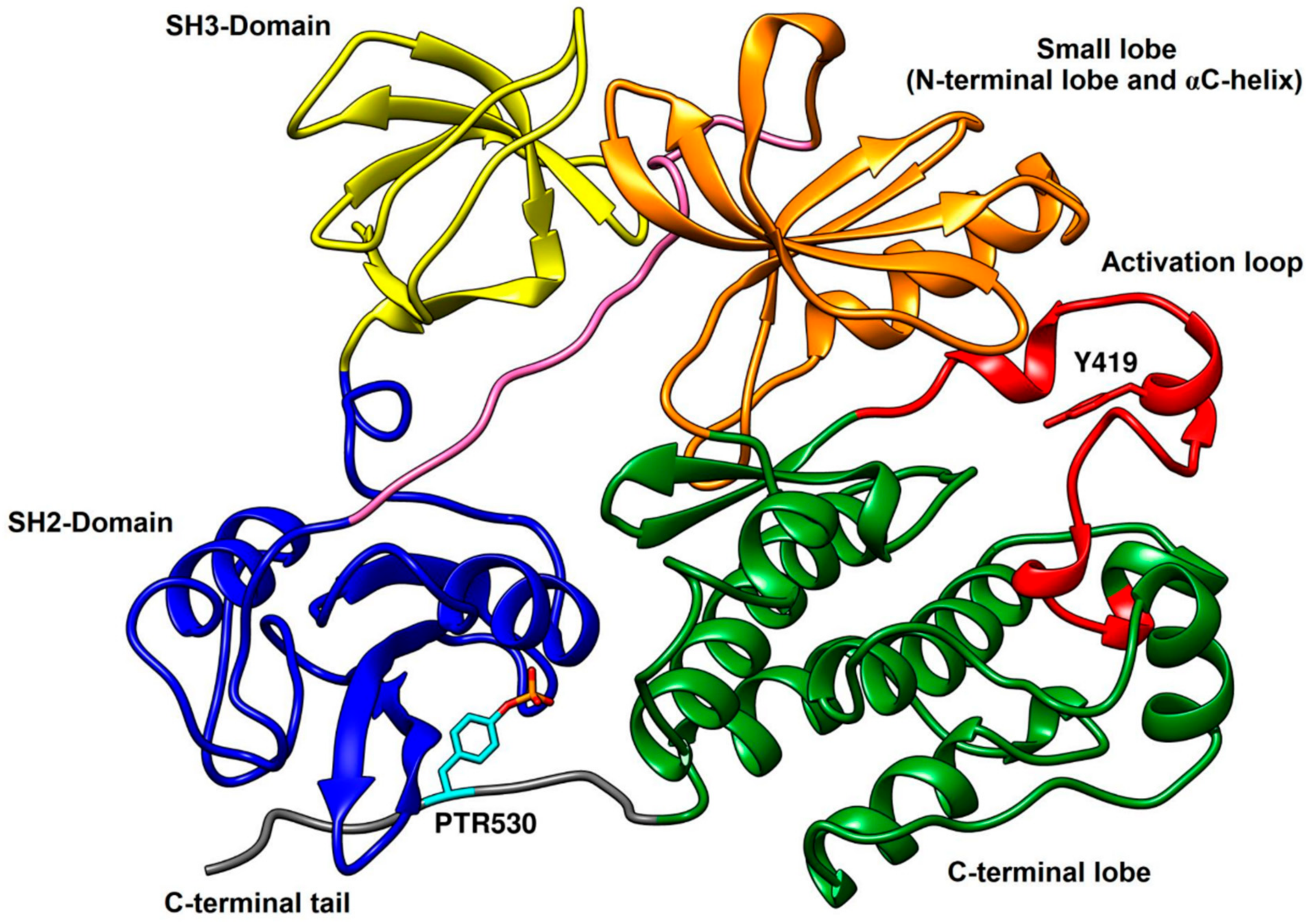
2.3. Type-I Inhibitors
The development of type-I inhibitors has led to successful results, since some outstanding candidates from type-I category were advanced to clinical trials and few of them, such as dasatinib, bosutinib and ruxolitinib, have been accepted as actual drugs. These compounds and other type-I inhibitors have been co-crystallized with c-Src (
Table S2) to understand the structural bases of their affinity for the target.
The first attempt to develop selective type-I inhibitors, which could competitively occupy the ATP-binding site of c-Src kinase was made by Lombardo and coworkers in 2004 [
38]. The Lck kinase activity of 2-acylamino-5-carboxamidothiazoles had previously been reported by the same research group [
16]. Based on the considerable results on Lck kinase activity, carboxamidothiazoles were rationally designed and substituted 2-(aminopyridyl) and 2-(aminopyrimidinyl) thiazole-5-carboxamides were synthesized and tested for c-Src inhibition properties. The first compound of the series, which is a 2-aminopyrimidine derivative, impressively displayed biochemical potency with a
Ki of 96 pM for c-Src and an IC
50 below 1.0 nM for Bcr-Abl kinase [
39]. In general, both pyridine-based and pyrimidine-based analogues of the series demonstrated nanomolar to sub-nanomolar inhibitory potency against c-Src and Bcr-Abl kinase. The two 2-amino-thiazole pyrimidine derivatives with hydroxymethyl piperidine and hydroxyethyl piperazine tail at the fourth position of the pyrimidine ring were found to show the best balance in terms of broad spectrum antiproliferative activity against K562, PC3, MDA-MB-231 human breast tumor and WiDr human colon tumor cell lines. Both derivatives showed low-to-mid nanomolar inhibition potency against all cell lines and one of the two derivatives (compound
1) was subjected to in vivo pharmacological evaluation through K562 xenograft mouse model. Compound
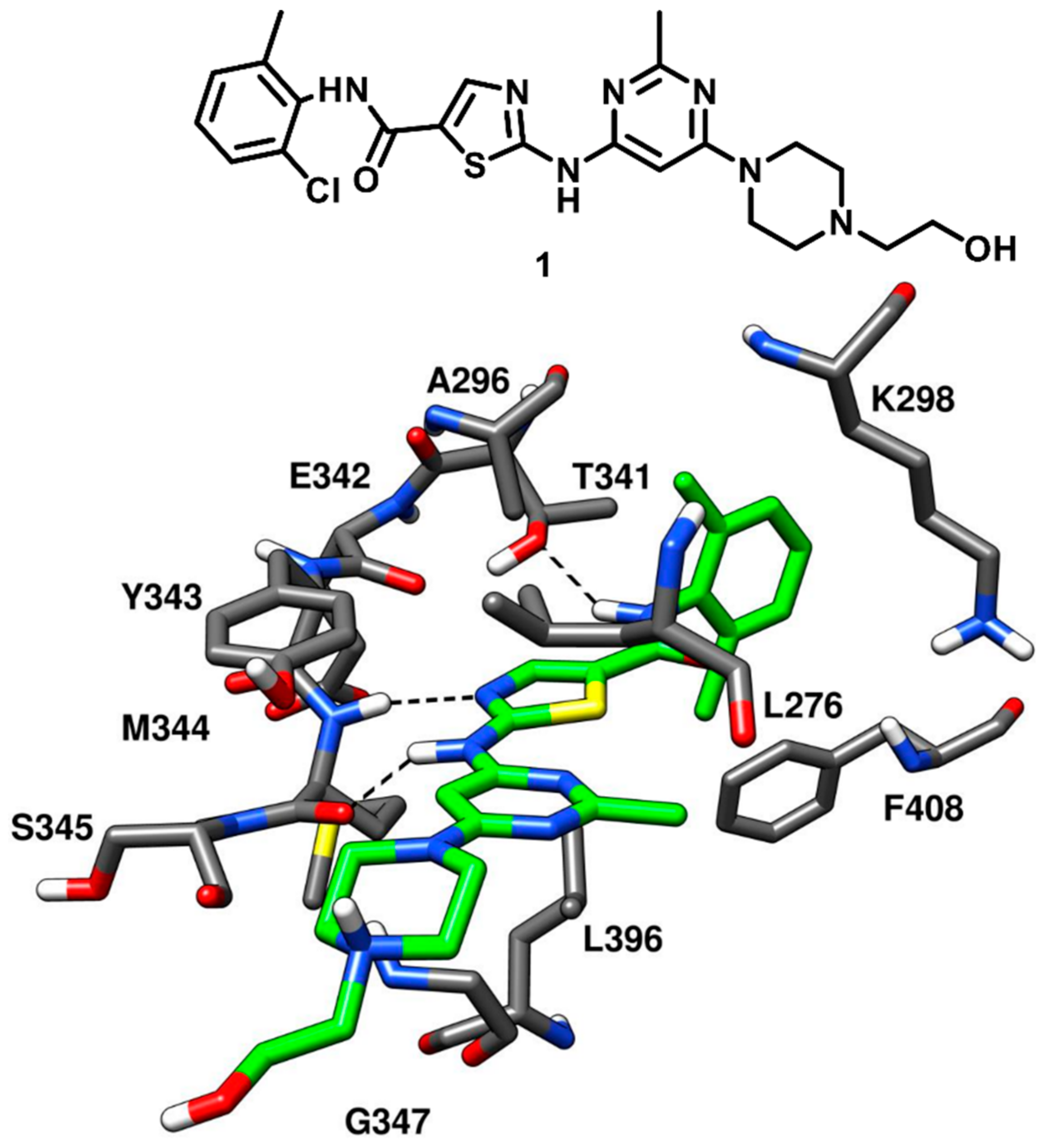
1 (BMS-354825, later known as dasatinib) which demonstrated an impressive IC
50 of 0.50 nM for c-Src kinase, was co-crystallized with Abl kinase, revealing the key ligand–protein interactions with the enzyme. Compound
1 was expected to show a binding mode into c-Src comparable to that observed into the X-ray complex with Abl kinase. This was confirmed in 2009, when Getlik and coworkers solved the X-ray structure of active c-Src kinase in complex with compound
1 (PDB ID: 3G5D,
Figure 3). Three H-bond interactions were formed by the ligand in the ATP-binding site of c-Src kinase. In particular, a pair of hydrogen bonds was observed in the highly conserved hinge region: a first one between the central NH group of compound
