
Epigenetic Effects of Benzene in Hematologic Neoplasms: The Altered Gene Expression

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. General Considerations on Benzene and Neoplasms
1.2. Absorption and Metabolism of Benzene
1.3. Benzene and Cancer
1.4. Benzene and Hematological Malignancies
1.5. Benzene and Leukemia
1.6. Benzene and Lymphoproliferative Diseases
1.7. Benzene and Stem Cells
2. Mechanisms of Benzene Carcinogenesis
Benzene and Immune System
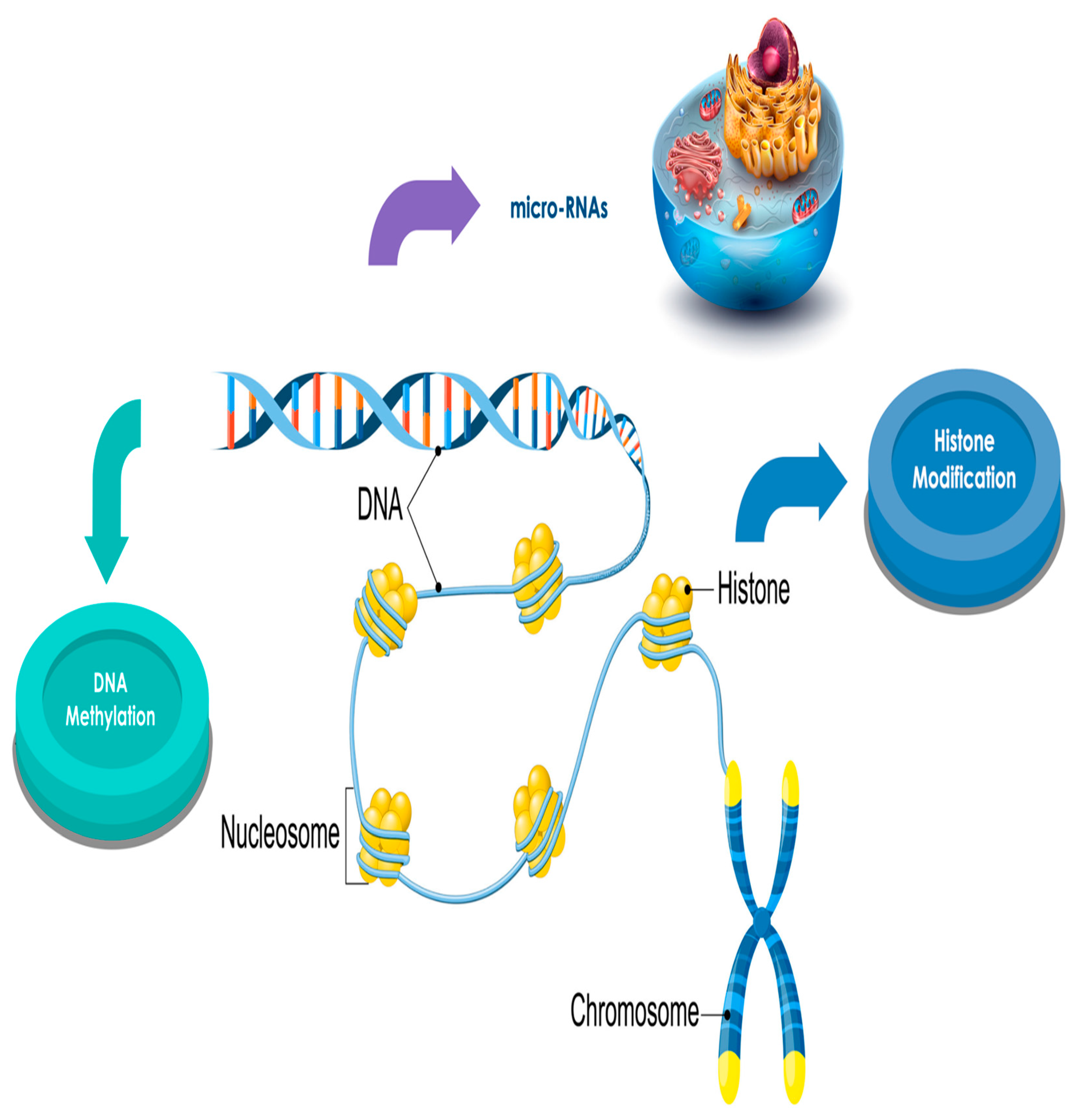
3. Epigenetic and Cancer
4. Epigenetics, Hematological Neoplasms, and Benzene
Benzene and Hemopoietic System: In Vitro and In Vivo Studies
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- International Agency for Research on Cancer (IARC). Monographs on the Identification of Carcinogenic Hazards to Human. Benzene. 2018. Available online: https://publications.iarc.fr/576 (accessed on 10 May 2021).
- ATSDR Toxicological Profile for Benzene. Toxicological Profile. 2007. Available online: https://www.atsdr.cdc.gov/toxprofiles/tp3.pdf (accessed on 9 March 2021).
- Arnold, S.M.; Angerer, J.; Boogaard, P.J.; Hughes, M.F.; O’Lone, R.B.; Robison, S.H.; Schnatter, A.R. The use of biomonitoring data in exposure and human health risk assessment: Benzene case study. Crit. Rev. Toxicol. 2013, 43, 119–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masiol, M.; Agostinelli, C.; Formenton, G.; Tarabotti, E.; Pavoni, B. Thirteen years of air pollution hourly monitoring in a large city: Potential sources, trends, cycles and effects of car-free days. Sci. Total Environ. 2014, 494–495, 84–96. [Google Scholar] [CrossRef]
- Wallace, L. Environmental exposure to benzene: An update. Environ. Health Perspect. 1996, 104, 1129–1136. [Google Scholar]
- Williams, P.R.; Paustenbach, D.J. Reconstruction of benzene exposure for the Pliofilm cohort (1936–1976) using Monte Carlo techniques. J. Toxicol. Environ. Health A 2003, 66, 677–781. [Google Scholar] [CrossRef]
- Fracasso, M.E.; Doria, D.; Bartolucci, G.B.; Carrieri, M.; Lovreglio, P.; Ballini, A.; Soleo, L.; Tranfo, G.; Manno, M. Low air levels of benzene: Correlation between biomarkers of exposure and genotoxic effects. Toxicol. Lett. 2010, 192, 22–28. [Google Scholar] [CrossRef]
- Carrieri, M.; Tranfo, G.; Pigini, D.; Paci, E.; Salamon, F.; Scapellato, M.L.; Fracasso, M.E.; Manno, M.; Bartolucci, G.B. Correlation between environmental and biological monitoring of exposure to benzene in petrochemical industry operators. Toxicol. Lett. 2010, 192, 17–21. [Google Scholar] [CrossRef]
- Campo, L.; Rossella, F.; Mercadante, R.; Fustinoni, S. Exposure to BTEX and Ethers in Petrol Station Attendants and Proposal of Biological Exposure Equivalents for Urinary Benzene and MTBE. Ann. Occup. Hyg. 2016, 60, 318–333. [Google Scholar] [CrossRef] [PubMed]
- Lovreglio, P.; Maffei, F.; Carrieri, M.; D’Errico, M.N.; Drago, I.; Hrelia, P.; Bartolucci, G.B.; Soleo, L. Evaluation of chromosome aberration and micronucleus frequencies in blood lymphocytes of workers exposed to low concentrations of benzene. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2014, 770, 55–60. [Google Scholar] [CrossRef]
- Periago, J.F.; Prado, C. Evolution of occupational Exposure to Environmental Levels of Aromatic Hydrocarbons in Service Stations. Ann. Occup. Hyg. 2005, 49, 233–240. [Google Scholar] [PubMed] [Green Version]
- Almerud, P.; Akerstrom, M.; Andersson, E.M.; Stranberg, B.; Sallsten, G. Low personal exposure to benzene and 1,3-butadiene in the Swedish petroleum refinery industry. Int. Arch. Occup. Environ. Health 2017, 90, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Rauma, M.; Boman, A.; Johanson, G. Predicting the absorption of chemical vapours. Adv. Drug Deliv. Rev. 2013, 65, 306–314. [Google Scholar] [CrossRef]
- Hostynek, J.J.; Lamel, S.A.; Maibach, H.I. Benzene absorption in animals and man: An overview. Rev. Environ. Health 2012, 27, 85–101. [Google Scholar] [CrossRef]
- Maibach, H.I. Percutaneous Penetration of Benzene in Man; Report to the API; American Petroleum Institute: Washington, DC, USA, 1980. [Google Scholar]
- Hanke, J.; Dutkiewcz, T.; Pitrowski, J. The absorption of benzene through human skin. Int. J. Occup. Health 2000, 6, 104–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DECOS (Dutch Expert Committee on Occupational Safety). Benzene: Health-Based Recommended Occupational Exposure Limit, No. 2014/03. 21 February 2014. Available online: https://www.gezondheidsraad.nl/en/task-and-procedure/areas-of-activity/healthy-working-conditions/benzene-health-based-recommended (accessed on 22 May 2019).
