
Orai1 Channel Regulates Human-Activated Pancreatic Stellate Cell Proliferation and TGFβ1 Secretion through the AKT Signaling Pathway

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Functional Expression of Orai1 Channel in PS-1 and RLT Human-Activated PSCs
2.2. Orai1 Is Involved in Human-Activated PSC Proliferation and TGFβ1 Secretion
2.3. Orai1 Regulates the Activation of AKT But Not of ERK1/2 and SMAD2 in Human-Activated PSCs
2.4. AKT Signaling Pathway Mediates Human-Activated PSC Proliferation and TGFβ1 Secretion
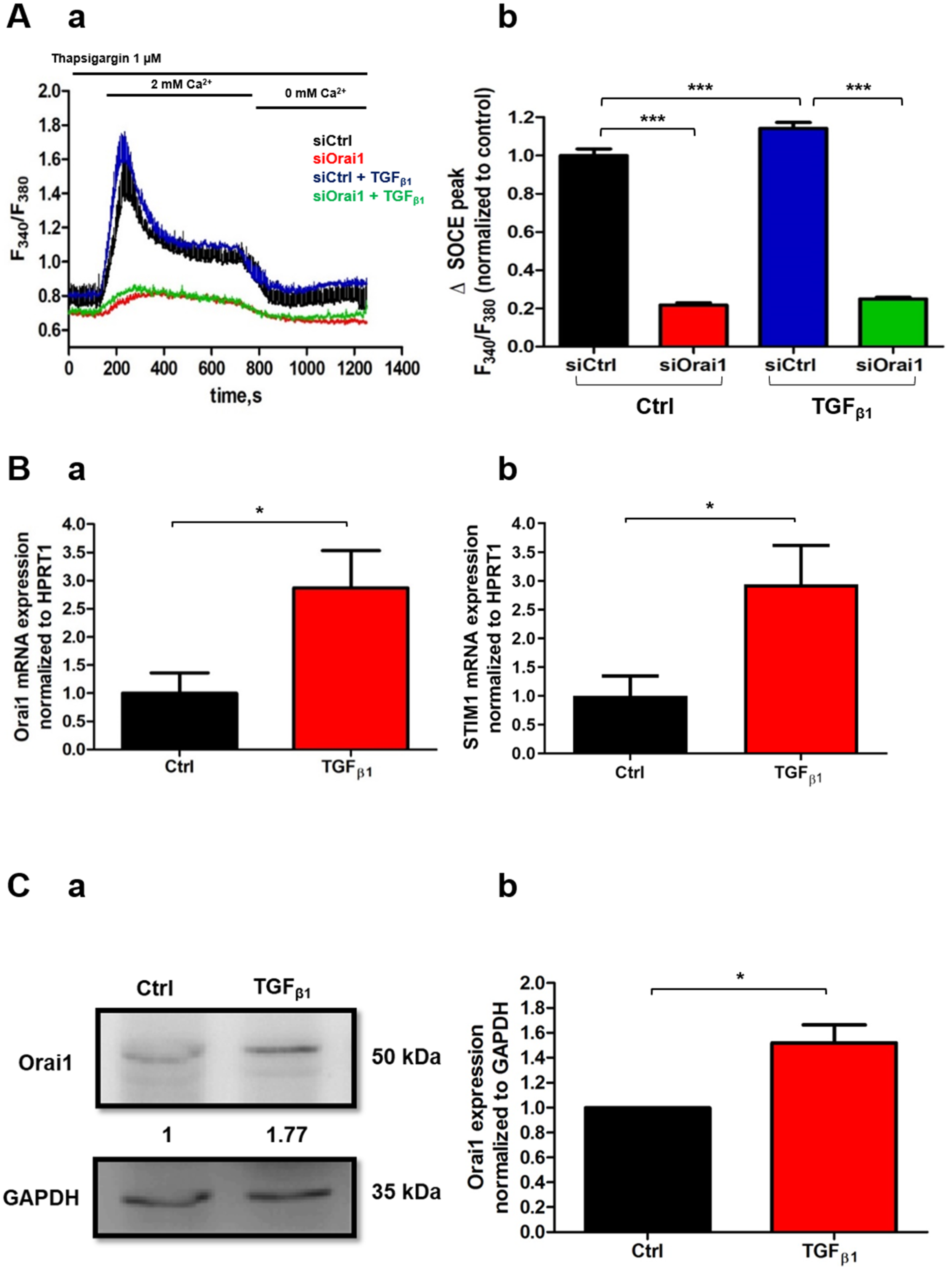
2.5. TGFβ1 Promotes Orai1-Mediated Ca2+ Entry and Increases Both Orai1 mRNA and Protein Expression in Human-Activated PSCs
2.6. TGFβ1 Enhances Orai1/AKT-Dependent Proliferation Through an Autocrine Positive Feedback Loop in Human-Activated PSCs
2.7. Ca2+ Entry Through Orai1 Regulates AKT-Dependent Proliferation and TGFβ1 Secretion in PS-1 Human-Activated PSCs
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Cell Culture
5.2. Cell Transfection and RNA Interference
5.3. Chemicals and Reagents
5.4. Cell Proliferation Assay
5.5. Cell Cycle Analysis
5.6. Cell Mortality
5.7. Apoptosis Analysis
5.8. Calcium Imaging
5.9. qRT-PCR
5.10. TGFβ1 Dosage Assay
5.11. Western Blotting and Co-Immunoprecipitation
5.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jaster, R.; Emmrich, J. Crucial role of fibrogenesis in pancreatic diseases. Best Pract. Res. Clin. Gastroenterol. 2008, 22, 17–29. [Google Scholar] [CrossRef]
- Apte, M.V.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells: A starring role in normal and diseased pancreas. Front. Physiol. 2012, 3, 344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norton, J.; Foster, D.; Chinta, M.; Titan, A.; Longaker, M. Pancreatic Cancer Associated Fibroblasts (CAF): Under-Explored Target for Pancreatic Cancer Treatment. Cancers 2020, 12, 1347. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Radhakrishnan, P. Pancreatic Stellate Cells: The Key Orchestrator of the Pancreatic Tumor Microenvironment. In Advances in Experimental Medicine and Biology; Birbrair, A., Ed.; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2020; Volume 1234, pp. 57–70. [Google Scholar]
- Ferdek, P.E.; Jakubowska, M.A. Biology of pancreatic stellate cells—more than just pancreatic cancer. Pflügers Arch. Eur. J. Physiol. 2017, 469, 1039–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masamune, A.; Shimosegawa, T. Pancreatic stellate cells–Multi-functional cells in the pancreas. Pancreatology 2013, 13, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Bynigeri, R.R.; Jakkampudi, A.; Jangala, R.; Subramanyam, C.; Sasikala, M.; Rao, G.V.; Reddy, D.N.; Talukdar, R. Pancreatic stellate cell: Pandora’s box for pancreatic disease biology. World J. Gastroenterol. 2017, 23, 382–405. [Google Scholar] [CrossRef]
- Masamune, A.; Watanabe, T.; Kikuta, K.; Shimosegawa, T. Roles of Pancreatic Stellate Cells in Pancreatic Inflammation and Fibrosis. Clin. Gastroenterol. Hepatol. 2009, 7, S48–S54. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef] [Green Version]
