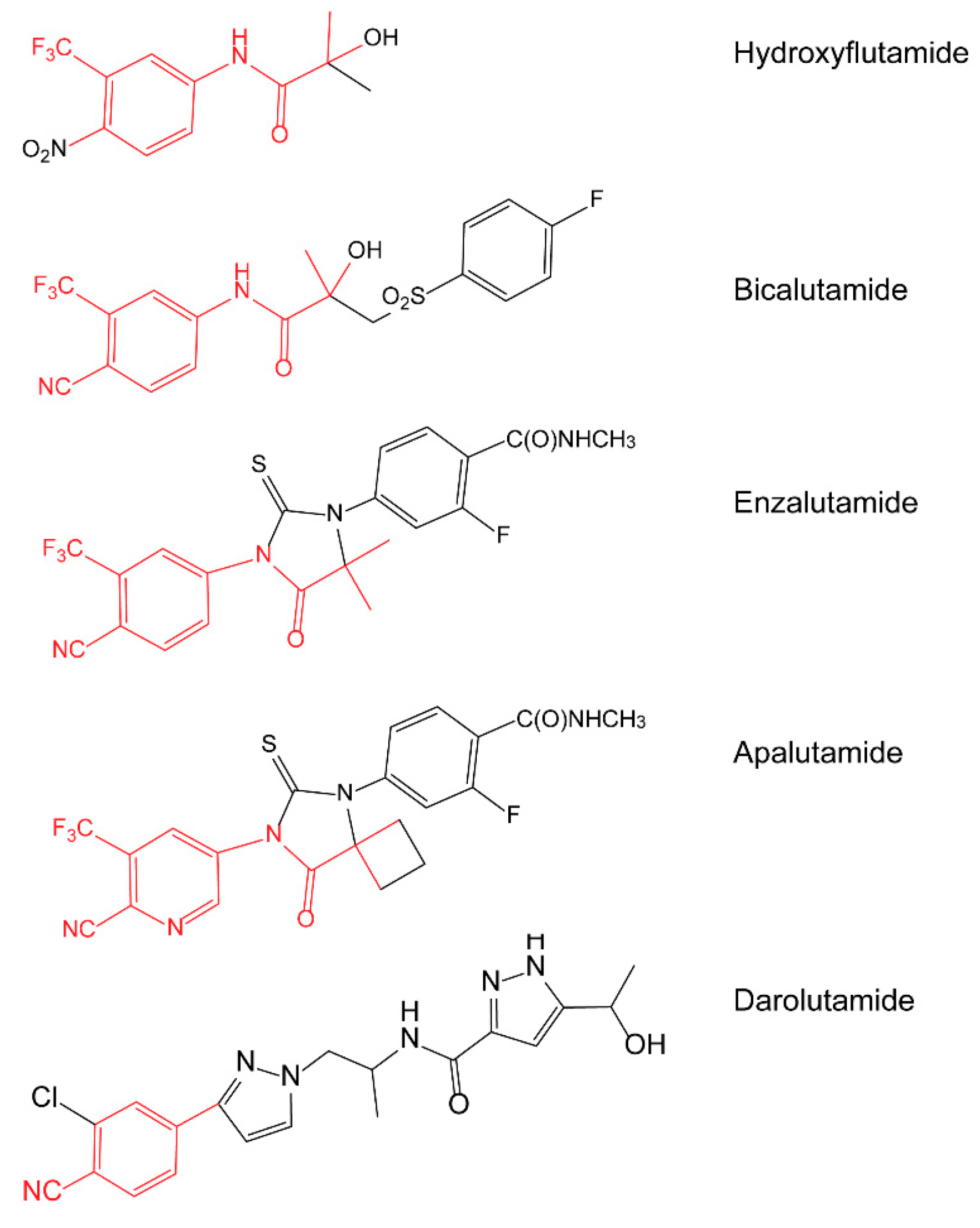
1. Introduction
2. Materials and Methods
2.1. Constructs
2.2. Steroid Activation Assay
2.3. AR Inhibition Assay
2.4. Western Blotting
3. Results
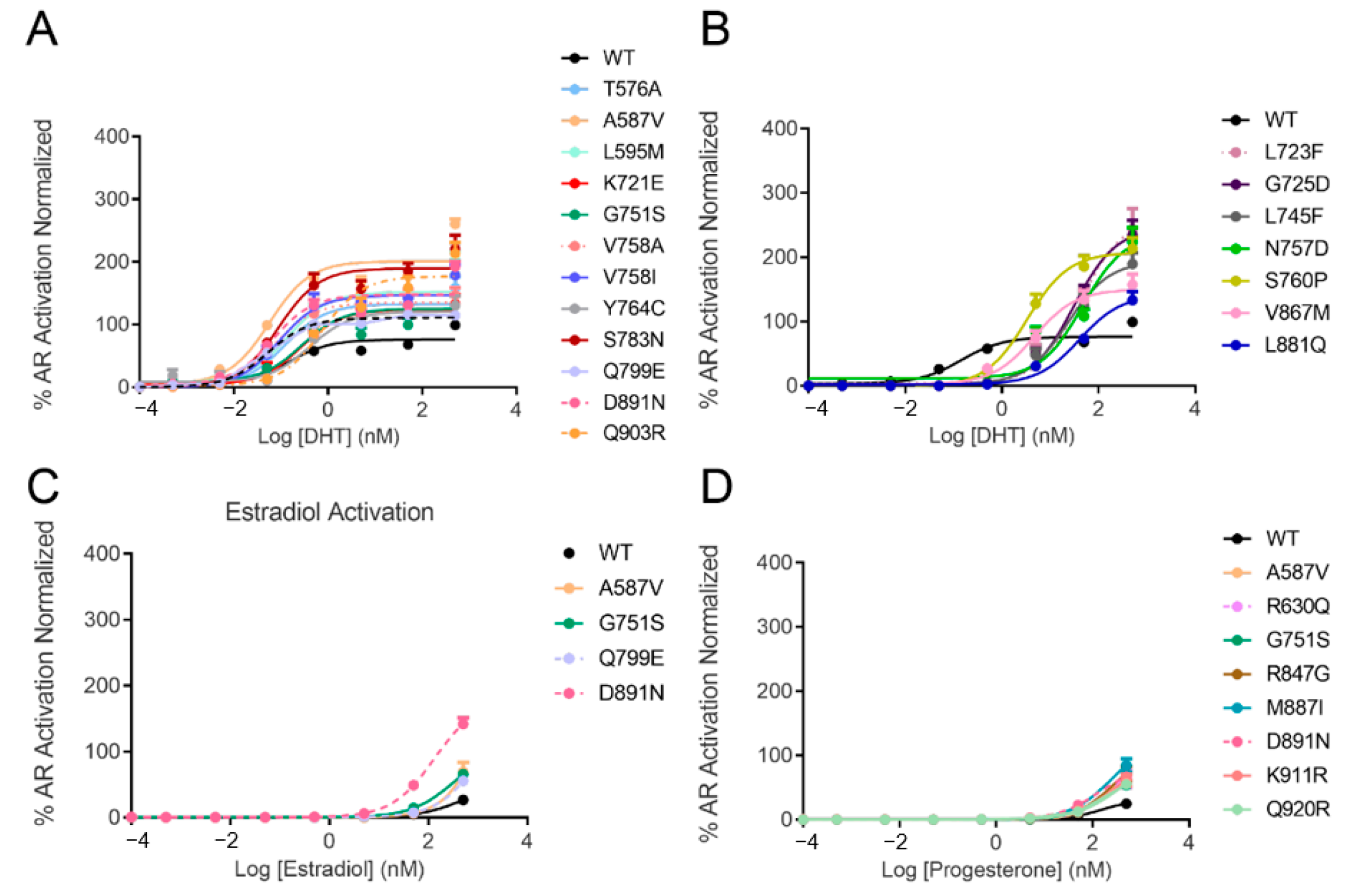
3.1. AR Transcriptional Activation by Steroids
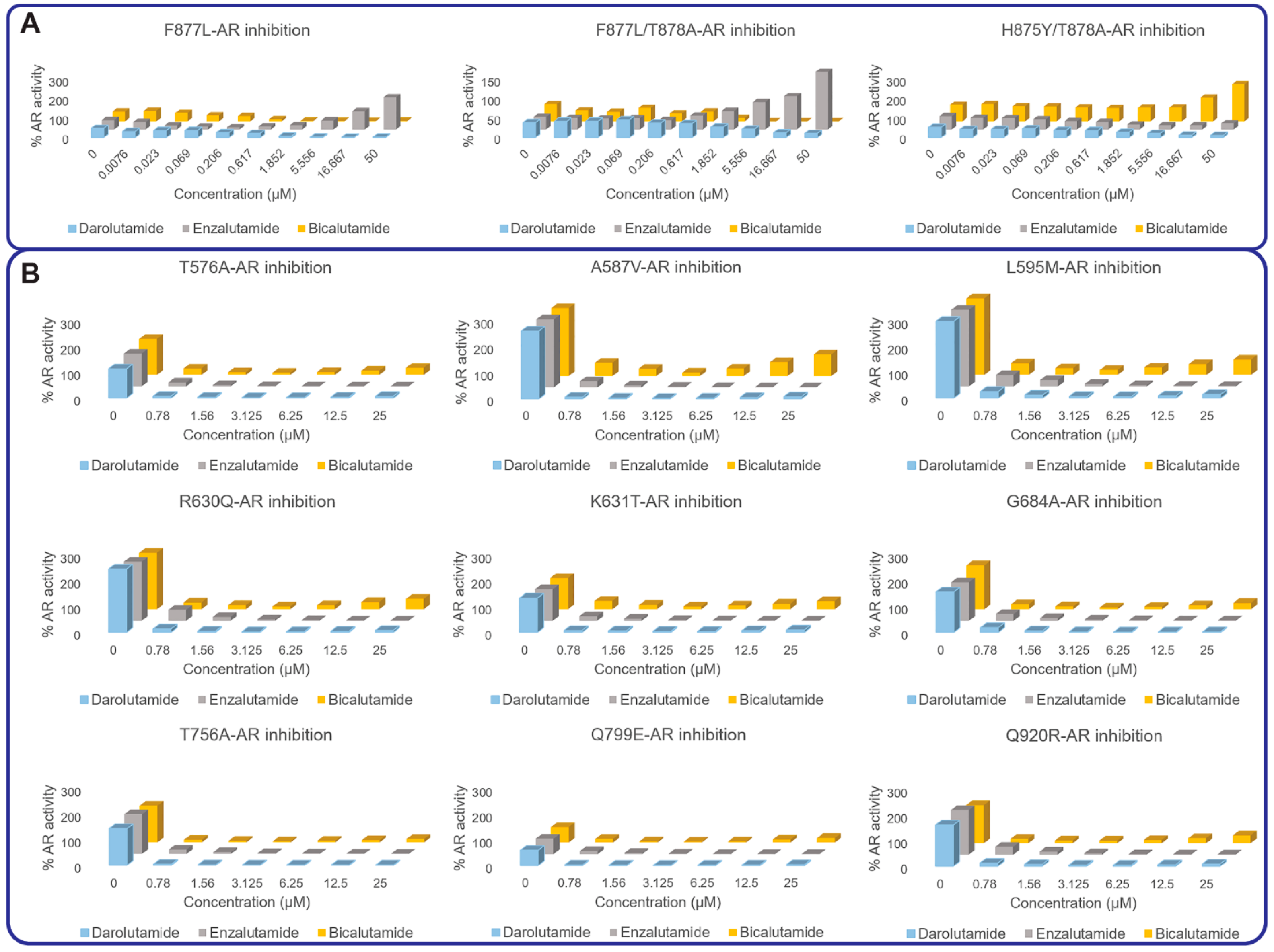
3.2. AR Transcriptional Inhibition by AR Antagonists
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feng, Q.; He, B. Androgen Receptor Signaling in the Development of Castration-Resistant Prostate Cancer. Front. Oncol. 2019, 9, 858. [Google Scholar] [CrossRef] [Green Version]
- Shore, N.D. Current and Future Management of Locally Advanced and Metastatic Prostate Cancer. Rev. Urol. 2020, 22, 110–123. [Google Scholar] [PubMed]
- Snow, O.; Lallous, N.; Singh, K.; Lack, N.; Rennie, P.; Cherkasov, A. Androgen receptor plasticity and its implications for prostate cancer therapy. Cancer Treat. Rev. 2019, 81, 101871. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montgomery, R.B.; Mostaghel, E.A.; Vessella, R.; Hess, D.L.; Kalhorn, T.F.; Higano, C.S.; True, L.D.; Nelson, P.S. Maintenance of Intratumoral Androgens in Metastatic Prostate Cancer: A Mechanism for Castration-Resistant Tumor Growth. Cancer Res. 2008, 68, 4447–4454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sailer, V.; Eng, K.W.; Zhang, T.; Bareja, R.; Pisapia, D.J.; Sigaras, A.; Bhinder, B.; Romanel, A.; Wilkes, D.; Sticca, E.; et al. Integrative Molecular Analysis of Patients With Advanced and Metastatic Cancer. JCO Precis. Oncol. 