- Meek, M.E.; Klaunig, J.E. Proposed mode of action of benzene-induced leukemia: Interpreting available data and identifying critical data gaps for risk assessment. Chem. Biol. Interact. 2010, 184, 279–285. [Google Scholar]
- McHale, C.M.; Zhang, L.; Smith, M.T. MCurrent understanding of the mechanism of benzene-induced leukemia in humans: Implications for risk assessment. Carcinogenesis 2012, 33, 240–252. [Google Scholar] [CrossRef] [Green Version]
- Luijten, M.; Ball, N.S.; Dearfield, K.L.; Gollapudi, B.B.; Johnson, G.E.; Madia, F.; Peel, L.; Pfuhler, S.; Settivari, R.S.; Ter Burg, W.; et al. Utility of a next generation framework for assessment of genomic damage: A case study using the industrial chemical benzene. Environ. Mol. Mutagen. 2020, 61, 94–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusch, G.M.; Leong, B.K.J.; Laskin, S. Benzene metabolism. J. Toxicol. Environ. Health 1997, 2, 23–36. [Google Scholar]
- ECHA (European Chemicals Agency). Annex 1. Background Document in support of the Committee for Risk Assessment (RAC) Evaluation of Limit Values for Benzene in the Workplace; European Chemicals Agency: Helsinki, Finland, 2018; Available online: https://echa.europa.eu/documents/10162/13641/benzene_bg_annex1_en.pdf/37b38de4-0e36-6058-eaa4-1ffc56938831 (accessed on 12 May 2021).
- Carbonari, D.; Chiarella, P.; Mansi, A.; Pigini, D.; Iavicoli, S.; Tranfo, G. Biomarkers of susceptibility following benzene exposure: Influence of genetic polymorphisms on benzene metabolism and health effects. Biomark. Med. 2016, 10, 145–163. [Google Scholar] [CrossRef] [PubMed]
- Petralia, S.A.; Vena, J.E.; Freudenheim, J.L.; Dosemeci, M.; Michalek, A.; Goldberg, M.S.; Brasure, J.; Graham, S. Risk of premenopausal breast cancer in association with occupational exposure to polycyclic aromatic hydrocarbons and benzene. Scand. J. Work Environ. Health 1999, 25, 215–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolff, M.S.; Collman, G.W.; Barrett, J.C.; Huff, J. Breast cancer and environmental risk factors: Epidemiological and experimental findings. Annu. Rev. Pharmacol. Toxicol. 1996, 36, 573–596. [Google Scholar] [CrossRef] [PubMed]
- Costantini, A.S.; Gorini, G.; Consonni, D.; Miligi, L.; Giovannetti, L.; Quinn, M. Exposure to benzene and risk of breast cancer among shoe factory workers in Italy. Tumori 2009, 95, 8–12. [Google Scholar] [CrossRef] [PubMed]
- White, A.J.; Chen, J.; McCullough, L.E.; Xu, X.; Cho, Y.H.; Teitelbaum, S.L.; Neugut, A.I.; Terry, M.B.; Hibshoosh, H.; Santella, R.M.; et al. Polycyclic aromatic hydrocarbon (PAH)-DNA adducts and breast cancer: Modification by gene promoter methylation in a population-based study. Cancer Causes Control 2015, 26, 1791–1802. [Google Scholar] [CrossRef] [PubMed]
- Fenga, C. Occupational exposure and risk of breast cancer. Biomed. Rep. 2016, 4, 282–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steineck, G.; Plato, N.; Gerhardsson, M.; Norell, S.E.; Hogstedt, C. Increased risk of urothelial cancer in Stockholm during 1985–1987 after exposure to benzene and exhausts. Int. J. Cancer 1990, 45, 1012–1017. [Google Scholar] [CrossRef]
- Axelsson, G.; Barregard, L.; Holmberg, E.; Sallsten, G. Cancer incidence in a petrochemical industry area in Sweden. Sci. Total Environ. 2010, 408, 4482–4487. [Google Scholar] [CrossRef]
- Lan, Q.; Zhang, L.; Li, G.; Vermeulen, R.; Weinberg, R.S.; Dosemeci, M.; Rappaport, S.M.; Shen, M.; Alter, B.P.; Wu, Y.; et al. Hematotoxicity in workers exposed to low levels of benzene. Science 2004, 306, 1774–1776. [Google Scholar] [CrossRef] [Green Version]
- Lamm, S.H.; Grunwald, H.W. Benzene exposure and hematotoxicity. Science 2006, 312, 998. [Google Scholar] [CrossRef]
- Jephcote, C.; Brown, D.; Verbeek, T.; Mah, A. A systematic review and meta-analysis of haematological malignancies in residents living near petrochemical facilities. Environ. Health. 2020, 19, 53. [Google Scholar] [CrossRef] [PubMed]
- Khalade, A.; Jaakkola, M.S.; Pukkala, E.; Jaakkola, J.J. Exposure to benzene at work and the risk of leukemia: A systematic review and meta-analysis. Environ. Health 2010, 9, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante, P.F.; Rinsky, R.A.; Wagoner, J.K.; Young, R.J. Leukaemia in benzene workers. Lancet 1977, 310, 76–78. [Google Scholar] [CrossRef]
- Hayes, R.B.; Yin, S.N.; Dosemeci, M.; Li, G.L.; Wacholder, S.; Travis, L.B.; Li, C.Y.; Rothman, N.; Hoover, R.N.; Linet, M.S. Benzene and the dose-related incidence of hematologic neoplasms in China. J. Natl. Cancer Inst. 1997, 89, 1065–1071. [Google Scholar] [CrossRef] [Green Version]
- Wong, O.; Harris, F.; Armstrong, T.W.; Fu, H. A hospital based case-control study of acute myeloid leukemia in Shanghai: Analysis of environmental and occupational risk factors by subtypes of the WHO classification. Chem. Biol. Interact. 2010, 184, 112–128. [Google Scholar] [CrossRef]
- Stenehjem JS, Kjærheim K, Bråtveit M, Samuelsen SO, Barone-Adesi F, Rothman N, Lan Q, Grimsrud TK. Benzene exposure and risk of lymphohaematopoietic cancers in 25 000 offshore oil industry workers. Br. J. Cancer 2015, 112, 1603–1612. [Google Scholar] [CrossRef] [Green Version]
- Yin, S.N.; Li, G.L.; Tain, F.D.; Fu, Z.I.; Jin, C.; Chen, Y.J.; Luo, S.J.; Ye, P.Z.; Zhang, J.Z.; Wang, G.C.; et al. Leukaemia in Benzene Workers: A Retrospective Cohort Study. Br. J. Ind. Med. 1987, 44, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Hayes, R.B.; Yin, S.N.; Dosemeci, M.; Li, G.L.; Wacholder, S.; Chow, W.H.; Rothman, N.; Wang, Y.Z.; Dai, T.R.; Chao, X.J.; et al. Mortality among Benzene-Exposed Workers in China. Environ. Health Perspect. 1996, 104, 1349–1352. [Google Scholar]
- Yin, S.N.; Hayes, R.B.; Linet, M.S.; Li, G.L.; Dosemeci, M.; Travis, L.B.; Zhang, Z.N.; Li, D.G.; Chow, W.H.; Wacholder, S.; et al. An Expanded Cohort Study of Cancer among Benzene-Exposed Workers in China. Benzene Study Group. Environ. Health Perspect. 1996, 104, 1339–1341. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.C.; Gray, C.N.; Jolley, D.J.; Gibbons, C.; Sim, M.R.; Fritschi, L.; Adams, G.G.; Bisby, J.A.; Manuell, R. Leukemia Risk Associated with Low-Level Benzene Exposure. Epidemiology 2003, 14, 569–577. [Google Scholar] [CrossRef]