- Jin, G.; Hong, W.; Guo, Y.; Bai, Y.; Chen, B. Molecular Mechanism of Pancreatic Stellate Cells Activation in Chronic Pancreatitis and Pancreatic Cancer. J. Cancer 2020, 11, 1505–1515. [Google Scholar] [CrossRef] [Green Version]
- Schneider, E.; Schmid-Kotsas, A.; Zhao, J.; Weidenbach, H.; Schmid, R.M.; Menke, A.; Adler, G.; Waltenberger, J.; Grünert, A.; Bachem, M.G. Identification of mediators stimulating proliferation and matrix synthesis of rat pancreatic stellate cells. Am. J. Physiol. Physiol. 2001, 281, C532–C543. [Google Scholar] [CrossRef]
- Kulkarni, A.A.; Thatcher, T.H.; Olsen, K.C.; Maggirwar, S.B.; Phipps, R.P.; Sime, P.J. PPAR-γ Ligands Repress TGFβ-Induced Myofibroblast Differentiation by Targeting the PI3K/Akt Pathway: Implications for Therapy of Fibrosis. PLoS ONE 2011, 6, e15909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-β1 Signaling and Tissue Fibrosis. Cold Spring Harb. Perspect. Biol. 2018, 10, a022293. [Google Scholar] [CrossRef] [Green Version]
- Omary, M.B.; Lugea, A.; Lowe, A.W.; Pandol, S.J. The pancreatic stellate cell: A star on the rise in pancreatic diseases. J. Clin. Investig. 2007, 117, 50–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apte, M.V.; Haber, P.S.; Darby, S.J.; Rodgers, S.C.; McCaughan, G.W.; Korsten, M.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells are activated by proinflammatory cytokines: Implications for pancreatic fibrogenesis. Gut 1999, 44, 534–541. [Google Scholar] [CrossRef] [Green Version]
- Kruse, M.-L.; Hildebrand, P.B.; Timke, C.; Fölsch, U.R.; E Schmidt, W. TGFβ1 autocrine growth control in isolated pancreatic fibroblastoid cells/stellate cells in vitro. Regul. Pept. 2000, 90, 47–52. [Google Scholar] [CrossRef]
- Finnson, K.W.; Almadani, Y.; Philip, A. Non-canonical (non-SMAD2/3) TGF-β signaling in fibrosis: Mechanisms and targets. Semin. Cell Dev. Biol. 2020, 101, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Masamune, A.; Shimosegawa, T. Signal transduction in pancreatic stellate cells. J. Gastroenterol. 2009, 44, 249–260. [Google Scholar] [CrossRef]
- Kusiak, A.A.; Szopa, M.D.; Jakubowska, M.A.; Ferdek, P.E. Signaling in the Physiology and Pathophysiology of Pancreatic Stellate Cells—A Brief Review of Recent Advances. Front. Physiol. 2020, 11, 78. [Google Scholar] [CrossRef]
- Schwer, C.I.; Stoll, P.; Rospert, S.; Fitzke, E.; Schallner, N.; Bürkle, H.; Schmidt, R.; Humar, M. Carbon monoxide releasing molecule-2 CORM-2 represses global protein synthesis by inhibition of eukaryotic elongation factor eEF2. Int. J. Biochem. Cell Biol. 2013, 45, 201–212. [Google Scholar] [CrossRef]
- Zhang, X.; Jin, T.; Huang, X.; Liu, X.; Liu, Z.; Jia, Y.; Hao, J. Effects of the tumor suppressor PTEN on biological behaviors of activated pancreatic stellate cells in pancreatic fibrosis. Exp. Cell Res. 2018, 373, 132–144. [Google Scholar] [CrossRef]