2019, 3, 1–12. [Google Scholar] [CrossRef]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, M.; Crisafulli, G.; Sogari, A.; Reilly, N.M.; Arena, S.; Lamba, S.; Bartolini, A.; Amodio, V.; Magrì, A.; Novara, L.; et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science 2019, 366, 1473–1480. [Google Scholar] [CrossRef]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.-E.C.; Radhakrishna, V.K.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef]
- Tandefelt, D.G.; de Bono, J. Circulating cell-free DNA: Translating prostate cancer genomics into clinical care. Mol. Asp. Med. 2020, 72, 100837. [Google Scholar] [CrossRef] [PubMed]
- Ritch, E.; Fu, S.Y.; Herberts, C.; Wang, G.; Warner, E.W.; Schönlau, E.; Taavitsainen, S.; Murtha, A.J.; Vandekerkhove, G.; Beja, K.; et al. Identification of Hypermutation and Defective Mismatch Repair in ctDNA from Metastatic Prostate Cancer. Clin. Cancer Res. 2019, 26, 1114–1125. [Google Scholar] [CrossRef]
- Lallous, N.; Volik, S.V.; Awrey, S.; Leblanc, E.; Tse, R.; Murillo, J.; Singh, K.; Azad, A.A.; Wyatt, A.W.; LeBihan, S.; et al. Functional analysis of androgen receptor mutations that confer anti-androgen resistance identified in circulating cell-free DNA from prostate cancer patients. Genome Biol. 2016, 17, 10. [Google Scholar] [CrossRef] [Green Version]
- Bohl, C.E.; Gao, W.; Miller, D.D.; Bell, C.E.; Dalton, J.T. Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 6201–6206. [Google Scholar] [CrossRef] [Green Version]
- Bohl, C.E.; Miller, D.D.; Chen, J.; Bell, C.E.; Dalton, J.T. Structural Basis for Accommodation of Nonsteroidal Ligands in the Androgen Receptor. J. Biol. Chem. 2005, 280, 37747–37754. [Google Scholar] [CrossRef] [Green Version]