1 and the backbone oxygen of M344, and a second one between the nitrogen of the ligand thiazole ring and the backbone nitrogen of M344. Another important H-bond was formed between the gatekeeper residue T341 and the amide nitrogen of the inhibitor. As a type-I inhibitor, the ligand was bound to the active DFG-in conformation of the kinase with the 2-chloro-6-methylbenzamide moiety extending into hydrophobic pocket I, lined by the hydrophobic portions of T341 and K298. The orientation of the 2-chloro-6-methylbenzamide moiety provided important data for the structure-based design of new type-II inhibitors, which could bind to the DFG-out conformation and protrude deeper into hydrophobic pocket I (
vide infra). The substituted pyrimidine moiety of compound
1 was located within hydrophobic pocket II, establishing van der Waals contacts with the sidechains of L276, G347 and L396 that account for the binding affinity towards the kinase. The hydroxyethylpiperazine tail of the inhibitor did not make any key contact with the protein residues and was mainly exposed to the solvent. Also, the ribose binding pocket of the catalytic domain was mostly unoccupied [
38]. Compound
1, was orally active in a K562 xenograft model of chronic myelogenous leukemia with high safety margin, later promoted into clinical trials due to its outstanding pharmacokinetic and in vivo profile. Today, compound
1 is an oral drug known as dasatinib and employed in a targeted therapy used for the treatment of chronic myelogenous leukemia (CML) and acute lymphoblastic leukemia (ALL) [
40].
Significant efforts were made to exploit new heterocyclic scaffolds once the first aminothiazole-pyrimidine inhibitors of c-Src kinase with subnanomolar affinity were discovered. Cowan-Jacob and coworkers in 2004 employed a well-known antileukemia drug imatinib (STI571; Gleevec), with strong affinity for c-Abl but low inhibitory potency against c-Src kinase, as a starting point to develop new c-Src inhibitors [
41]. During the structural optimization of imatinib, it was found that the incorporation of a methyl group in the lead compound from which imatinib was derived, reduced its inhibitory activity against c-Src kinase. The loss of activity could be probably due to the steric clash with the hinge-region produced by the methyl group on the central scaffold of the compound. In addition, imatinib showed high affinity for a specific inactive conformation of c-Abl but demonstrated very low affinity for the active conformation of c-Src kinase. Later in 2007, Seeliger and coworkers solved the X-ray structure of c-Src kinase in complex with imatinib [
42] (compound
19, discussed in the next section) to identify the binding mode of the ligand into c-Src and rationalize its weak inhibitory potency against c-Src.