- Glass, D.C.; Sim, M.R.; Fritschi, L.; Gray, C.N.; Jolley, D.J.; Gibbons, C. Leukemia Risk and Relevant Benzene Exposure Period-Re: Follow-up Time on Risk Estimates. Am. J. Ind. Med. 2004, 42, 481–489. [Google Scholar]
- Glass, D.C.; Gray, C.N.; Jolley, D.J.; Gibbons, C.; Sim, M.R. Health Watch Exposure Estimates: Do They Underestimate Benzene Exposure? Chem. Biol. Interact. 2005, 153–154, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.A.; Paustenbach, D.J. Shanghai health study (2001–2009): What was learned about benzene health effects? Crit. Rev. Toxicol. 2018, 48, 217–251. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.C.W.; Shahbaz, S.; Winn, L.M. Benzene and its effects on cell signaling pathways related to hematopoiesis and leukemia. J. Appl. Toxicol. 2020, 40, 1018–1032. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Profiling aberrant DNA methylation in hematologic neoplasms: A view from the tip of the iceberg. Clin. Immunol. 2003, 109, 80–88. [Google Scholar] [CrossRef]
- Goldstein, B.D. Benzene as a Cause of Lymphoproliferative Disorders. Chem. Biol. Interact. 2010, 184, 147–150. [Google Scholar] [CrossRef]
- Steinmaus, C.; Smith, A.H.; Jones, R.M.; Smith, M.T. Meta-Analysis of Benzene Exposure and Non-Hodgkin Lymphoma: Biases Could Mask an Important Association. Occup. Environ. Med. 2008, 65, 371–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decoufle, P.; Blattner, W.A.; Blair, A. Mortality among Chemical Workers Exposed to Benzene and Other Agents. Environ. Res. 1983, 30, 16–25. [Google Scholar] [CrossRef]
- Infante, P.F. Benzene Exposure and Multiple Myeloma: A Detailed Meta-Analysis of Benzene Cohort Studies. Ann. N. Y. Acad. Sci. 2006, 1076, 90–109. [Google Scholar] [CrossRef]
- Goldstein, B.D. Is Exposure to Benzene a Cause of Human Multiple Myeloma? Ann. N. Y. Acad. Sci. 1990, 609, 225–230. [Google Scholar] [CrossRef]
- Brosselin, P.; Rudant, J.; Orsi, L.; Leverger, G.; Baruchel, A.; Bertrand, Y.; Nelken, B.; Robert, A.; Michel, G.; Margueritte, G.; et al. Acute Childhood Leukaemia and Residence Next to Petrol Stations and Automotive Repair Garages: The Escale Study (Sfce). Occup. Environ. Med. 2009, 66, 598–606. [Google Scholar] [CrossRef]
- Whitworth, K.W.; Symanski, E.; Coker, A.L. Childhood Lymphohematopoietic Cancer Incidence and Hazardous Air Pollutants in Southeast Texas 1995–2004. Environ. Health Perspect. 2008, 116, 1576–1580. [Google Scholar] [CrossRef] [Green Version]
- Pyatt, D.; Hays, S. A Review of the Potential Association between Childhood Leukemia and Benzene. Chem. Biol. Interact. 2010, 184, 151–164. [Google Scholar] [CrossRef]
- Smith, M.T.; Zhang, L.; McHale, C.M.; Skibola, C.F.; Rappaport, S.M. Benzene, the Exposome and Future Investigations of Leukemia Etiology. Chem. Biol. Interact. 2011, 192, 155–159. [Google Scholar] [CrossRef] [Green Version]
- Badham, H.J.; Winn, L.M. In Utero and in Vitro Effects of Benzene and Its Metabolites on Erythroid Differentiation and the Role of Reactive Oxygen Species. Toxicol. Appl. Pharmacol. 2010, 244, 273–279. [Google Scholar] [CrossRef]
- Lau, A.; Belanger, C.L.; Winn, L.M. In Utero and Acute Exposure to Benzene: Investigation of DNA Double-Strand Breaks and DNA Recombination in Mice. Mutat. Res. 2009, 676, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Beelte, S.; Haas, R.; Germing, U.; Jansing, P.J. Paradigm Change in The Assessment of Myeloid and Lymphoid Neoplasms Associated with Occupational Benzene Exposure. Med. Klin. 2009, 104, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; He, X.; Bi, Y.; Ma, Q. Stem cell and benzene-induced malignancy and hematotoxicity. Chem. Res. Toxicol. 2012, 25, 1303–1315. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Zhang, J.; Wei, H.; Meng, X.; Ding, Q.; Sun, F.; Cao, M.; Yin, L.; Pu, Y. Acetyl-l-carnitine partially prevents benzene-induced hematotoxicity and oxidative stress in C3H/He mice. Environ. Toxicol. Pharmacol. 2017, 51, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Zhang, J.; Xiong, M.; Wei, H.; Tan, K.; Yin, L.; Pu, Y. Altered expression of genes in signaling pathways regulating proliferation of hematopoietic stem and progenitor cells in mice with subchronic benzene exposure. Int. J. Environ. Res. Public Health 2015, 12, 9298–9313. [Google Scholar] [CrossRef] [Green Version]
- Yoon, B.I.; Hirabayashi, Y.; Kawasaki, Y.; Kodama, Y.; Kaneko, T.; Kim, D.Y.; Inoue, T. Mechanism of action of benzene toxicity: Cell cycle suppression in hemopoietic progenitor cells (CFU-GM). Exp. Hematol. 2001, 29, 278–285. [Google Scholar]
- Giver, C.R.; Wong, R.; Moore II, D.H.; Pallavicini, M.G. Persistence of aneuploid immature/primitive hemopoietic sub-populations in mice 8 months after benzene exposure in vivo. Mutat. Res. 2001, 491, 127–138. [Google Scholar] [CrossRef]
- Chow, P.W.; Abdul Hamid, Z.; Chan, K.M.; Inayat-Hussain, S.H.; Rajab, N.F. Lineage-related cytotoxicity and clonogenic profile of 1,4-benzoquinone-exposed hematopoietic stem and progenitor cells. Toxicol. Appl. Pharmacol. 2015, 284, 8–15. [Google Scholar] [CrossRef]