- Korpal, M.; Korn, J.M.; Gao, X.; Rakiec, D.P.; Ruddy, D.A.; Doshi, S.; Yuan, J.; Kovats, S.G.; Kim, S.; Cooke, V.G.; et al. An F876L Mutation in Androgen Receptor Confers Genetic and Phenotypic Resistance to MDV3100 (Enzalutamide). Cancer Discov. 2013, 3, 1030–1043. [Google Scholar] [CrossRef] [Green Version]
- Ledet, E.M.; Lilly, M.B.; Sonpavde, G.; Lin, E.; Nussenzveig, R.H.; Barata, P.C.; Yandell, M.; Nagy, R.J.; Kiedrowski, L.; Agarwal, N.; et al. Comprehensive Analysis of AR Alterations in Circulating Tumor DNA from Patients with Advanced Prostate Cancer. Oncologist 2019, 25, 327–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgmann, H.; Lallous, N.; Ozistanbullu, D.; Beraldi, E.; Paul, N.; Dalal, K.; Fazli, L.; Haferkamp, A.; Lejeune, P.; Cherkasov, A.; et al. Moving Towards Precision Urologic Oncology: Targeting Enzalutamide-resistant Prostate Cancer and Mutated Forms of the Androgen Receptor Using the Novel Inhibitor Darolutamide (ODM-201). Eur. Urol. 2018, 73, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.A.; Volik, S.V.; Wyatt, A.W.; Haegert, A.; Le Bihan, S.; Bell, R.H.; Anderson, S.A.; McConeghy, B.; Shukin, R.; Bazov, J.; et al. Androgen Receptor Gene Aberrations in Circulating Cell-Free DNA: Biomarkers of Therapeutic Resistance in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2015, 21, 2315–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, S.-Y.; Gleave, M.; Beltran, H. Towards precision oncology in advanced prostate cancer. Nat. Rev. Urol. 2019, 16, 645–654. [Google Scholar] [CrossRef]