- Won, J.H.; Zhang, Y.; Ji, B.; Logsdon, C.D.; Yule, D.I. Phenotypic changes in mouse pancreatic stellate cell Ca2+ signaling events following activation in culture and in a disease model of pancreatitis. Mol. Biol. Cell 2011, 22, 421–436. [Google Scholar] [CrossRef]
- Son, A.; Park, S.; Shin, D.M.; Muallem, S. Orai1 and STIM1 in ER/PM junctions: Roles in pancreatic cell function and dysfunction. Am. J. Physiol. Physiol. 2016, 310, C414–C422. [Google Scholar] [CrossRef] [PubMed]
- Gryshchenko, O.; Gerasimenko, J.V.; Gerasimenko, O.V.; Petersen, O.H. Ca2+signals mediated by bradykinin type 2 receptors in normal pancreatic stellate cells can be inhibited by specific Ca2+channel blockade. J. Physiol. 2015, 594, 281–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldron, R.T.; Chen, Y.; Pham, H.; Go, A.; Su, H.; Hu, C.; Wen, L.; Husain, S.Z.; Sugar, C.A.; Roos, J.; et al. The Orai Ca 2+ channel inhibitor CM4620 targets both parenchymal and immune cells to reduce inflammation in experimental acute pancreatitis. J. Physiol. 2019, 597, 3085–3105. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.; Xu, Y.; Jiang, W.; Zhang, H.; Zhu, P.; Wu, J. EGF receptor-mediated intracellular calcium increase in human hepatoma BEL-7404 cells. Cell Res. 1994, 4, 145–153. [Google Scholar] [CrossRef]
- Wang, L.; Wormstone, I.M.; Reddan, J.R.; Duncan, G. Growth factor receptor signalling in human lens cells: Role of the calcium store. Exp. Eye Res. 2005, 80, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.M.; Peters, A.A.; Grice, D.M.; Cabot, P.J.; Parat, M.-O.; Roberts-Thomson, S.J.; Monteith, G.R. Non-Stimulated, Agonist-Stimulated and Store-Operated Ca2+ Influx in MDA-MB-468 Breast Cancer Cells and the Effect of EGF-Induced EMT on Calcium Entry. PLoS ONE 2012, 7, e36923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azimi, I.; Bong, A.H.; Poo, G.X.H.; Armitage, K.; Lok, D.; Roberts-Thomson, S.J.; Monteith, G.R. Pharmacological inhibition of store-operated calcium entry in MDA-MB-468 basal A breast cancer cells: Consequences on calcium signalling, cell migration and proliferation. Cell. Mol. Life Sci. 2018, 75, 4525–4537. [Google Scholar] [CrossRef]
- Aoki, H.; Ohnishi, H.; Hama, K.; Shinozaki, S.; Kita, H.; Yamamoto, H.; Osawa, H.; Sato, K.; Tamada, K.; Sugano, K. Existence of autocrine loop between interleukin-6 and tranforming growth factor-β1 in activated rat pancreatic stellate cells. J. Cell. Biochem. 2006, 99, 221–228. [Google Scholar] [CrossRef]
- Masamune, A.; Kikuta, K.; Satoh, M.; Kume, K.; Shomosegawa, T. Differential Roles of Signaling Pathways for Proliferation and Migration of Rat Pancreatic Stellate Cells. Tohoku J. Exp. Med. 2003, 199, 69–84. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, H.; Miyata, T.; Yasuda, H.; Satoh, Y.; Hanatsuka, K.; Kita, H.; Ohashi, A.; Tamada, K.; Makita, N.; Iiri, T.; et al. Distinct roles of Smad2-, Smad3-, and ERK-dependent pathways in transforming growth factor-beta1 regulation of pancreatic stellate cellular functions. J. Biol. Chem. 2004, 279, 8873–8878. [Google Scholar] [CrossRef] [Green Version]