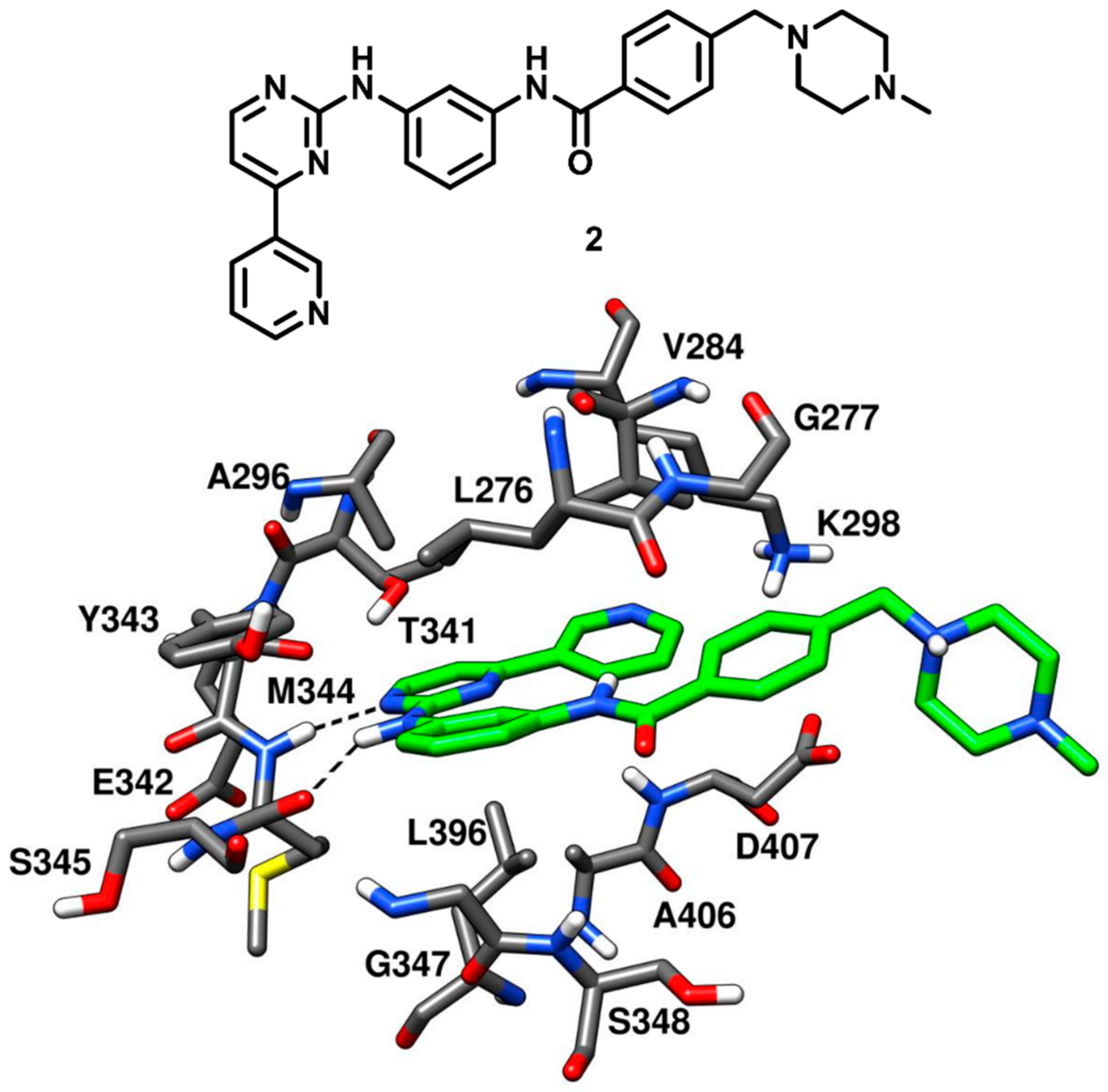
The des-methyl analog of imatinib (compound
2) was tested against c-Src kinase, revealing an IC
50 value of 1.6 nM in kinase inhibition assays. Compound
2 was co-crystallized with c-Src (PDB ID: 1Y57,
Figure 4) that showed a new and partially assembled state in which the kinase domain adopted an active conformation. The X-ray structure was aimed to be utilized for the structure-based design of new c-Src inhibitors. Two of the four aromatic rings of the ligand, i.e., the terminal pyridine ring and the pyrimidinyl moiety, occupied the adenine binding site and were almost coplanar. Compound
2 formed two H-bonds with the backbone of M344 within the hinge-region. One H-bond was formed between the anilino-group of the inhibitor and the backbone oxygen of M344, while the other H-bond was observed between a pyrimidine nitrogen of compound
2 and the backbone nitrogen of M344. This X-ray structure confirmed that the presence of the methyl group in the inhibitor (like for imatinib) would have weakened these H-bonds due to the steric clashes with M344. The pyridine ring of the inhibitor established van der Waals contacts with V284, while the pyrimidine scaffold formed hydrophobic contacts with A296 and L396. The central phenyl moiety of the inhibitor laid on hydrophobic pocket II and established van der Waals interactions with L276 and G347. The phenyl ring of the lateral tail was located near G277 in the ribose pocket, while the terminal piperazine ring was largely exposed to the solvent [
43].
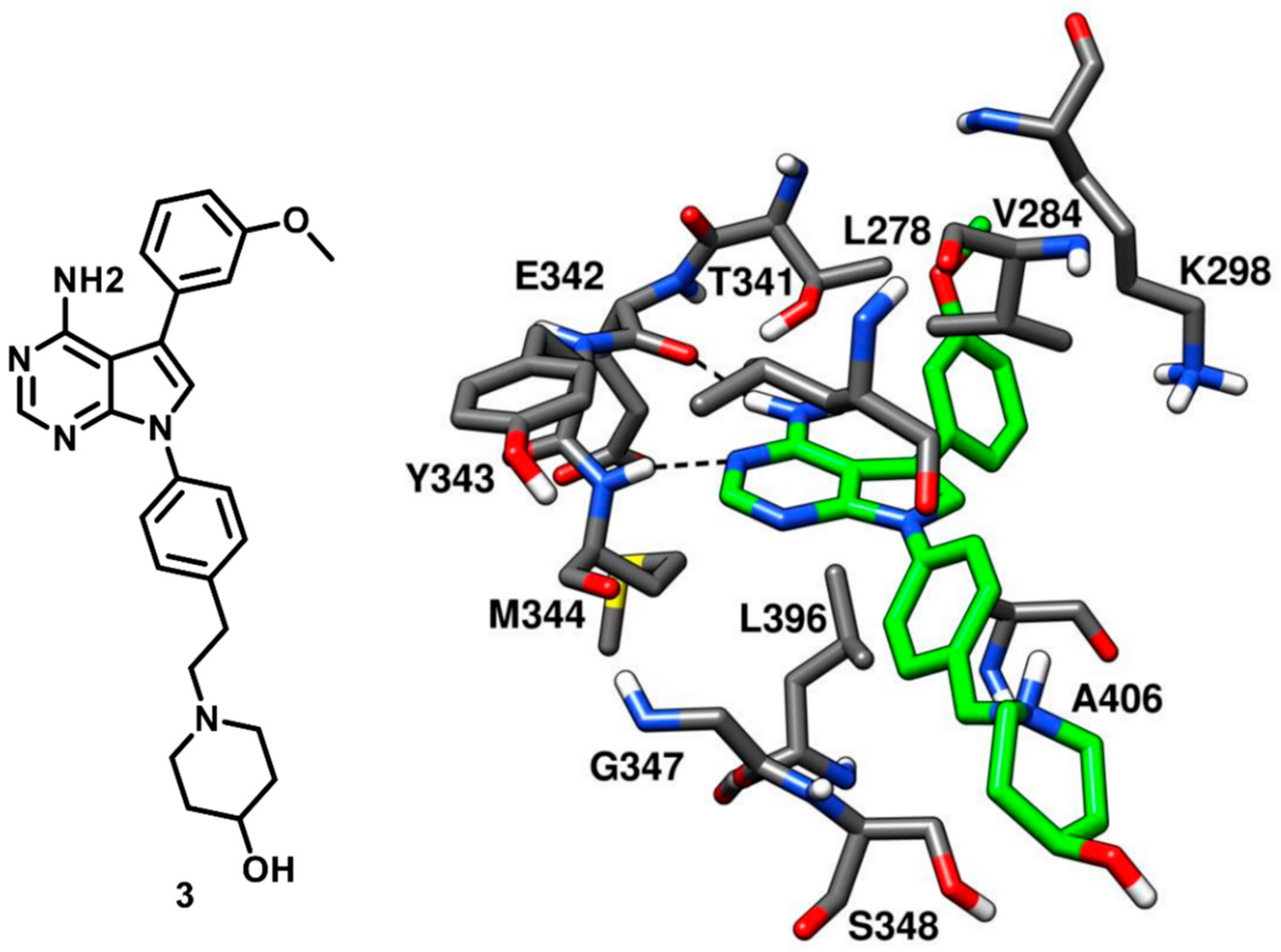
With the ongoing efforts in the development of c-Src kinase inhibitors, Breitenlechneret and coworkers in 2005 crystallized c-Src in active conformation and revealed how the kinase domain, in absence of the rigidifying interdomain interactions of the inactive state, adopts a more open and flexible conformation. The inhibitor CGP77675 (compound
3) was co-crystallized with c-Src kinase (PDB ID: 1YOL,
Figure 5) [
20]. Compound 3 is a pyrrolopyrimidine derivative developed for the treatment of osteoporosis, with IC
50 values for c-Src inhibition between 5 and 20 nM, depending on the substrate used [
