
Inhibition of DNA Repair in Combination with Temozolomide or Dianhydrogalactiol Overcomes Temozolomide-Resistant Glioma Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture, Creation of TMZ-Resistant Cells, and Reagents
2.2. Genetic Suppression of RAD51, or MSH6
2.3. Protein Extraction and Immunoblot Analyses
2.4. Cell Cycle Studies
2.5. Immunofluorescence Studies
2.6. Colony Formation Efficiency
2.7. Measurement of HR Efficiency
2.8. Statistical Analyses
3. Results
3.1. TMZ-Resistant U251 Clones Showed Different Response to TMZ
3.2. Inhibition of HR Resensitized U251TMZR#3 Clone, But Not #8 Clone, to TMZ
3.3. PARP Inhibitor Resensitized U251TMZR#8 Clone to TMZ
3.4. PARP Inhibitor Resensitized Cells with High Expression of MGMT to TMZ
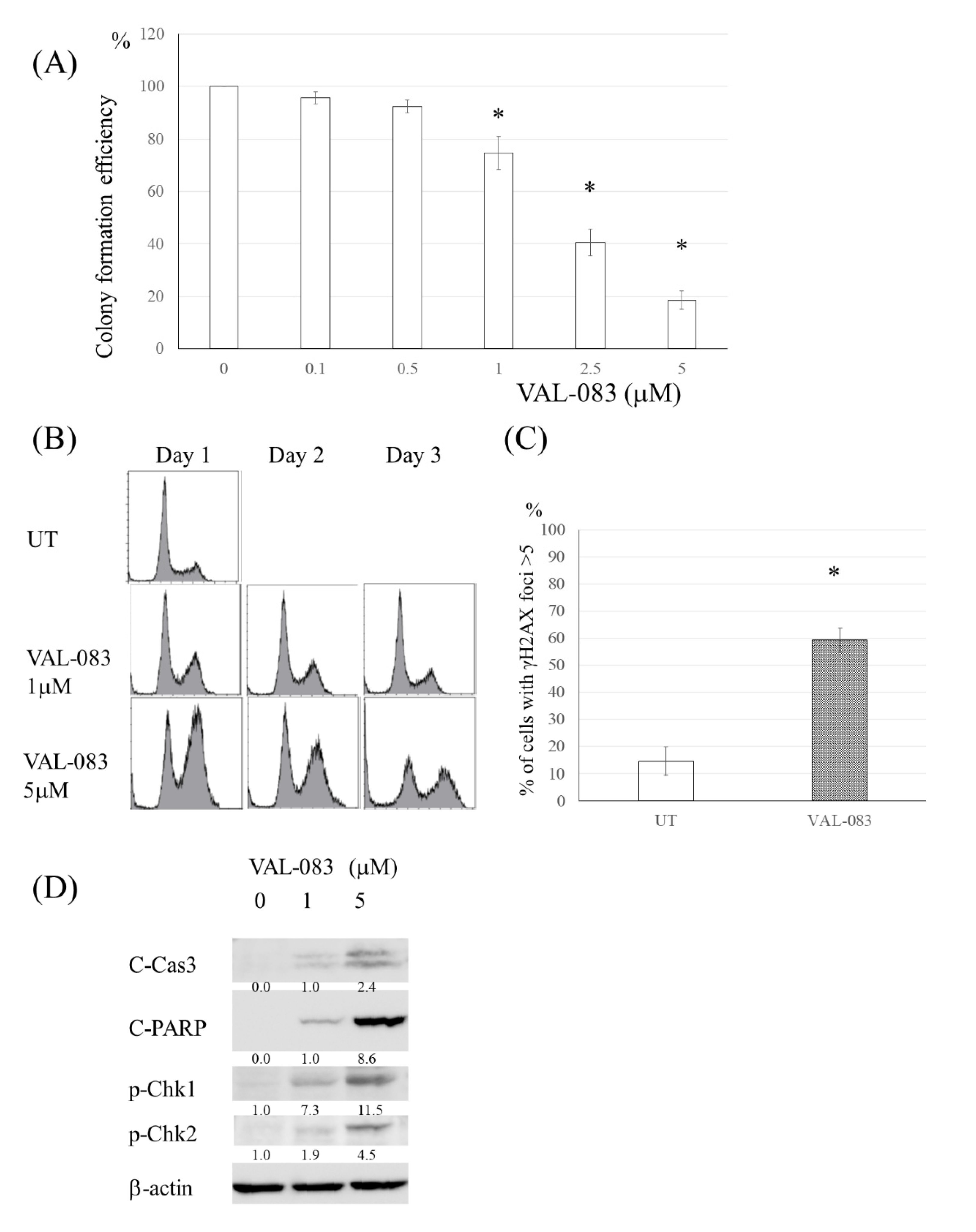
3.5. DAG Induced Cytotoxicity in TMZ-Resistant Glioma Cells Independent of MMR Deficiency or MGMT Expression
3.6. Chk1 Inhibitor Enhanced the Cytotoxicity Induced by DAG in TMZ-Resistant Glioma Cells
3.7. Inhibition of HR Enhanced the Cytotoxicity Induced by DAG in TMZ-Resistant Glioma Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Ohba, S.; Hirose, Y. Current and Future Drug Treatments for Glioblastomas. Curr. Med. Chem. 2016, 23, 4309–4316. [Google Scholar] [CrossRef]
- Barazzuol, L.; Jena, R.; Burnet, N.G.; Meira, L.B.; Jeynes, J.C.; Kirkby, K.J.; Kirkby, N.F. Evaluation of poly (ADP-ribose) polymerase inhibitor ABT-888 combined with radiotherapy and temozolomide in glioblastoma. Radiat. Oncol. 2013, 8, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, J.; Ramos, A.A.; Almeida, T.; Azqueta, A.; Rocha, E. Drug resistance in glioblastoma and cytotoxicity of seaweed compounds, alone and in combination with anticancer drugs: A mini review. Phytomedicine 2018, 48, 84–93. [Google Scholar] [CrossRef]
- Hirose, Y.; Berger, M.S.; Pieper, R.O. p53 effects both the duration of G2/M arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Res. 2001, 61, 1957–1963. [Google Scholar]
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar] [CrossRef] [Green Version]
- Hilton, J.F.; Hadfield, M.J.; Tran, M.T.; Shapiro, G.I. Poly(ADP-ribose) polymerase inhibitors as cancer therapy. Front. Biosci. 2013, 18, 1392–1406. [Google Scholar] [CrossRef]
- Horton, J.K.; Stefanick, D.F.; Prasad, R.; Gassman, N.R.; Kedar, P.S.; Wilson, S.H. Base excision repair defects invoke hypersensitivity to PARP inhibition. Mol. Cancer Res. 2014, 12, 1128–1139. [Google Scholar] [CrossRef] [Green Version]