- Jaster, R.; Sparmann, G.; Emmrich, J.; Liebe, S. Extracellular signal regulated kinases are key mediators of mitogenic signals in rat pancreatic stellate cells. Gut 2002, 51, 579–584. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.-K.; Zhang, C.-X.; Zhao, S.-S.; Zhang, S.-H.; Zhang, H.; Cai, S.-Y.; Shao, R.-G.; He, H.-W. The anti-fibrotic effects of epigallocatechin-3-gallate in bile duct-ligated cholestatic rats and human hepatic stellate LX-2 cells are mediated by the PI3K/Akt/Smad pathway. Acta Pharmacol. Sin. 2015, 36, 473–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Wang, G.; Zhou, H.; Cai, J.; Li, P.; Zhou, M.; Lu, Y.; Jiang, X.; Huang, H.; Zhang, Y.; et al. TGF-β1-miR-200a-PTEN induces epithelial–mesenchymal transition and fibrosis of pancreatic stellate cells. Mol. Cell. Biochem. 2017, 431, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Alevizopoulos, A.; Dusserre, Y.; Rüegg, U.; Mermod, N. Regulation of the Transforming Growth Factor β-responsive Transcription Factor CTF-1 by Calcineurin and Calcium/ Calmodulin-dependent Protein Kinase IV. J. Biol. Chem. 1997, 272, 23597–23605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, J.Y.C.; Dong, H.; Quach, K.T.; Van Nguyen, P.N.; Chen, K.; Carethers, J.M. TGF-β mediates PTEN suppression and cell motility through calcium-dependent PKC-α activation in pancreatic cancer cells. Am. J. Physiol. Liver Physiol. 2008, 294, G899–G905. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Shim, K.-N.; Li, J.M.J.; Estrema, C.; Ornelas, T.A.; Nguyen, F.; Liu, S.; Ramamoorthy, S.L.; Ho, S.; Carethers, J.M.; et al. Molecular mechanisms underlying Ca2+-mediated motility of human pancreatic duct cells. Am. J. Physiol. Physiol. 2010, 299, C1493–C1503. [Google Scholar] [CrossRef] [Green Version]
- Iyer, S.C.; Kannan, A.; Gopal, A.; Devaraj, N.; Halagowder, D. Receptor channel TRPC6 orchestrate the activation of human hepatic stellate cell under hypoxia condition. Exp. Cell Res. 2015, 336, 66–75. [Google Scholar] [CrossRef]
- Kurahara, L.H.; Sumiyoshi, M.; Aoyagi, K.; Hiraishi, K.; Nakajima, K.; Nakagawa, M.; Hu, Y.; Inoue, R. Intestinal Myofibroblast TRPC6 Channel May Contribute to Stenotic Fibrosis in Crohn’s Disease. Inflamm. Bowel Dis. 2015, 21, 496–506. [Google Scholar] [CrossRef]
- Hodeify, R.; Yu, F.; Courjaret, R.; Nader, N.; Dib, M.; Sun, L.; Adap, E.; Hubrack, S.; Machaca, K.; Kozak, J.A.; et al. Regulation and Role of Store-Operated Ca2+ Entry in Cellular Proliferation. In Calcium Entry Channels in Non-Excitable Cells; Kozak, J.A., Putney, J.W., Eds.; CRC Press: Boca Raton, FL, USA, 2017; pp. 215–240. [Google Scholar]
- Gao, X.; Xia, J.; Munoz, F.M.; Manners, M.T.; Pan, R.; Meucci, O.; Dai, Y.; Hu, H. STIMs and Orai1 regulate cytokine production in spinal astrocytes. J. Neuroinflamm. 2016, 13, 126. [Google Scholar] [CrossRef] [Green Version]