44]. The pyrrolopyrimidine moiety bound to the adenine pocket, analogously to the adenine group of ATP, and formed two H-bonds with the hinge region. One H-bond was formed between the pyrimidine nitrogen and the backbone nitrogen of M344 while the other H-bond was formed between the 4-amino group of the pyrrolopyrimidine ring and the backbone oxygen of E342. The inhibitor showed an additional H-bond between the 3-methoxy phenyl ring of the inhibitor and the hydroxyl group of the gatekeeper residue T341, which is believed to contribute to the selectivity for the kinase. Extensive van der Waals contacts with T341 and K298 were also formed by the methoxyphenyl substituent of the ligand, located into hydrophobic pocket I. The second phenyl ring occupied the ribose pocket and was partially exposed to the solvent, forming hydrophobic interactions with V284 and L396. The 4-hydroxypiperidine moiety was distant from the adenine binding pocket and mainly solvent exposed, similarly to the polar tail of compound
2 (
Figure 4).
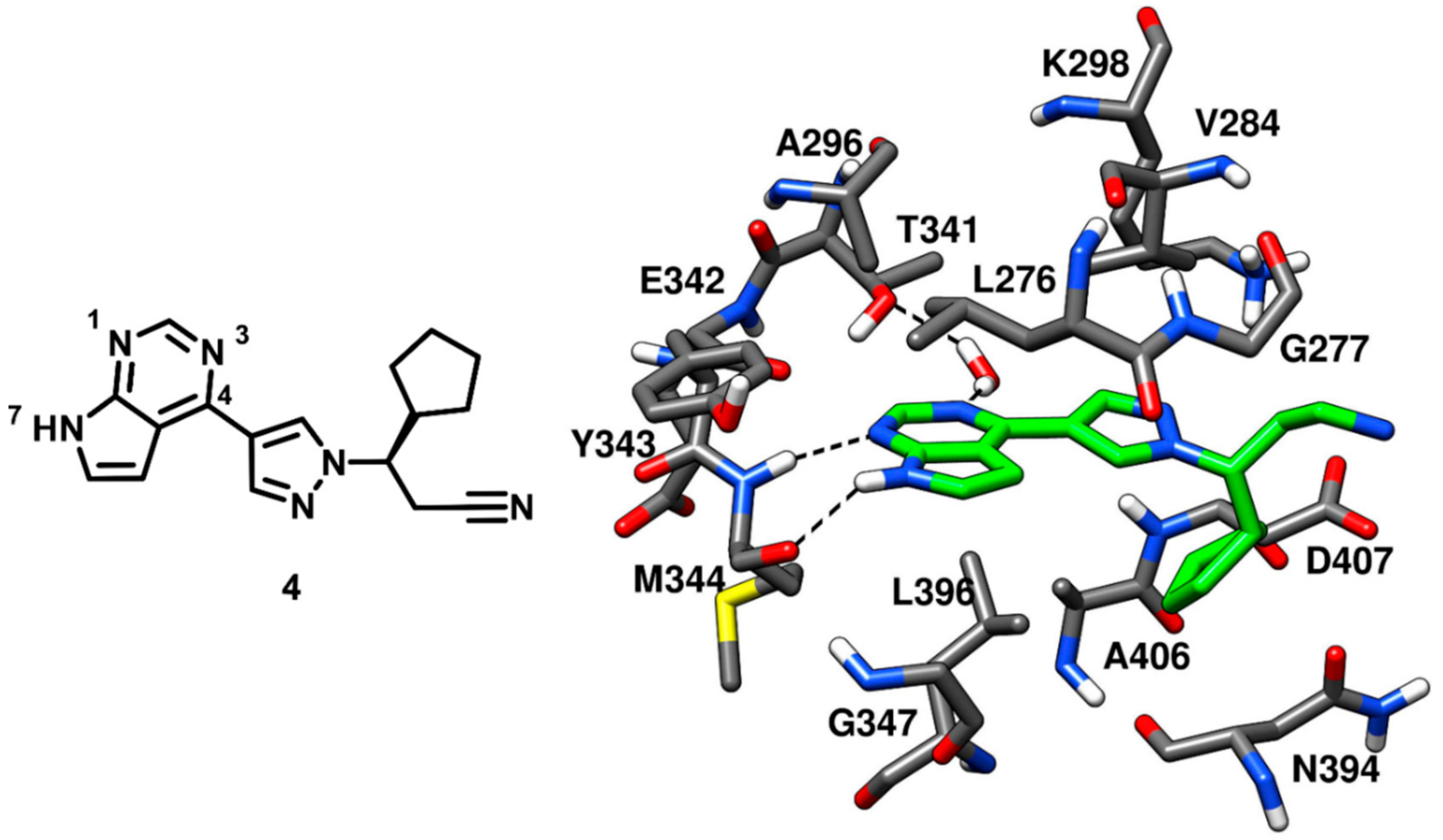
Ruxolitinib is an established anticancer drug, a potent janus kinase inhibitor approved for the treatment of myeloproliferative neoplasms. Ruxolitinib, which is a pyrrolopyrimidine inhibitor, demonstrated IC
50 values of 3.3 nM and 2.8 nM against JAK1 and JAK2, respectively, but a weak inhibitory potency against c-Src kinase (with an IC
50 value of 2.8 µM) [
45,
46]. Although the inhibitor was weakly potent against c-Src, no structural information about the bioactive conformation of ruxolitinib in respect to any kinase, was available. This led Duan and coworkers, in 2014, to further extend the investigation on the pyrrolopyrimidine scaffolds. Ruxolitinib (compound
4) was co-crystallized with c-Src kinase (PDB ID: 4U5J,
Figure 6) to identify its binding mode. The pyrrolopyrimidine ring was placed into the highly conserved ATP binding pocket and displayed two H-bonds within the hinge-region; one H-bond between N-1 and the backbone nitrogen of M344 and another H-bond between N-7 and the backbone oxygen of M344. Moreover, the N-3 of the pyrrolopyrimidine ring formed a water-mediated H-bond with the gatekeeper residue T341. The pyrrolopyrimidine central core demonstrated van der Waals contacts with L276 and A296 of the N-terminal lobe and L396 of the C-terminal lobe. The inhibitor was not found in the vicinity of the ribose pocket, which was left unoccupied. However, the cyclopentyl ring of the inhibitor was lined by N394 and D407, forming van der Waals contacts with these residues, while the pyrazole ring of compound
4 established additional van der Waals interactions with V284 of the N-terminal lobe. Unlike compound
3, compound
4 did not present any groups that could occupy hydrophobic pocket I; thus suggesting that modifications of ruxolitinib aimed at occupying this pocket could lead to the development of more potent and promising c-Src inhibitors [
47].