- Zhou, H.; Kepa, J.K.; Siegel, D.; Miura, S.; Hiraki, Y.; Ross, D. Benzene metabolite hydroquinone up-regulates chondromodulin-I and inhibits tube formation in human bone marrow endothelial cells. Mol. Pharmacol. 2009, 76, 579–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivedal, E.; Witz, G.; Leithe, E. Gap junction intercellular communication and benzene toxicity. Chem. Biol. Interact. 2010, 184, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Dehn, D.; Kepa, J.K.; Siegel, D.; Scott, D.E.; Tan, W.; Ross, D. NAD(P)H: Quinone oxidoreductase 1-compromised human bone marrow endothelial cells exhibit decreased adhesion molecule expression and CD34+ hematopoietic cell adhesion. J. Pharmacol. Exp. Ther. 2010, 334, 260–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guyton, K.Z.; Kyle, A.D.; Aubrecht, J.; Cogliano, V.J.; Eastmond, D.A.; Jackson, M.; Keshava, N.; Sandy, M.S.; Sonawane, B.; Zhang, L.; et al. Improving prediction of chemical carcinogenicity by considering multiple mechanisms and applying toxico-genomic approaches. Mutat Res. 2009, 681, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.T.; Guyton, K.Z.; Gibbons, C.F.; Fritz, J.M.; Portier, C.J.; Rusyn, I.; DeMarini, D.M.; Caldwell, J.C.; Kavlock, R.J.; Lambert, P.F.; et al. Key Characteristics of Carcinogens as a Basis for Organizing Data on Mechanisms of Carcinogenesis. Environ. Health Perspect. 2016, 124, 713–721. [Google Scholar] [CrossRef] [Green Version]
- Haro-García, L.C.; Juárez-Pérez, C.A.; Aguilar-Madrid, G.; Vélez-Zamora, N.M.; Muñoz-Navarro, S.; Chacón-Salinas, R.; González-Bonilla, C.R.; Iturbe-Haro, C.R.; Estrada-García, I.; Borja-Aburto, V.H. Production of IL-10, TNF and IL-12 by peripheral blood mononuclear cells in Mexican workers exposed to a mixture of benzene-toluene-xylene. Arch. Med. Res. 2012, 43, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, M.; Spatari, G.; Tranfo, G.; Sapienza, D.; Scapellato, M.L.; Bartolucci, G.B.; Manno, M. Biological monitoring of low level exposure to benzene in an oil refinery: Effect of modulating factors. Toxicol. Lett. 2018, 298, 70–75. [Google Scholar] [CrossRef]
- Bird, M.G.; Wetmore, B.A.; Letinski, D.J.; Nicolich, M.; Chen, M.; Schnatter, A.R.; Whitman, F.T. Influence of toluene co-exposure on the metabolism and genotoxicity of benzene in mice using continuous and intermittent exposures. Chem. Biol. Interact. 2010, 184, 233–239. [Google Scholar] [CrossRef]
- Sarma, S.N.; Kim, Y.J.; Song, M.; Ryu, J.C. Induction of apoptosis in human leukemia cells through the production of reactive oxygen species and activation of HMOX1 and Noxa by benzene, toluene, and o-xylene. Toxicology 2011, 280, 109–117. [Google Scholar] [CrossRef]
- Guo, H.; Ahn, S.; Zhang, L. Benzene-associated immunosuppression and chronic inflammation in humans: A systematic review. Occup. Environ. Med. 2020, 78. [Google Scholar] [CrossRef]
- Spatari, G.; Saitta, S.; Giorgianni, C.; Cristani, M.T.; Quattrocchi, P.; Abbate, A.; Carrieri, M.; Ferraro, G.; Saija, A.; Gangemi, S. Interleukin-10 involvement in exposure to low dose of benzene. Toxicol. Ind. Health. 2015, 31, 351–354. [Google Scholar] [CrossRef]
- Minciullo PL, Navarra M, Calapai G, Gangemi, S. Cytokine network involvement in subjects exposed to benzene. J. Immunol. Res. 2014, 2014, 937987. [Google Scholar] [CrossRef] [Green Version]
- Hwang, K.A.; Kim, H.R.; Kang, I. Aging and human CD4(þ) regulatory T cells. Mech. Ageing Dev. 2009, 130, 509–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romer-Seibert, Y.J.; Meyer, S.E. Genetic heterogeneity and clonal evolution in acute myeloid leukemia. Curr. Opin. Hematol. 2021, 28, 64–70. [Google Scholar] [CrossRef]
- Kim, Y.J.; Choi, J.Y.; Paek, D.; Chung, H.W. Association of the NQO1, MPO, and XRCC1 polymorphisms and chromosome damage among workers at a petroleum refinery. J. Toxicol. Environ. Health 2008, 71, 333–341. [Google Scholar] [CrossRef]
- Kim, Y.J.; Choi, J.Y.; Cho, Y.H.; Woo, H.D.; Chung, H.W. Micronucleus-centromere assay in workers occupationally exposed to low level of benzene. Hum. Exp. Toxicol. 2010, 29, 343–350. [Google Scholar]
- Pitarque, M.; Carbonell, E.; Lapeña, N.; Marsá, M.; Torres, M.; Creus, A.; Xamena, N.; Marcos, R. No increase in micronuclei frequency in cultured blood lymphocytes from a group of filling station attendants. Mutat. Res. 1996, 367, 161–167. [Google Scholar] [CrossRef]
- Basso, E.; Cevoli, C.; Papacchini, M.; Tranfo, G.; Mansi, A.; Testa, A. Cytogenetic biomonitoring on a group of petroleum refinery workers. Environ. Mol. Mutagen. 2011, 52, 440–447. [Google Scholar] [CrossRef]
- Sha, Y.; Zhou, W.; Yang, Z.; Zhu, X.; Xiang, Y.; Li, T.; Zhu, D.; Yang, X. Changes in poly(ADP-ribosyl)ation patterns in workers exposed to BTX. PLoS ONE 2014, 9, e106146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Eastmond, D.A.; Smith, M.T. The nature of chromosomal aberrations detected in humans exposed to benzene. Crit. Rev. Toxicol. 2002, 32, 1–42. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lan, Q.; Ji, Z.; Li, G.; Shen, M.; Vermeulen, R.; Guo, W.; Hubbard, A.E.; McHale, C.M.; Rappaport, S.M.; et al. Leukemia-related chromosomal loss detected in hematopoietic progenitor cells of benzene-exposed workers. Leukemia 2012, 26, 2494–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Z.; Zhang, L.; Peng, V.; Ren, X.; McHale, C.M.; Smith, M.T. A comparison of the cytogenetic alterations and global DNA hypomethylation induced by the benzene metabolite, hydroquinone, with those induced by melphalan and etoposide. Leukemia 2010, 24, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.T.; Zhang, L.; Jeng, M.; Wang, Y.; Guo, W.; Duramad, P.; Hubbard, A.E.; Hofstadler, G.; Holland, N.T. Hydroquinone, a benzene metabolite, increases the level of aneusomy of chromosomes 7 and 8 in human CD34-positive blood progenitor cells. Carcinogenesis 2000, 21, 1485–1490. [Google Scholar] [CrossRef]
- Stillman, W.S.; Varella-Garcia, M.; Irons, R.D. The benzene metabolite, hydroquinone, selectively induces 5q31- and -7 in human CD34+CD19- bone marrow cells. Exp. Hematol. 2000, 28, 169–176. [Google Scholar] [CrossRef]
- Smith, M.T. The mechanism of benzene-induced leukemia: A hypothesis and speculations on the causes of leukemia. Environ. Health Perspect. 1996, 104, 1219–1225. [Google Scholar] [CrossRef]
- Faiola, B.; Fuller, E.S.; Wong, V.A.; Recio, L. Gene expression profile in bone marrow and hematopoietic stem cells in mice exposed to inhaled benzene. Mutat. Res. 2004, 549, 195–212. [Google Scholar] [CrossRef] [PubMed]