- Kondratska, K.; Kondratskyi, A.; Yassine, M.; Lemonnier, L.; Lepage, G.; Morabito, A.; Skryma, R.; Prevarskaya, N. Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance of pancreatic adenocarcinoma. Biochim. Biophys. Acta (BBA) Bioenerg. 2014, 1843, 2263–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somasundaram, A.; Shum, A.K.; McBride, H.J.; Kessler, J.A.; Feske, S.; Miller, R.J.; Prakriya, M. Store-Operated CRAC Channels Regulate Gene Expression and Proliferation in Neural Progenitor Cells. J. Neurosci. 2014, 34, 9107–9123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Hughes, J.D.; Rollins, S.; Chen, B.; Perkins, E. Calcium entry via ORAI1 regulates glioblastoma cell proliferation and apoptosis. Exp. Mol. Pathol. 2011, 91, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhang, H.; Jin, F.; Fang, M.; Huang, M.; Yang, C.S.; Chen, T.; Fu, L.; Pan, Z. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget 2014, 5, 3455–3471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, D.; Park, E.; Jung, E.; Seong, Y.J.; Hong, M.; Lee, S.; Burford, J.; Gyarmati, G.; Peti-Peterdi, J.; Srikanth, S.; et al. ORAI1 Activates Proliferation of Lymphatic Endothelial Cells in Response to Laminar Flow through Krüppel-Like Factors 2 and 4. Circ. Res. 2017, 120, 1426–1439. [Google Scholar] [CrossRef]
- Capiod, T. Cell proliferation, calcium influx and calcium channels. Biochimie 2011, 93, 2075–2079. [Google Scholar] [CrossRef]
- Kim, J.-H.; Lkhagvadorj, S.; Lee, M.-R.; Hwang, K.-H.; Chung, H.C.; Jung, J.H.; Cha, S.-K.; Eom, M. Orai1 and STIM1 are critical for cell migration and proliferation of clear cell renal cell carcinoma. Biochem. Biophys. Res. Commun. 2014, 448, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Capiod, T. The Need for Calcium Channels in Cell Proliferation. Recent Pat. Anti-Cancer Drug Discov. 2012, 8, 4–17. [Google Scholar] [CrossRef]
- El Boustany, C.; Katsogiannou, M.; Delcourt, P.; Dewailly, E.; Prevarskaya, N.; Borowiec, A.-S.; Capiod, T. Differential roles of STIM1, STIM2 and Orai1 in the control of cell proliferation and SOCE amplitude in HEK293 cells. Cell Calcium 2010, 47, 350–359. [Google Scholar] [CrossRef]
- Abdullaev, I.F.; Bisaillon, J.M.; Potier, M.; Gonzalez, J.C.; Motiani, R.K.; Trebak, M. Stim1 and Orai1 Mediate CRAC Currents and Store-Operated Calcium Entry Important for Endothelial Cell Proliferation. Circ. Res. 2008, 103, 1289–1299. [Google Scholar] [CrossRef]
- Borowiec, A.-S.; Bidaux, G.; Tacine, R.; Dubar, P.; Pigat, N.; Delcourt, P.; Mignen, O.; Capiod, T. Are Orai1 and Orai3 channels more important than calcium influx for cell proliferation? Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2014, 1843, 464–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Hao, J.; Zhang, Y.; Yang, Z.; Cao, Y.; Lu, W.; Shu, Y.; Jiang, L.; Hu, Y.; Lv, W.; et al. Orai1 mediates tumor-promoting store-operated Ca2+ entry in human gastrointestinal stromal tumors via c-KIT and the extracellular signal–regulated kinase pathway. Tumor Biol. 2017, 39, 1010428317691426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-W.; Chen, Y.-F.; Chen, Y.-T.; Chiu, W.-T.; Shen, M.