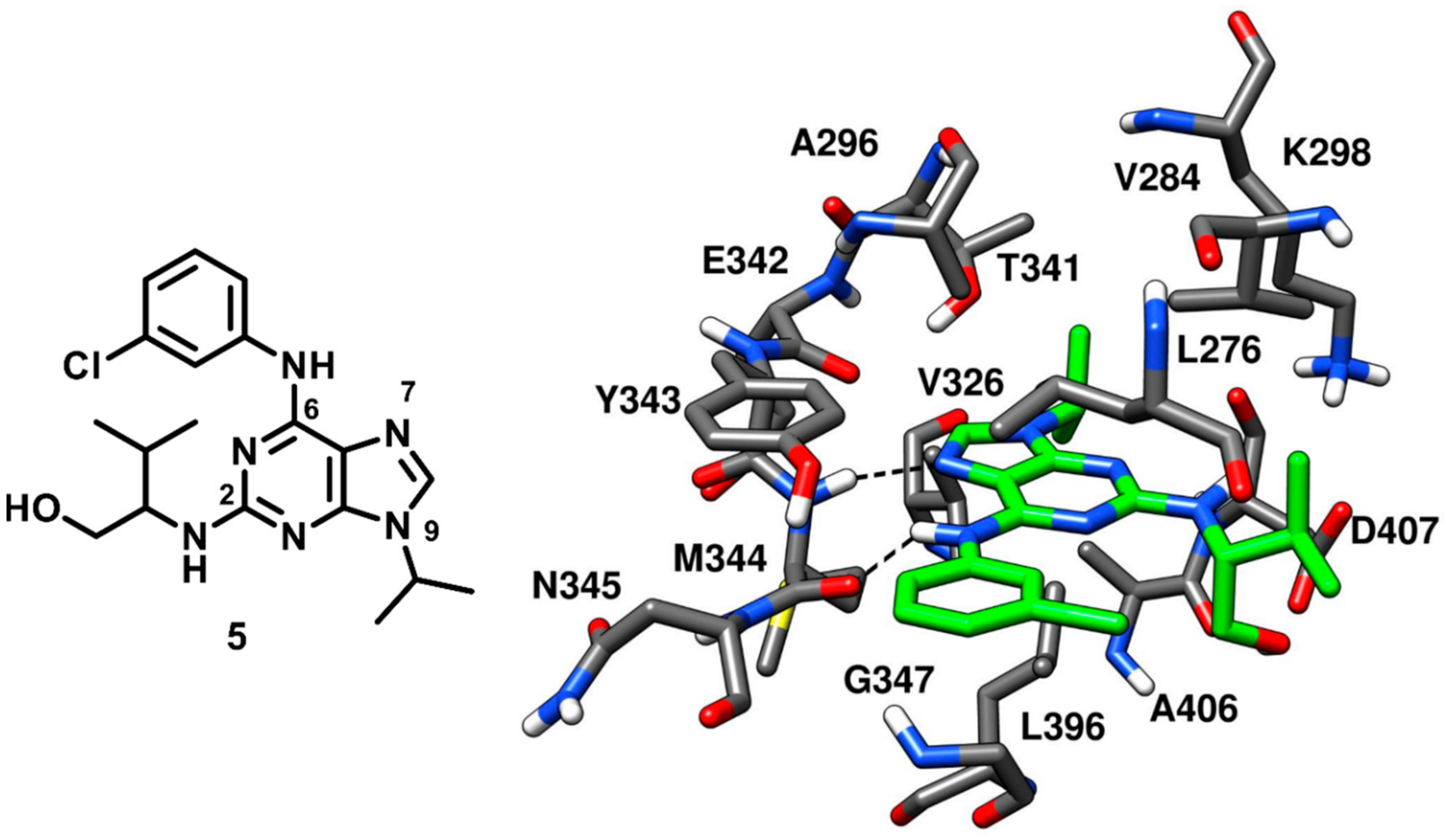
A tri-substituted purine-based inhibitor was investigated by the research group that already identified the pyrrolopyrimidine inhibitor
3. Purvalanol A, reported to cause an arrest of G1 and G2 phase in cancer cell cycle progression in vitro, is a known low nanomolar inhibitor of cyclin-dependent protein kinases (CDKs) that also shows submicromolar affinity (with an IC
50 of 0.24 µM) for c-Src kinase [
48]. Purvalanol A (compound
5) was co-crystallized with c-Src (
Figure 7, PDB ID: 1YOM) and bound to the active kinase conformation. The inhibitor occupied the ATP-binding site of c-Src and established two H-bonds in the hinge-region; one H-bond between N-7 of the purine central core and the backbone nitrogen of M344, while the other H-bond between its 6-anilino group and the backbone oxygen of M344. The central purine ring of the molecule displayed van der Waals contacts with A296 and L396, while the N-9 isopropyl group of the inhibitor was found to interact with V326 of the C-terminal lobe, establishing additional hydrophobic contacts also with L396. The 3-chlorophenyl ring of compound
5 formed lipophilic interactions with L276 and G347 within hydrophobic pocket II, while the 3-methylbutanol chain occupied the ribose pocket of the binding site and showed van der Waals contacts with V284 of the N-terminal lobe [
20]. The low nanomolar activity of compound
5 and its crystallographic structure served as a template for the structure-based design of more potent purine-based analogues which are discussed below.