- Gottlieb, B.; Beitel, L.K.; Nadarajah, A.; Paliouras, M.; Trifiro, M. The androgen receptor gene mutations database: 2012 update. Hum. Mutat. 2012, 33, 887–894. [Google Scholar] [CrossRef]
- Fizazi, K.; Shore, N.; Tammela, T.L.; Ulys, A.; Vjaters, E.; Polyakov, S.; Jievaltas, M.; Luz, M.; Alekseev, B.; Kuss, I.; et al. Darolutamide in Nonmetastatic, Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2019, 380, 1235–1246. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AR Construct | EC50 of DHT (nM) | EC50 of Estradiol (nM) | EC50 of Progesterone (nM) | EC50 of Hydrocortisone (nM) |
|---|---|---|---|---|
| WT | 0.14 | >500 | 144 | >500 |
| T576A | 0.12 | >500 | 115 | >500 |
| K581R | weak | >500 | >500 | >500 |
| A587V | 0.06 | >500 | 168 | >500 |
| A588S | 1.15 | >500 | >500 | >500 |
| L595M | 0.14 | >500 | 167 | >500 |
| C620Y | >500 | >500 | >500 | >500 |
| R630Q | 0.10 | >500 | 151 | >500 |
| K631T | 0.05 | >500 | 118 | >500 |
| E666D | 0.11 | >500 | 269 | >500 |
| Q671R | 0.05 | >500 | 170 | >500 |
| G684A | 0.17 | >500 | 148 | >500 |
| K721E | 0.27 | >500 | >500 | >500 |
| A722T | 0.67 | >500 | >500 | >500 |
| L723F | 42.66 | >500 | >500 | >500 |
| G725D | 31.12 | >500 | >500 | >500 |
| L745F | 23.27 | >500 | >500 | >500 |
| A749T | 17.56 | >500 | >500 | >500 |
| A749V | 140.3 | >500 | >500 | >500 |
| M750I | >500 | >500 | >500 | >500 |
| G751S | 0.11 | >500 | 134 | >500 |
| F755L | 0.66 | >500 | >500 | >500 |
| T756A | 0.43 | >500 | >500 | >500 |
| N757D | 2.67 | >500 | >500 | >500 |
| V758A | 0.07 | >500 | >500 | >500 |
| V758I | 0.09 | >500 | >500 | >500 |
| S760P | 3.33 | >500 | >500 | >500 |
| Y764C | 0.40 | >500 | >500 | >500 |
| S783N | 0.09 | >500 | 162 | >500 |