-R. The STIM1-Orai1 pathway of store-operated Ca2+ entry controls the checkpoint in cell cycle G1/S transition. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Jairaman, A.; Yamashita, M.; Schleimer, R.P.; Prakriya, M. Store-Operated Ca2+ Release-Activated Ca2+ Channels Regulate PAR2-Activated Ca2+ Signaling and Cytokine Production in Airway Epithelial Cells. J. Immunol. 2015, 195, 2122–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, D.K.; Lim, H.M.; Nam, J.H.; Lee, M.G.; Kim, J.Y. Regulation of phagocytosis and cytokine secretion by store-operated calcium entry in primary isolated murine microglia. Cell. Signal. 2015, 27, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Murthy, D.; Attri, K.S.; Singh, P.K. Phosphoinositide 3-Kinase Signaling Pathway in Pancreatic Ductal Adenocarcinoma Progression, Pathogenesis, and Therapeutics. Front. Physiol. 2018, 9, 335. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.-H.; Li, C.-X.; Zhuo, Y.-Z.; Yang, L.; Cui, N.-Q.; Zhang, S.-K. Saikosaponin d ameliorates pancreatic fibrosis by inhibiting autophagy of pancreatic stellate cells via PI3K/Akt/mTOR pathway. Chem. Interact. 2019, 300, 18–26. [Google Scholar] [CrossRef]
- Ramakrishnan, P.; Loh, W.M.; Gopinath, S.C.B.; Bonam, S.R.; Fareez, I.M.; Mac Guad, R.; Sim, M.S.; Wu, Y.S. Selective phytochemicals targeting pancreatic stellate cells as new anti-fibrotic agents for chronic pancreatitis and pancreatic cancer. Acta Pharm. Sin. B 2020, 10, 399–413. [Google Scholar] [CrossRef]
- Deng, W.; Wang, J.; Zhang, J.; Cai, J.; Bai, Z.; Zhang, Z. Orai1, a Direct Target of microRNA-519, Promotes Progression of Colorectal Cancer via Akt/GSK3β Signaling Pathway. Dig. Dis. Sci. 2016, 61, 1553–1560. [Google Scholar] [CrossRef]
- Zhan, Z.-Y.; Zhong, L.-X.; Feng, M.; Wang, J.-F.; Liu, D.-B.; Xiong, J.-P. Over-expression of Orai1 mediates cell proliferation and associates with poor prognosis in human non-small cell lung carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 5080–5088. [Google Scholar]
- Khan, H.Y.; Mpilla, G.B.; Sexton, R.; Viswanadha, S.; Penmetsa, K.V.; Aboukameel, A.; Diab, M.; Kamgar, M.; Al-Hallak, M.N.; Szlaczky, M.; et al. Calcium Release-Activated Calcium (CRAC) Channel Inhibition Suppresses Pancreatic Ductal Adenocarcinoma Cell Proliferation and Patient-Derived Tumor Growth. Cancers 2020, 12, 750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.-F.; Liu, F.; Xin, J.-Q.; Fan, J.-W.; Wu, N.; Zhu, L.-J.; Duan, L.-F.; Li, Y.-Y.; Zhang, H. Respective roles of the mitogen-activated protein kinase (MAPK) family members in pancreatic stellate cell activation induced by transforming growth factor-β1 (TGF-β1). Biochem. Biophys. Res. Commun. 2018, 501, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.-D.; Zheng, J.-W.; Li, P.; Cheng, M.; Yang, J. Store-Operated Ca2+Entry is involved in Transforming Growth Factor-β1 Facilitated Proliferation of Rat Airway Smooth Muscle Cells. J. Asthma 2013, 50, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Roth-Eichhorn, S.; Eberheim, A.; Bode, H.-P.; Gressner, A.M. Transformation-dependent calcium influx by voltage-operated calcium channels in stellate cells of rat liver. J. Hepatol. 1999, 30, 612–620. [Google Scholar] [CrossRef]