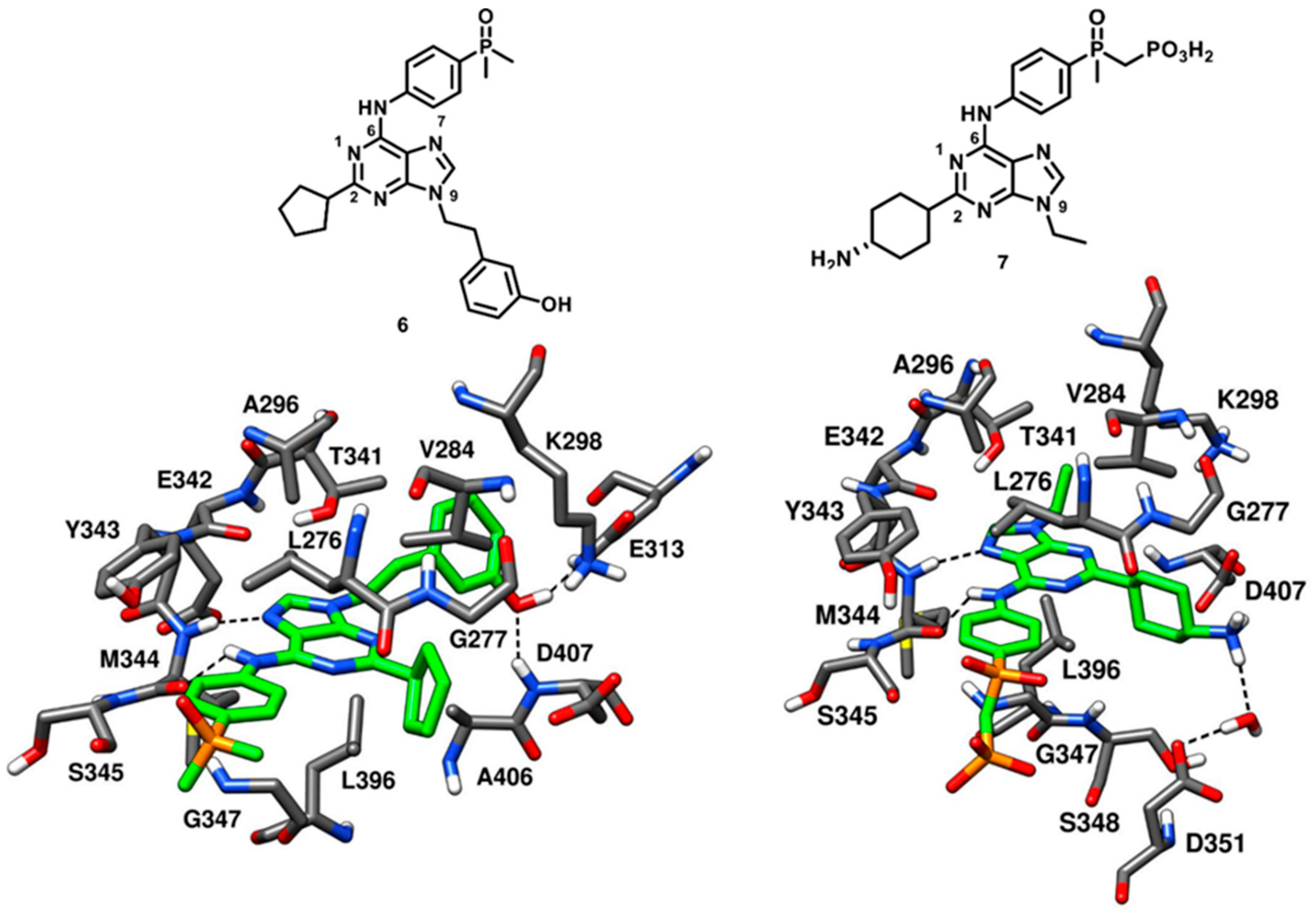
Dalgarno and coworkers in 2006 adopted an approach for exploiting rationally designed compound libraries of 2,6,9-trisubstituted purines to achieve selective inhibition of c-Src kinase. The compound optimization was aided by molecular docking on c-Src X-ray structure, followed by synthesis and kinase inhibition assays. Purvalanol A (compound
5) was used as starting point in the chemical optimization focused on synthetic modifications at the 2-, 6- and 9-positions of the purine nucleus with the aim of obtaining new SAR data [
49]. Two lead compounds AP23464 (compound
6) and AP23451 (compound
7) were identified. Compound
6 displayed an exceptional and enhanced c-Src inhibitory potency, with an IC
50 of 0.45 nM, and a weak CDK2 inhibitory potency, with an IC
50 of 20.9 µM, thus demonstrating more than 46000-fold selectivity for c-Src kinase over CDK2. Compound
7 was designed with the aim of inhibiting c-Src kinase-dependent bone resorption [
50]. For this purpose, a bone-targeting phosphonomethylphosphinyl (PCP) chemical moiety was introduced into compound
7 at the para-position of its aniline ring. The incorporation of the PCP moiety favored the localization of the compound into to the bone, although the cellular permeability of the inhibitor was limited due to the presence of a charged moiety [
51]. The inhibitor showed a remarkable inhibitory activity against c-Src (IC
50 of 0.067 µM) and displayed a 30-fold selectivity over CDK2 (IC
50 = 2.01 µM). Both compounds
6 and
7 were co-crystallized with c-Src kinase (PDB IDs: 2BDJ and 2BDF, respectively,
Figure 8). The inhibitors well occupied the ligand binding site between the N-terminal and C-terminal lobes, interacting with the kinase in active conformation. Though both inhibitors are ATP site binders, the orientation of the tri-substituted purine ring significantly differed from ATP, as observed for Purvalanol A. Both inhibitors established H-bonds with the backbone nitrogen and oxygen of M344 through their N-7 and aniline nitrogen, respectively. The purine nucleus of both inhibitors was sandwiched between A296 and L396, forming van der Waals interactions with these residues. The C-2 substituents of compounds
6 and
7, corresponding to a 4-amino cyclohexyl and a cyclopentyl moiety, respectively, were located at the ribose pocket, contributing to fill the wide cleft between the N-terminal and C-terminal lobes of the kinase and showing van der Waals contacts predominantly with V284 and L396. The introduction of the 3-hydroxyphenethyl substituent into compound
6 allowed the formation of interactions with the binding site, which were considered critical for its picomolar affinity. This unique structural component of compound
6 penetrated deeply into hydrophobic pocket I and formed H-bonds with the carboxylic group of E313 and the backbone nitrogen of D407. Unlike compound
6, compound
7 bore a shorter N-9 ethyl substituent which was projected between A296 and T341, and only showed van der Waals contacts with the two residues. The shorter N-9 substituent of compound
7 lacked the ability to deeply occupy hydrophobic pocket I and thus could not form H-bonds with E313 and D407. Nevertheless, the terminal amino group of the cyclohexyl moiety of compound
7 showed a water-mediated H-bond interaction with the carboxylic group of D351. Both compounds
6 and
7 were characterized by the presence of an aniline ring at C-6 position, endowed with a para-substituted phosphine oxide and a phosphonate group, respectively. The anilino-moiety of both inhibitors established van der Waals contacts mainly with L276 and G347 in the hydrophobic pocket II. However, the terminal dimethyl phosphine oxide (compound
6) and phosphonate group (compound
7) did not make any contact with the catalytic domain and were largely exposed to solvent [
50]. The identification of compounds
6 and
7 was regarded as a breakthrough in the development of c-Src kinase inhibitors, especially in relation to compound
6, which is one of the most potent reversible inhibitors reported so far.