- Wickliffe, J.K.; Dertinger, S.D.; Torous, D.K.; Avlasevich, S.L.; Simon-Friedt, B.R.; Wilson, M.J. Diet-induced obesity increases the frequency of Pig-a mutant erythrocytes in male C57BL/6J mice. Environ. Mol. Mutagen. 2016, 57, 668–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.T.; Zhang, L.P. Biomarkers of leukemia risk: Benzene as a model. Environ. Health Perspect. 1998, 106, 937–946. [Google Scholar]
- Zhang, L.; Lan, Q.; Guo, W.; Hubbard, A.E.; Li, G.; Rappaport, S.M.; McHale, C.M.; Shen, M.; Ji, I.Z.; Vermeulen, R.; et al. Chromosome-wide aneuploidy study (CWAS) in workers exposed to an established leukemogen, benzene. Carcinogenesis 2011, 32, 605–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccone, G.; Mirabelli, D.; Levis, A.; Gavarotti, P.; Rege-Cambrin, G.; Davico, L.; Vineis, P. Myeloid leukemias and myelodysplastic syndromes: Chemical exposure, histologic subtype and cytogenetics in a case-control study. Cancer Genet. Cytogenet. 1993, 68, 135–139. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Razin, A.; Riggs, A.D. DNA methylation and gene function. Science 1980, 210, 604–610. [Google Scholar] [CrossRef]
- Reik, W.; Dean, W.; Walter, J. Epigenetic reprogramming in mammalian development. Science 2001, 293, 1089–1093. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Herceg, Z.; Lambert, M.P.; van Veldhoven, K.; Demetriou, C.; Vineis, P.; Smith, M.T.; Straif, K.; Wild, C.P. Towards incorporating epigenetic mechanisms into carcinogen identification and evaluation. Carcinogenesis 2013, 34, 1955–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pogribny, I.P.; Rusyn, I. Environmental toxicants, epigenetics, and cancer. Adv. Exp. Med. Biol. 2013, 754, 215–232. [Google Scholar]
- Sanjuan-Pla, A.; Bueno, C.; Prieto, C.; Acha, P.; Stam, R.W.; Marschalek, R.; Menéndez, P. Revisiting the biology of infant t(4;11)/MLL-AF4+ B-cell acute lymphoblastic leukemia. Blood 2015, 126, 2676–2685. [Google Scholar] [CrossRef] [Green Version]
- Das, P.M.; Singal, R. DNA methylation and cancer. J. Clin. Oncol. 2004, 22, 4632–4642. [Google Scholar] [CrossRef] [Green Version]
- Chappell, G.; Pogribny, I.P.; Guyton, K.Z.; Rusyn, I. Epigenetic alterations induced by genotoxic occupational and environmental human chemical carcinogens: A systematic literature review. Mutat. Res. Rev. Mutat. Res. 2016, 768, 27–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukushima, S.; Kinoshita, A.; Puatanachokchai, R.; Kushida, M.; Wanibuchi, H.; Morimura, K. Hormesis and dose-response-mediated mechanisms in carcinogenesis: Evidence for a threshold in carcinogenicity of non-genotoxic carcinogens. Carcinogenesis 2005, 26, 1835–1845. [Google Scholar] [CrossRef] [Green Version]
- Karpinets, T.V.; Foy, B.D. Tumorigenesis: The adaptation of mammalian cells to sustained stress environment by epigenetic alterations and succeeding matched mutations. Carcinogenesis 2005, 26, 1323–1334. [Google Scholar] [CrossRef] [Green Version]
- Franco, R.; Schoneveld, O.; Georgakilas, A.G.; Panayiotidis, M.I. Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett. 2008, 266, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Iacobuzio-Donahue, C.A. Epigenetic changes in cancer. Annu. Rev. Pathol. 2009, 4, 229–249. [Google Scholar] [CrossRef] [PubMed]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Shallis, R.M.; Ahmad, R.; Zeidan, A.M. The genetic and molecular pathogenesis of myelodysplastic syndromes. Eur. J. Haematol. 2018, 101, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Cui, J.P.; Luo, M.; Liu, H.; Hao, P.; Wang, X.; Zhang, G.H. The prevalence and persistence of aberrant promoter DNA methylation in benzene-exposed Chinese workers. PLoS ONE 2019, 14, e0220500. [Google Scholar] [CrossRef] [Green Version]
- Irons, R.D.; Chen, Y.; Wang, X.; Ryder, J.; Kerzic, P.J. Acute myeloid leukemia following exposure to benzene more closely resembles de novo than therapy related-disease. Genes Chromosomes Cancer. 2013, 52, 887–894. [Google Scholar] [CrossRef]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef]
- Benbrahim-Tallaa, L.; Waterland, R.A.; Dill, A.L.; Webber, M.M.; Waalkes, M.P. Tumor suppressor gene inactivation during cadmium-induced malignant transformation of human prostate cells correlates with overexpression of de novo DNA methyltransferase. Environ. Health Perspect. 2007, 115, 1454–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischle, W.; Franz, H.; Jacobs, S.A.; Allis, C.D.; Khorasanizadeh, S. Specificity of the chromodomain Y chromosome family of chromodomains for lysine-methylated ARK(S/T) motifs. J. Biol. Chem. 2008, 283, 19626–19635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Chapman-Rothe, N.; Curry, E.; Zeller, C.; Liber, D.; Stronach, E.; Gabra, H.; Ghaem-Maghami, S.; Brown, R. Chromatin H3K27me3/H3K4me3 histone marks define gene sets in high grade serous ovarian cancer that distinguish malignant, tumour-sustaining and chemoresistant ovarian tumour cells. Oncogene 2013, 32, 4586–4592. [Google Scholar] [CrossRef] [Green Version]
- Hahn, M.A.; Li, A.X.; Wu, X.; Yang, R.; Drew, D.A.; Rosenberg, D.W.; Pfeifer, G.P. Loss of the polycomb mark from bivalent promoters leads to activation of cancer-promoting genes in colorectal tumors. Cancer Res. 2014, 74, 3617–3629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, E.L.; Beese-Sims, S.E.; Brookes, E.; Spadafora, R.; Zhu, Y.; Rothbart, S.B.; Aristizábal-Corrales, D.; Chen, S.; Badeaux, A.I.; Jin, Q.; et al. A histone methylation network regulates transgenerational epigenetic memory in C. elegans. Cell Rep. 2014, 7, 113–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronner, C.; Krifa, M.; Mousli, M. Increasing role of UHRF1 in the reading and inheritance of the epigenetic code as well as in tumorogenesis. Biochem. Pharmacol. 2013, 86, 1643–1649. [Google Scholar] [CrossRef] [PubMed]