- Tsang, S.W.; Bian, Z.-X. Anti-fibrotic and Anti-tumorigenic Effects of Rhein, a Natural Anthraquinone Derivative, in Mammalian Stellate and Carcinoma Cells. Phytother. Res. 2014, 29, 407–414. [Google Scholar] [CrossRef]
- Berna, M.J.; Seiz, O.; Nast, J.F.; Benten, D.; Bläker, M.; Koch, J.; Lohse, A.W.; Pace, A. CCK1 and CCK2 Receptors Are Expressed on Pancreatic Stellate Cells and Induce Collagen Production. J. Biol. Chem. 2010, 285, 38905–38914. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Alexander, P.B.; Wang, X.-F. TGF-β Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb. Perspect. Biol. 2016, 9, a022145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsner, A.; Lange, F.; Fitzner, B.; Heuschkel, M.; Krause, B.J.; Jaster, R. Distinct antifibrogenic effects of erlotinib, sunitinib and sorafenib on rat pancreatic stellate cells. World J. Gastroenterol. 2014, 20, 7914–7925. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2016, 9, a022129. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad pathways in TGF-β signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Froeling, F.E.; Mirza, T.A.; Feakins, R.M.; Seedhar, A.; Elia, G.; Hart, I.R.; Kocher, H.M. Organotypic Culture Model of Pancreatic Cancer Demonstrates that Stromal Cells Modulate E-Cadherin, β-Catenin, and Ezrin Expression in Tumor Cells. Am. J. Pathol. 2009, 175, 636–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jesnowski, R.; Fürst, D.; Ringel, J.; Chen, Y.; Schrödel, A.; Kleeff, J.; Kolb, A.; Schareck, W.D.; Löhr, M. Immortalization of pancreatic stellate cells as an in vitro model of pancreatic fibrosis: Deactivation is induced by matrigel and N-acetylcysteine. Lab. Investig. 2005, 85, 1276–1291. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radoslavova, S.; Folcher, A.; Lefebvre, T.; Kondratska, K.; Guénin, S.; Dhennin-Duthille, I.; Gautier, M.; Prevarskaya, N.; Ouadid-Ahidouch, H. Orai1 Channel Regulates Human-Activated Pancreatic Stellate Cell Proliferation and TGFβ1 Secretion through the AKT Signaling Pathway. Cancers 2021, 13, 2395. https://doi.org/10.3390/cancers13102395
Radoslavova S, Folcher A, Lefebvre T, Kondratska K, Guénin S, Dhennin-Duthille I, Gautier M, Prevarskaya N, Ouadid-Ahidouch H. Orai1 Channel Regulates Human-Activated Pancreatic Stellate Cell Proliferation and TGFβ1 Secretion through the AKT Signaling Pathway. Cancers. 2021; 13(10):2395. https://doi.org/10.3390/cancers13102395
Chicago/Turabian StyleRadoslavova, Silviya, Antoine Folcher, Thibaut Lefebvre, Kateryna Kondratska, Stéphanie Guénin, Isabelle Dhennin-Duthille, Mathieu Gautier, Natalia Prevarskaya, and Halima Ouadid-Ahidouch. 2021. "Orai1 Channel Regulates Human-Activated Pancreatic Stellate Cell Proliferation and TGFβ1 Secretion through the AKT Signaling Pathway" Cancers 13, no. 10: 2395. https://doi.org/10.3390/cancers13102395