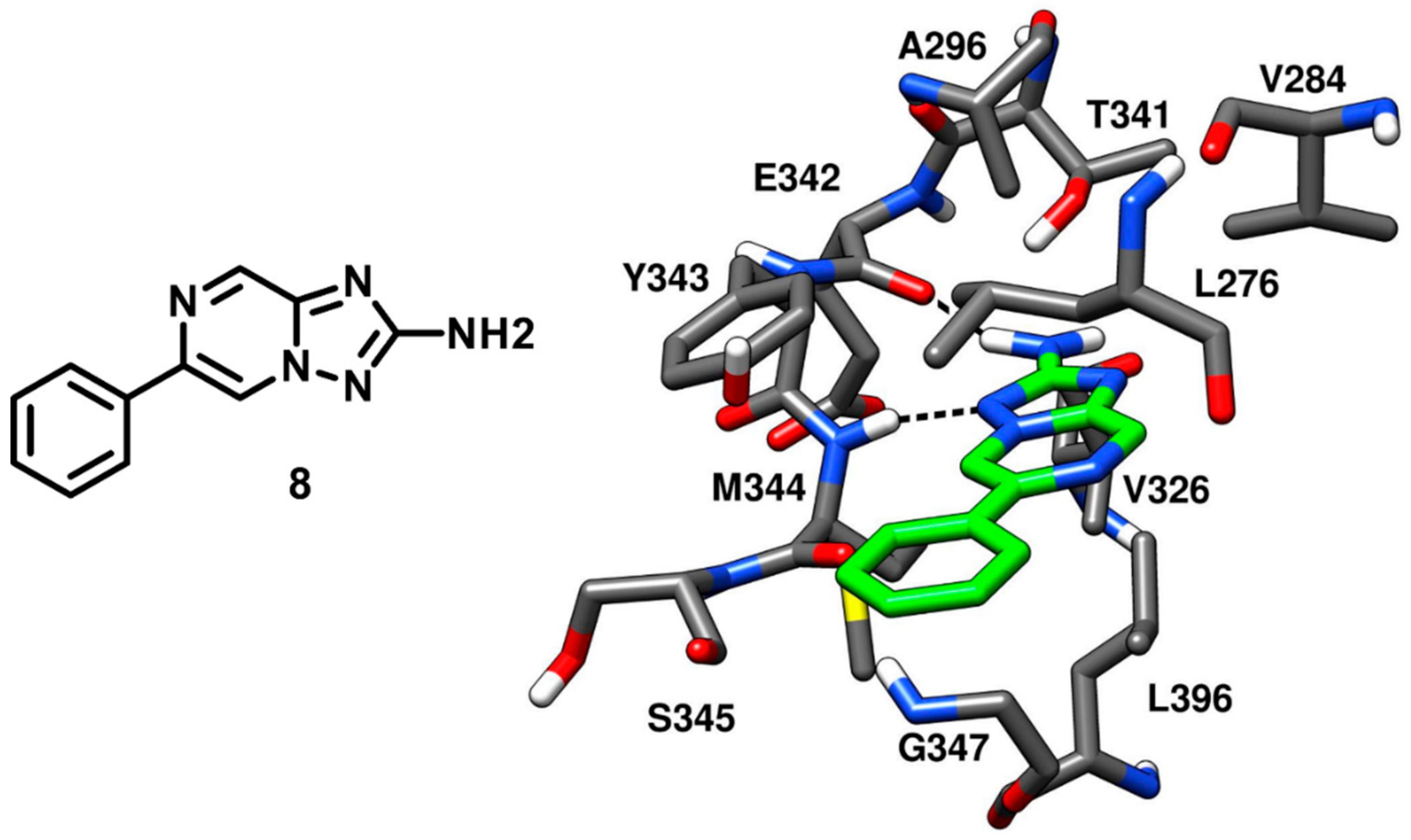
Robert and coworkers in 2013 performed an extensive structure-based de novo design of protein kinase inhibitors. A virtual screening (VS) protocol was implemented with the following steps: (a) core fragment extraction out of commercially available compounds, (b) selection of candidate core fragments by filtering out unwanted functionalities, (c) 3D-pharmacophore search to screen fragments with hinge-region binding moieties, (d) docking of core fragments into X-ray structures of several different kinases and (e) fragment expansion by synthesis. At the end of the VS study, six new compounds were obtained and tested against a panel of different kinases, showing c-Src inhibitory activity in the micromolar range. One of the best inhibitors of the series (compound
8), containing a triazolopyrazine core, demonstrated an IC
50 of 20 µM against c-Src and was co-crystallized with the kinase (PDB ID: 4FIC,
Figure 9) to validate the proposed binding mode [
52]. The 2-amino group of the bicyclic triazolopyrazine ring formed an H-bond with the backbone oxygen of E342, while the adjacent nitrogen of the triazole established another H-bond with the backbone nitrogen of M344. These two H-bond interactions well mimicked those displayed by ATP in the hinge-region. Moreover, the phenyl substituent of compound
8 formed extensive van der Waals contacts with L276 and G347 within hydrophobic pocket II that certainly contributed to its binding affinity for the target. However, compound
8 lacked a group that could occupy hydrophobic pocket I like other type-I inhibitors, which justified its micromolar potency. Although the inhibitor being weakly potent, it satisfied the Lipinski rule of 5 [
53] and the triazolopyrazine ring was identified as a new scaffold for optimization studies where hydrophobic groups could be incorporated for the discovery of new c-Src inhibitors with improved selectivity and affinity.