- Lubbert, M.; Oster, W.; Ludwig, W.D.; Ganser, A.; Mertelsmann, R.; Herrmann, F. A switch toward demethylation is associated with the expression of myeloperoxidase in acute myeloblastic and promyelocytic leukemias. Blood 1992, 80, 2066–2073. [Google Scholar] [CrossRef] [Green Version]
- Greiner, J.; Ringhoffer, M.; Simikopinko, O.; Szmaragowska, A.; Huebsch, S.; Maurer, U.; Bergmann, L.; Schmitt, M. Simultaneous expression of different immunogenic antigens in acute myeloid leukemia. Exp. Hematol. 2000, 28, 1413–1422. [Google Scholar] [CrossRef]
- Chambost, H.; Brasseur, F.; Coulie, P.; de Plaen, E.; Stoppa, A.M.; Baume, D.; Mannoni, P.; Boon, T.; Maraninchi, D.; Olive, D. A tumour associated antigen expression in human haematological malignancies. Br. J. Haematol. 1993, 84, 524–526. [Google Scholar] [CrossRef]
- Zumkeller, W. The insulin-like growth factor system in hematopoietic cells. Leuk. Lymphoma 2002, 43, 487–491. [Google Scholar] [CrossRef]
- Ernst, P.; Wang, J.; Korsmeyer, S.J. The role of MLL in hematopoiesis and leukemia. Curr. Opin. Hematol. 2002, 9, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, F.; Girardi, K.; Avvisati, G. Pathogenetic, Clinical, and Prognostic Features of Adult t(4;11)(q21;q23)/MLL-AF4 positive B-cell acute lymphoblastic leukemia. Adv. Hematol. 2011, 2011, 621627. [Google Scholar] [CrossRef]
- Nanya, M.; Sato, M.; Tanimoto, K.; Tozuka, M.; Mizutani, S.; Takagi, M. Dysregulation of the DNA damage response and KMT2A rearrangement in fetal liver hematopoietic cells. PLoS ONE 2015, 10, e0144540. [Google Scholar] [CrossRef] [PubMed]
- Emerenciano, M.; Koifman, S.; Pombo-de-Oliveira, M.S. Acute leukemia in early childhood. Braz. J. Med. Biol. Res. 2007, 40, 749–760. [Google Scholar] [CrossRef] [Green Version]
- Strick, R.; Strissel, P.L.; Borgers, S.; Smith, S.L.; Rowley, J.D. Dietary bioflavonoids induce cleavage in the MLL gene and may contribute to infant leukemia. Proc. Natl. Acad. Sci. USA 2000, 97, 4790–4795. [Google Scholar] [CrossRef] [Green Version]
- Andersson, A.K.; Ma, J.; Wang, J.; Chen, X.; Gedman, A.L.; Dang, J.; Nakitandwe, J.; Holmfeldt, L.; Parker, M.; Easton, J.; et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat. Genet. 2015, 47, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Bardini, M.; Woll, P.S.; Corral, L.; Luc, S.; Wittmann, L.; Ma, Z.; Lo Nigro, L.; Basso, G.; Biondi, A.; Cazzaniga, G.; et al. Clonal variegation and dynamic competition of leukemia-initiating cells in infant acute lymphoblastic leukemia with MLL rearrangement. Leukemia 2015, 29, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Stumpel, D.J.; Schneider, P.; van Roon, E.H.; Pieters, R.; Stam, R.W. Absence of global hypomethylation in promoter hypermethylated Mixed Lineage Leukemia-rearranged infant acute lymphoblastic leukemia. Eur. J. Cancer 2013, 49, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Stam, R.W.; Schneider, P.; Hagelstein, J.A.; van der Linden, M.H.; Stumpel, D.J.; de Menezes, R.X.; de Lorenzo, P.; Valsecchi, M.G.; Pieters, R. Gene expression profiling-based dissection of MLL translocated and MLL germline acute lymphoblastic leukemia in infants. Blood 2010, 115, 2835–2844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhojwani, D.; Yang, J.J.; Pui, C.H. Biology of childhood acute lymphoblastic leukemia. Pediatr. Clin. N. Am. 2015, 62, 47–60. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.; Xu, K.; Ji, S.; Pu, Y.; Yu, L.; Yin, L.; Zhang, J.; Pu, Y. Toxicity in hematopoietic stem cells from bone marrow and peripheral blood in mice after benzene exposure: Single-cell transcriptome sequencing analysis. Ecotoxicol. Environ. Saf. 2021, 207, 111490. [Google Scholar] [CrossRef]
- Bollati, V.; Baccarelli, A.; Hou, L.; Bonzini, M.; Fustinoni, S.; Cavallo, D.; Byun, H.M.; Jiang, J.; Marinelli, B.; Pesatori, A.C.; et al. Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res. 2007, 67, 876–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez-Garza, O.; Guo, L.; Byun, H.M.; Carrieri, M.; Bartolucci, G.B.; Barrón-Vivanco, B.S.; Baccarelli, A.A. Aberrant promoter methylation in genes related to hematopoietic malignancy in workers exposed to a VOC mixture. Toxicol. Appl. Pharmacol. 2018, 339, 65–72. [Google Scholar] [CrossRef]
- Mancini, M.; Mandruzzato, M.; Garzia, A.C.; Sahnane, N.; Magnani, E.; Macchi, F.; Oulad-Abdelghani, M.; Oudet, P.; Bollati, V.; Fustinoni, S.; et al. In vitro hydroquinone-induced instauration of histone bivalent mark on human retroelements (LINE-1) in HL60 cells. Toxicol. In Vitro 2017, 40, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Tabish, A.M.; Poels, K.; Hoet, P.; Godderis, L. Epigenetic factors in cancer risk: Effect of chemical carcinogens on global DNA methylation pattern in human TK6 cells. PLoS ONE 2012, 7, e34674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Ma, H.; Zhang, W.; Yu, Z.; Sheng, G.; Fu, J. Effects of benzene and its metabolites on global DNA methylation in human normal hepatic L02 cells. Environ. Toxicol. 2014, 29, 108–116. [Google Scholar] [CrossRef]
- Nishikawa, T.; Izumo, K.; Miyahara, E.; Horiuchi, M.; Okamoto, Y.; Kawano, Y.; Takeuchi, T. Benzene induces cytotoxicity without metabolic activation. J. Occup. Health. 2011, 53, 84–92. [Google Scholar] [CrossRef] [Green Version]
- Carnero, A.; Hannon, G.J. The INK4 family of CDK inhibitors. Curr. Top. Microbiol. Immunol. 1998, 227, 43–55. [Google Scholar]
- Xing, E.P.; Nie, Y.; Song, Y.; Yang, G.Y.; Cai, Y.C.; Wang, L.D.; Yang, C.S. Mechanisms of inactivation of p14ARF, p15INK4b, and p16INK4a genes in human esophageal squamous cell carcinoma. Clin. Cancer Res. 1999, 5, 2704–2713. [Google Scholar]
- Robertson, K.D.; Jones, P.A. The human ARF cell cycle regulatory gene promoter is a CpG island which can be silenced by DNA methylation and down-regulated by wild-type p53. Mol. Cell Biol. 1998, 18, 6457–6473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savic, S.; Franco, N.; Grilli, B.; Barascud Ade, V.; Herzog, M.; Bode, B.; Loosli, H.; Spieler, P.; Schönegg, R.; Zlobec, I.; et al. Fluorescence in situ hybridization in the definitive diagnosis of malignant mesothelioma in effusion cytology. Chest 2010, 138, 137–144. [Google Scholar] [CrossRef]