| S792P | >500 | >500 | >500 | >500 |
| Q799E | 0.05 | >500 | 147 | >500 |
| I800T | 0.55 | >500 | >500 | >500 |
| R847G | 0.17 | >500 | 267 | >500 |
| S866P | >500 | >500 | >500 | >500 |
| V867M | 4.62 | >500 | >500 | >500 |
| E873Q | weak | >500 | >500 | >500 |
| D880G | 0.85 | >500 | >500 | >500 |
| L881Q | 40.58 | >500 | >500 | >500 |
| M887I | 0.17 | >500 | 253 | >500 |
| D891N | 0.07 | 134.2 | 135 | >500 |
| A897T | 0.15 | >500 | 154 | >500 |
| Q903R | 0.74 | >500 | >500 | >500 |
| G910E | 0.25 | >500 | >500 | >500 |
| K911R | 0.11 | >500 | 295 | >500 |
| Q920R | 0.15 | >500 | 320 | >500 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lallous, N.; Snow, O.; Sanchez, C.; Parra Nuñez, A.K.; Sun, B.; Hussain, A.; Lee, J.; Morin, H.; Leblanc, E.; Gleave, M.E.; et al. Evaluation of Darolutamide (ODM201) Efficiency on Androgen Receptor Mutants Reported to Date in Prostate Cancer Patients. Cancers 2021, 13, 2939. https://doi.org/10.3390/cancers13122939
Lallous N, Snow O, Sanchez C, Parra Nuñez AK, Sun B, Hussain A, Lee J, Morin H, Leblanc E, Gleave ME, et al. Evaluation of Darolutamide (ODM201) Efficiency on Androgen Receptor Mutants Reported to Date in Prostate Cancer Patients. Cancers. 2021; 13(12):2939. https://doi.org/10.3390/cancers13122939
Chicago/Turabian StyleLallous, Nada, Oliver Snow, Christophe Sanchez, Ana Karla Parra Nuñez, Bei Sun, Ahmed Hussain, Joseph Lee, Helene Morin, Eric Leblanc, Martin E. Gleave, and et al. 2021. "Evaluation of Darolutamide (ODM201) Efficiency on Androgen Receptor Mutants Reported to Date in Prostate Cancer Patients" Cancers 13, no. 12: 2939. https://doi.org/10.3390/cancers13122939