- Institóris, E.; Szikla, K.; Otvös, L.; Gál, F. Absence of cross-resistance between two alkylating agents: BCNU vs bifunctional galactitol. Cancer Chemother. Pharmacol. 1989, 24, 311–313. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, S.; Csetényi, J.; Horváth, I.P.; Kerpel-Fronius, S.; Számel, I.; Institóris, L.; Szlovik, F.; Pásztor, E.; Afra, D. Uptake of labeled dianhydrogalactitol into human gliomas and nervous tissue. Cancer Treat. Rep. 1977, 61, 841–847. [Google Scholar]
- Jiang, X.; Huang, Y.; Wang, X.; Liang, Q.; Li, Y.; Li, F.; Fu, X.; Huang, C.; Liu, H. Dianhydrogalactitol, a potential multitarget agent, inhibits glioblastoma migration, invasion, and angiogenesis. Biomed Pharmacother. 2017, 91, 1065–1107. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Adachi, K.; Ohba, S.; Hirose, Y. The Cdk inhibitor flavopiridol enhances temozolomide-induced cytotoxicity in human glioma cells. J. Neurooncol. 2013, 115, 169–178. [Google Scholar] [CrossRef]
- Ohba, S.; Mukherjee, J.; See, W.L.; Pieper, R.O. Mutant IDH1-driven cellular transformation increases RAD51-mediated homologous recombination and temozolomide resistance. Cancer Res. 2014, 74, 4836–4844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohba, S.; Hirose, Y.; Yoshida, K.; Kawase, T. Inhibition of 90-kD heat shock protein potentiates the cytotoxicity of chemotherapeutic agents in human glioma cells. J. Neurosurg. 2010, 112, 33–42. [Google Scholar] [CrossRef]
- Felsberg, J.; Thon, N.; Eigenbrod, S.; Hentschel, B.; Sabel, M.C.; Westphal, M.; Schackert, G.; Kreth, F.W.; Pietsch, T.; Löffler, M.; et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int. J. Cancer 2011, 129, 659–670. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, M.; Gan, H.; Wang, H.; Lee, J.H.; Fang, D.; Kitange, G.J.; He, L.; Hu, Z.; Parney, I.F.; et al. A novel enhancer regulates MGMT expression and promotes temozolomide resistance in glioblastoma. Nat. Commun. 2018, 9, 2949. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Han, E.K.; Anderson, M.; Shi, Y.; Semizarov, D.; Wang, G.; McGonigal, T.; Roberts, L.; Lasko, L.; Palma, J.; et al. Acquired resistance to combination treatment with temozolomide and ABT-888 is mediated by both base excision repair and homologous recombination DNA repair pathways. Mol. Cancer Res. 2009, 7, 1686–1692. [Google Scholar] [CrossRef] [Green Version]
- Roos, W.P.; Nikolova, T.; Quiros, S.; Naumann, S.C.; Kiedron, O.; Zdzienicka, M.Z.; Kaina, B. Brca2/Xrcc2 dependent HR, but not NHEJ, is required for protection against O(6)-methylguanine triggered apoptosis, DSBs and chromosomal aberrations by a process leading to SCEs. DNA Repair 2009, 8, 72–86. [Google Scholar] [CrossRef]