- Jarmalaite, S.; Kannio, A.; Anttila, S.; Lazutka, J.R.; Husgafvel-Pursiainen, K. Aberrant p16 promoter methylation in smokers and former smokers with nonsmall cell lung cancer. Int. J. Cancer 2003, 106, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Jamebozorgi, I.; Majidizadeh, T.; Pouryagoub, G.; Mahjoubi, F. Aberrant DNA Methylation of Two Tumor Suppressor Genes, p14ARF and p15INK4b, after Chronic Occupational Exposure to Low Level of Benzene. Int. J. Occup. Environ. Med. 2018, 9, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.T. Advances in understanding benzene health effects and susceptibility. Annu. Rev. Public Health 2010, 31, 133–148. [Google Scholar] [CrossRef] [Green Version]
- Rinsky, R.A.; Hornung, R.W.; Silver, S.R.; Tseng, C.Y. Benzene exposure and hematopoietic mortality: A long-term epidemiologic risk assessment. Am. J. Ind. Med. 2002, 42, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, B.D. Benzene toxicity. Occup. Med. 1988, 3, 541–554. [Google Scholar]
- Richardson, D.B. Temporal variation in the association between benzene and leukemia mortality. Environ. Health Perspect. 2008, 116, 370–374. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Zhang, X.; Wang, D.; Baccarelli, A. Environmental chemical exposure and human epigenetics. Int. J. Epidemiol. 2012, 41, 79–105. [Google Scholar] [CrossRef] [Green Version]
- Esteller, M.; Tortola, S.; Toyota, M.; Capella, G.; Peinado, M.A.; Baylin, S.B.; Herman, J.G. Hypermethylation-associated inactivation of p14(ARF) is independent of p16(INK4a) methylation and p53 mutational status. Cancer Res. 2000, 60, 129–133. [Google Scholar]
- Bodoor, K.; Haddad, Y.; Alkhateeb, A.; Al-Abbadi, A.; Dowairi, M.; Magableh, A.; Bsoul, N.; Ghabkari, A. DNA hypermethylation of cell cycle (p15 and p16) and apoptotic (p14, p53, DAPK and TMS1) genes in peripheral blood of leukemia patients. Asian Pac. J. Cancer Prev. 2014, 15, 75–84. [Google Scholar] [CrossRef]
- Li, J.; Bi, L.; Lin, Y.; Lu, Z.; Hou, G. Clinicopathological significance and potential drug target of p15INK4B in multiple myeloma. Drug Des. Devel. Ther. 2014, 8, 2129–2136. [Google Scholar] [CrossRef] [Green Version]
- Seow, W.J.; Pesatori, A.C.; Dimont, E.; Farmer, P.B.; Albetti, B.; Ettinger, A.S.; Bollati, V.; Bolognesi, C.; Roggieri, P.; Panev, T.I.; et al. Urinary benzene biomarkers and DNA methylation in Bulgarian petrochemical workers: Study findings and comparison of linear and beta regression models. PLoS ONE 2012, 7, e50471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fustinoni, S.; Rossella, F.; Polledri, E.; Bollati, V.; Campo, L.; Byun, H.M.; Agnello, L.; Consonni, D.; Pesatori, A.C.; Baccarelli, A.; et al. Global DNA methylation and low-level exposure to benzene. Med. Lav. 2012, 103, 84–95. [Google Scholar]
- Forrest, M.S.; Lan, Q.; Hubbard, A.E.; Zhang, L.; Vermeulen, R.; Zhao, X.; Li, G.; Wu, Y.Y.; Shen, M.; Yin, S.; et al. Discovery of novel biomarkers by microarray analysis of peripheral blood mononuclear cell gene expression in benzene-exposed workers. Environ. Health Perspect. 2005, 113, 801–807. [Google Scholar] [CrossRef] [PubMed]
- McHale, C.M.; Zhang, L.; Lan, Q.; Li, G.; Hubbard, A.E.; Forrest, M.S.; Vermeulen, R.; Chen, J.; Shen, M.; Rappaport, S.M.; et al. Changes in the peripheral blood transcriptome associated with occupational benzene exposure identified by cross-comparison on two microarray platforms. Genomics 2009, 93, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHale, C.M.; Zhang, L.; Lan, Q.; Vermeulen, R.; Li, G.; Hubbard, A.E.; Porter, K.E.; Thomas, R.; Portier, C.J.; Shen, M.; et al. Global gene expression profiling of a population exposed to a range of benzene levels. Environ. Health Perspect. 2011, 119, 628–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffman, C.; McHale, C.M. Identification of gene expression predictors of occupational benzene exposure. PLoS ONE 2018, 13, e0205427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Bai, W.; Niu, P.; Tian, L.; Gao, A. Aberrant hypomethylated STAT3 was identified as a biomarker of chronic benzene poisoning through integrating DNA methylation and mRNA expression data. Exp. Mol. Pathol. 2014, 96, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Xing, C.; Wang, Q.F.; Li, B.; Tian, H.; Ni, Y.; Yin, S.; Li, G. Methylation and expression analysis of tumor suppressor genes p15 and p16 in benzene poisoning. Chem. Biol. Interact. 2010, 184, 306–309. [Google Scholar] [CrossRef]
- Philbrook, N.A.; Winn, L.M. Investigating the effects of in utero benzene exposure on epigenetic modifications in maternal and fetal CD-1 mice. Toxicol. Appl. Pharmacol. 2015, 289, 12–19. [Google Scholar] [CrossRef]
- Gao, A.; Zuo, X.; Song, S.; Guo, W.; Tian, L. Epigenetic modification involved in benzene-induced apoptosis through regulating apoptosis-related genes expression. Cell Biol. Int. 2011, 35, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zuo, X.; Bai, W.; Niu, P.; Tian, L.; Gao, A. PTEN methylation involved in benzene-induced hematotoxicity. Exp. Mol. Pathol. 2014, 96, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.; Zuo, X.; Liu, Q.; Lu, X.; Guo, W.; Tian, L. Methylation of PARP-1 promoter involved in the regulation of benzene-induced decrease of PARP-1 mRNA expression. Toxicol. Lett. 2010, 195, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Heffler, E.; Allegra, A.; Pioggia, G.; Picardi, G.; Musolino, C.; Gangemi, S. MicroRNA Profiling in Asthma: Potential Biomarkers and Therapeutic Targets. Am. J. Respir. Cell Mol. Biol. 2017, 57, 642–650. [Google Scholar] [CrossRef]