- Del Alcazar, C.R.G.; Todorova, P.K.; Habib, A.A.; Mukherjee, B.; Burma, S. Augmented HR Repair Mediates Acquired Temozolomide Resistance in Glioblastoma. Mol. Cancer Res. 2016, 14, 928–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Happold, C.; Roth, P.; Wick, W.; Schmidt, N.; Florea, A.M.; Silginer, M.; Reifenberger, G.; Weller, M. Distinct molecular mechanisms of acquired resistance to temozolomide in glioblastoma cells. J. Neurochem. 2012, 122, 444–455. [Google Scholar] [CrossRef] [Green Version]
- Wiewrodt, D.; Nagel, G.; Dreimüller, N.; Hundsberger, T.; Perneczky, A.; Kaina, B. MGMT in primary and recurrent human glioblastomas after radiation and chemotherapy and comparison with p53 status and clinical outcome. Int. J. Cancer 2008, 122, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Quiros, S.; Roos, W.P.; Kaina, B. Rad51 and BRCA2--New molecular targets for sensitizing glioma cells to alkylating anticancer drugs. PLoS ONE 2011, 6, e27183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashiro, K.; Nakao, K.; Ohba, S.; Hirose, Y. Human glioma cells acquire temozolomide resistance after repeated drug exposure via DNA mismatch repair dysfunction. Anticancer. Res. 2020, 40, 1315–1323. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas (TCGA) Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Kizilbash, S.H.; Gupta, S.K.; Chang, K.; Kawashima, R.; Parrish, K.E.; Carlson, B.L.; Bakken, K.K.; Mladek, A.C.; Schroeder, M.A.; Decker, P.A.; et al. Restricted Delivery of Talazoparib Across the Blood-Brain Barrier Limits the Sensitizing Effects of PARP Inhibition on Temozolomide Therapy in Glioblastoma. Mol. Cancer Ther. 2017, 16, 2735–2746. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.K.; Mladek, A.C.; Carlson, B.L.; Boakye-Agyeman, F.; Bakken, K.K.; Kizilbash, S.H.; Schroeder, M.A.; Reid, J.; Sarkaria, J.N. Discordant in vitro and in vivo chemopotentiating effects of the PARP inhibitor veliparib in temozolomide-sensitive versus -resistant glioblastoma multiforme xenografts. Clin. Cancer Res. 2014, 20, 3730–3741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemasson, B.; Wang, H.; Galbán, S.; Li, Y.; Zhu, Y.; Heist, K.A.; Tsein, C.; Chenevert, T.L.; Rehemtulla, A.; Galbán, C.J.; et al. Evaluation of Concurrent Radiation, Temozolomide and ABT-888 Treatment Followed by Maintenance Therapy with Temozolomide and ABT-888 in a Genetically Engineered Glioblastoma Mouse Model. Neoplasia 2016, 18, 82–89. [Google Scholar] [CrossRef] [Green Version]
- Halford, S.E.R.; Cruickshank, G.; Dunn, L.; Erridge, S.; Godfrey, L.; Herbert, C.; Jefferies, S.; Lopez, J.S.; McBain, C.; Pittman, M.; et al. Results of the OPARATIC trial: A phase I dose escalation study of olaparib in combination with temozolomide (TMZ) in patients with relapsed glioblastoma (GBM). J. Clin. Oncol. 2017, 35 (Suppl. S15), 2022. [Google Scholar] [CrossRef]
- Lesueur, P.; Lequesne, J.; Grellard, J.M.; Dugué, A.; Coquan, E.; Brachet, P.E.; Geffrelot, J.; Kao, W.; Emery, E.; Berro, D.H.; et al. Phase I/IIa study of concomitant radiotherapy with olaparib and temozolomide in unresectable or partially resectable glioblastoma: OLA-TMZ-RTE-01 trial protocol. BMC Cancer 2019, 19, 198. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; de Gooijer, M.C.; Roig, E.M.; Buil, L.C.; Christner, S.M.; Beumer, J.H.; Würdinger, T.; Beijnen, J.H.; van Tellingen, O. ABCB1, ABCG2, and PTEN determine the response of glioblastoma to temozolomide and ABT-888 therapy. Clin. Cancer Res. 2014, 20, 2703–2713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijaya, J.; Fukuda, Y.; Schuetz, J.D. Obstacles to Brain Tumor Therapy: Key ABC Transporters. Int. J. Mol. Sci. 2017, 18, 2544. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.; Qi, X.M.; Miao, L.L.; Ren, J. 1,2:5,6-dianhydrogalactitol inhibits human glioma cell growth in vivo and in vitro by arresting the cell cycle at G2/M phase. Acta Pharmacol. Sin. 2017, 38, 561–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, B.; Steinø, A.; Bacha, J.; Brown, D.; Daugaard, M. Dianhydrogalactitol induces replication-dependent DNA damage in tumor cells preferentially resolved by homologous recombination. Cell Death Dis. 2018, 9, 1016. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohba, S.; Yamashiro, K.; Hirose, Y. Inhibition of DNA Repair in Combination with Temozolomide or Dianhydrogalactiol Overcomes Temozolomide-Resistant Glioma Cells. Cancers 2021, 13, 2570. https://doi.org/10.3390/cancers13112570
Ohba S, Yamashiro K, Hirose Y. Inhibition of DNA Repair in Combination with Temozolomide or Dianhydrogalactiol Overcomes Temozolomide-Resistant Glioma Cells. Cancers. 2021; 13(11):2570. https://doi.org/10.3390/cancers13112570
Chicago/Turabian StyleOhba, Shigeo, Kei Yamashiro, and Yuichi Hirose. 2021. "Inhibition of DNA Repair in Combination with Temozolomide or Dianhydrogalactiol Overcomes Temozolomide-Resistant Glioma Cells" Cancers 13, no. 11: 2570. https://doi.org/10.3390/cancers13112570