- Bai, W.; Chen, Y.; Yang, J.; Niu, P.; Tian, L.; Gao, A. Aberrant miRNA profiles associated with chronic benzene poisoning. Exp. Mol. Pathol. 2014, 96, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Herberth, G.; Bauer, M.; Gasch, M.; Hinz, D.; Order, S.; Olek, S.; Kohajda, T.; Rolle-Kampczyk, U.; von Bergen, M.; Sack, U.; et al. Maternal and cord blood miR-223 expression associates with prenatal tobacco smoke exposure and low regulatory T-cell numbers. J. Allergy Clin. Immunol. 2014, 133, 543–550. [Google Scholar] [CrossRef]
- Wei, H.; Zhang, J.; Tan, K.; Sun, R.; Yin, L.; Pu, Y. Benzene-Induced Aberrant miRNA Expression Profile in Hematopoietic Progenitor Cells in C57BL/6 Mice. Int. J. Mol. Sci. 2015, 16, 27058–27071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, W.; Yang, J.; Yang, G.; Niu, P.; Tian, L.; Gao, A. Long non-coding RNA NR_045623 and NR_028291 involved in benzene hematotoxicity in occupationally benzene-exposed workers. Exp. Mol. Pathol. 2014, 96, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.Y.; Wade, P.A. Cancer biology and NuRD: A multifaceted chromatin remodelling complex. Nat. Rev. 2011, 11, 588–596. [Google Scholar] [CrossRef]
- Cameron, E.E.; Bachman, K.E.; Myohanen, S.; Herman, J.G.; Baylin, S.B. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar] [CrossRef]
- Foulks, J.M.; Parnell, K.M.; Nix, R.N.; Chau, S.; Swierczek, K.; Saunders, M.; Wright, K.; Hendrickson, T.F.; Ho, K.K.; McCullar, M.V.; et al. Epigenetic drug discovery: Targeting DNA methyltransferases. J. Biomol. Screen. 2012, 17, 2–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahindra, A.; Laubach, J.; Raje, N.; Munshi, N.; Richardson, P.G.; Anderson, K. Latest advances and current challenges in the treatment of multiple myeloma. Nat. Rev. 2012, 9, 135–143. [Google Scholar] [CrossRef]
- Wyhs, N.; Walker, D.; Giovinazzo, H.; Yegnasubramanian, S.; Nelson, W.G. Time-Resolved Fluorescence Resonance Energy Transfer Assay for Discovery of Small-Molecule Inhibitors of Methyl-CpG Binding Domain Protein 2. J. Biomol. Screen. 2014, 19, 1060–1069. [Google Scholar] [CrossRef] [Green Version]
- Musolino, C.; Sant’antonio, E.; Penna, G.; Alonci, A.; Russo, S.; Granata, A.; Allegra, A. Epigenetic therapy in myelodysplastic syndromes. Eur. J. Haematol. 2010, 84, 463–473. [Google Scholar] [CrossRef]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [CrossRef]
- Sanchez, R.; Zhou, M.M. The role of human bromodomains in chromatin biology and gene transcription. Curr. Opin. Drug Discov. Dev. 2009, 12, 659–665. [Google Scholar]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [Green Version]
- Vidler, L.R.; Brown, N.; Knapp, S.; Hoelder, S. Druggability Analysis and Structural Classification of Bromodomain Acetyl-lysine Binding Sites. J. Med. Chem. 2012, 55, 7346–7359. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [Green Version]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef]
- Zuber, J.; Shi, J.W.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroder, S.; Cho, S.; Zeng, L.; Zhang, Q.; Kaehlcke, K.; Mak, L.; Lau, J.; Bisgrove, D.; Schnolzer, M.; Verdin, E.; et al. Two-pronged binding with bromodomain-containing protein 4 liberates positive transcription elongation factor b from inactive ribonucleoprotein complexes. J. Biol. Chem. 2012, 287, 1090–1099. [Google Scholar] [CrossRef] [Green Version]
- Hewings, D.S.; Fedorov, O.; Filippakopoulos, P.; Martin, S.; Picaud, S.; Tumber, A.; Wells, C.; Olcina, M.M.; Freeman, K.; Gill, A.; et al. Optimization of 3,5-Dimethylisoxazole Derivatives as Potent Bromodomain Ligands. J. Med. Chem. 2013, 56, 3217–3227. [Google Scholar] [CrossRef]
- Zhang, G.; Plotnikov, A.N.; Rusinova, E.; Shen, T.; Morohashi, K.; Joshua, J.; Zeng, L.; Mujtaba, S.; Ohlmeyer, M.; Zhou, M.M. Structure-guided design of potent diazobenzene inhibitors for the BET bromodomains. J. Med. Chem. 2013, 56, 9251–9264. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.R.; Lai, I.L.; Yang, H.C.; Lin, C.N.; Bai, L.Y. Identification of kazinol Q, a natural product from Formosan plants, as an inhibitor of DNA methyltransferase. Phytother. Res. 2014, 28, 49–54. [Google Scholar] [CrossRef]
- Fenga, C.; Gangemi, S.; Costa, C. Benzene exposure is associated with epigenetic changes (Review). Mol. Med. Rep. 2016, 13, 3401–3405. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Disease | Status | Target | Type of Study | Ref. |
|---|---|---|---|---|
| AML | Hypomethylation | MAGE-1 | In vitro | [122,123] |
| AML (HLA60 cell line) | Hypermethylation | H3K4mc3 | In vitro | [138] |
| ALL (TK6 cells lymphoblastoid cells) | Hypomethylation | DNA | In vitro | [140] |
| ALL (Children) | Hypermethylation | MLL-r | Ex vivo | [132] |
| Experimental model | Target | Type of study | Ref. | |
| Male C76B/6 mice | Leukemia stem cell quiescence and self renewal genes | In vivo | [135] | |
| Exposed subjects | Hypomethylation | p15, MAGE-1, Line-1 | Ex vivo | [136] |
| Exposed subjects | Hypomethylation | P15INK4b | Ex vivo | [157] |
| Exposed subjects | JUN, PF4, CXCL16, ZNF331 | Ex vivo | [158] | |
| Exposed subjects | Hypomethylation Hypomethylation | PRKG1, PARD3, EPHAS. STAT3, IFNGR1 | Ex vivo | [162] |
| Exposed subjects | Hypermethylation | p15, p16 | Ex vivo | [163] |
| Bone marrow rat cells, F32 lymphoblast cells | Hypermethylation | PTEN | In vitro and in vivo | [166] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spatari, G.; Allegra, A.; Carrieri, M.; Pioggia, G.; Gangemi, S. Epigenetic Effects of Benzene in Hematologic Neoplasms: The Altered Gene Expression. Cancers 2021, 13, 2392. https://doi.org/10.3390/cancers13102392
Spatari G, Allegra A, Carrieri M, Pioggia G, Gangemi S. Epigenetic Effects of Benzene in Hematologic Neoplasms: The Altered Gene Expression. Cancers. 2021; 13(10):2392. https://doi.org/10.3390/cancers13102392
Chicago/Turabian StyleSpatari, Giovanna, Alessandro Allegra, Mariella Carrieri, Giovanni Pioggia, and Sebastiano Gangemi. 2021. "Epigenetic Effects of Benzene in Hematologic Neoplasms: The Altered Gene Expression" Cancers 13, no. 10: 2392. https://doi.org/10.3390/cancers13102392