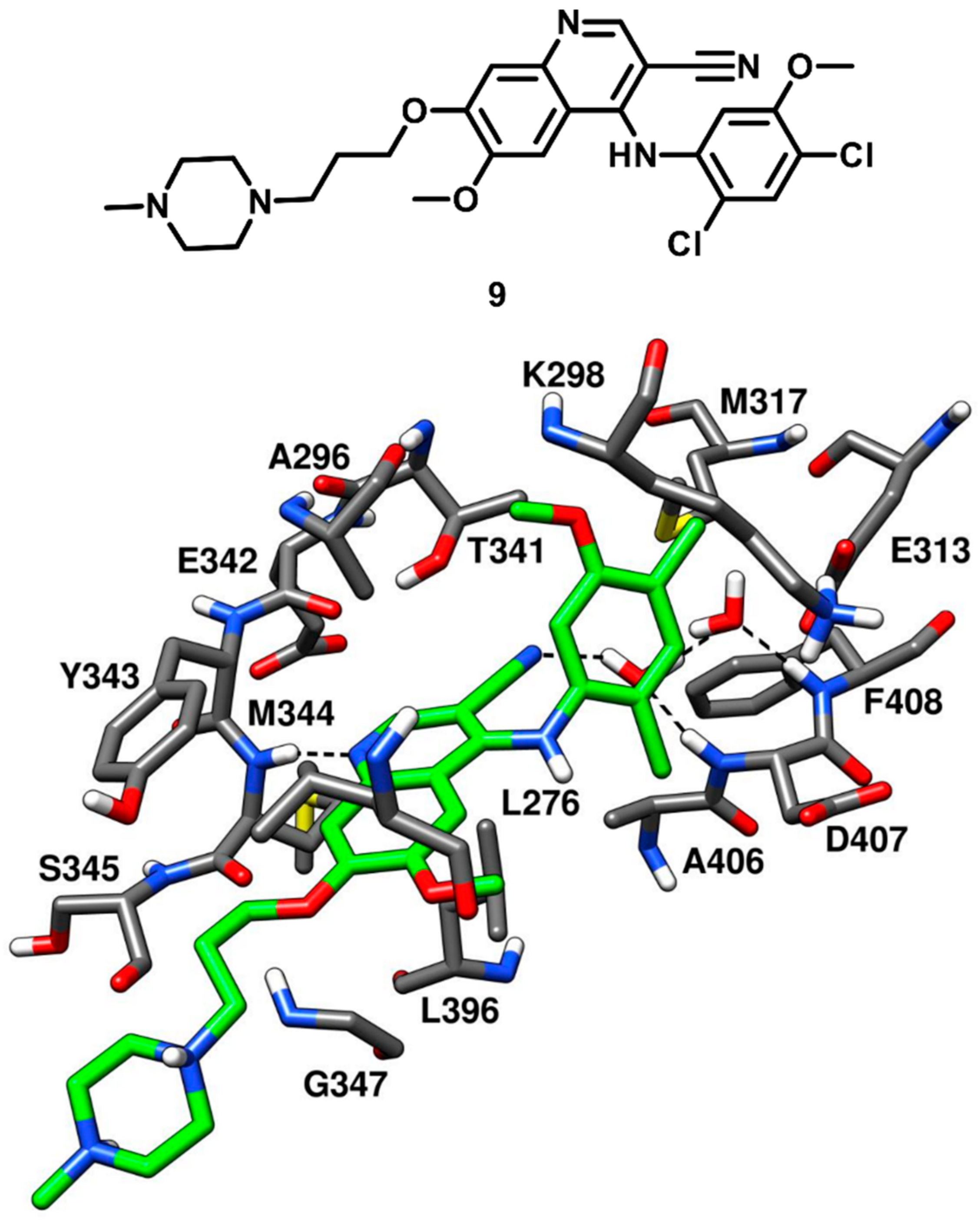
The benzopyridine inhibitor bosutinib (SKI-606) developed by Pfizer, was FDA-approved in 2017. Bosutinib is a Abl and c-Src tyrosine kinase inhibitor, used for the treatment of chronic myelogenous leukemia and marketed under the trade name Bosulif [
54]. In a cell-free assay, bosutinib demonstrated an IC
50 of 1.2 nM against c-Src kinase and was found to be selective for c-Src over non-Src family kinases. The compound potently inhibited Src-dependent cell proliferation in rat fibroblasts with an IC
50 of 0.1 µM. Back in 2014, Levinson and Boxer co-crystallized bosutinib (compound
9) with c-Src kinase (PDB ID: 4MXO,
Figure 10) for getting deeper insights on its selectivity for c-Src over other kinases [
55]. The same research group, in 2012, co-crystallized bosutinib with Abl kinase and revealed that the inhibitor bound to a DFG-out conformation of the kinase (PDB ID: 3UE4) [
56]. Similar results were expected from the X-ray complex of c-Src, but the kinase demonstrated a DFG-in active conformation when bound to the inhibitor. The benzopyridine core of compound
9 was encircled by the side chains of A296 and L276 of the N-terminal lobe and L396 of the C-terminal lobe, thus forming hydrophobic interactions with these residues. A classical H-bond interaction was observed in the ATP binding site, between the pyridine nitrogen of compound
9 and the backbone nitrogen of M344. Additional van der Waals interactions were established by the propyloxy-linker of compound
9 with G347 and L276 in the hydrophobic pocket II, while the piperazine tail was typically exposed to the solvent, similarly to other inhibitors. The ribose pocket of the binding site remained unoccupied, while hydrophobic pocket I was occupied by the dichloromethoxyphenyl ring of the inhibitor. The gatekeeper residue T341 of c-Src formed van der Waals contacts with the 3-nitrile group and the dichloromethoxyphenyl ring of the ligand, which was also seen in bosutinib-Abl complex. Levinson and Boxer also reported that most of the type I inhibitors left a small cavity next to the gatekeeper residue, adjacent to hydrophobic pocket I, unoccupied. Within this cavity, two structural water molecules are often found, showing a network of H-bonds in which the bound inhibitor often participates. Interestingly, a pair of conserved water molecules mediating H-bond interactions between the ligand and the activation loop of the kinase was identified in the c-Src-bosutinib X-ray complex. In particular, the DFG-in active conformation of the kinase revealed that the backbone NH groups of both D407 and F408 of the DFG motif pointed towards the small cavity adjacent to hydrophobic pocket I, anchoring two ordered water molecules. The 3-nitrile group of the inhibitor was oriented towards the same cavity and formed an H-bond with one of the two water molecules in the binding site, thus participating in a water-mediated H-bond network with D407, F408 and E313. Bosutinib-Abl kinase complex demonstrated that the structured-water molecules were oriented differently in the cavity, since the kinase adopted a DGF-out conformation where F408 was flipped outwards. These water-mediated interactions highlighted a crucial element for the inhibitor’s ability to differentiate between different kinases (e.g., c-Src and Abl) and suggested that a greater attention to structured water molecules within the kinase active site is required for the design of selective inhibitors.
As discussed in the introduction, the gatekeeper residue in the ATP-binding site is supposed to play a crucial role in the inhibitors’ selectivity profile, being very prone to undergo mutation and replacement by bigger residues like methionine or isoleucine. In fact, patients undergoing anticancer therapies based on kinase inhibitors frequently develop clinical resistance mediated by mutations at this position [
35,
36]. For this reason, bosutinib was co-crystallized with T341M-M317L double mutant (PDB ID: 4MXZ) and A406T single mutant (PDB ID: 4MXX) c-Src with the aim of investigating the role of the water-mediated interactions for bosutinib’s activity in presence of clinically relevant mutated c-Src residues, with respect to wild type c-Src (
Figure S1). Crystallographic observations from T341M/M317L c-Src structure highlighted a slight closure of the N-terminal and C-terminal lobes of the kinase domain, resulting in the rotation of the aniline ring of bosutinib towards the larger and hydrophobic gatekeeper residue. The 4-chloro group of the aniline ring hampered the disposition of the two structural water molecules within the pocket, which led to the loss of the water-mediated H-bonds with the nitrile group of bosutinib. In the A406T c-Src complex, the sidechain of T406 displaced the structural water molecules and consequently formed a direct H-bond with the nitrile group of bosutinib. The dissociation constants of bosutinib for wild-type c-Src and both mutant c-Src were measured using a fluorescence assay. The K
d value of bosutinib for wild type c-Src was found to be 0.73 nM whereas the K
d value obtained for T341M/M317L and A406T c-Src mutants were found to be 30-fold (21.2 nM) and 40-fold (29 nM) lower, respectively. These data confirmed that the mutation at the gatekeeper and A406 residues interferes with the water-mediated H-bond network of bosutinib and results in significant loss of binding affinities.
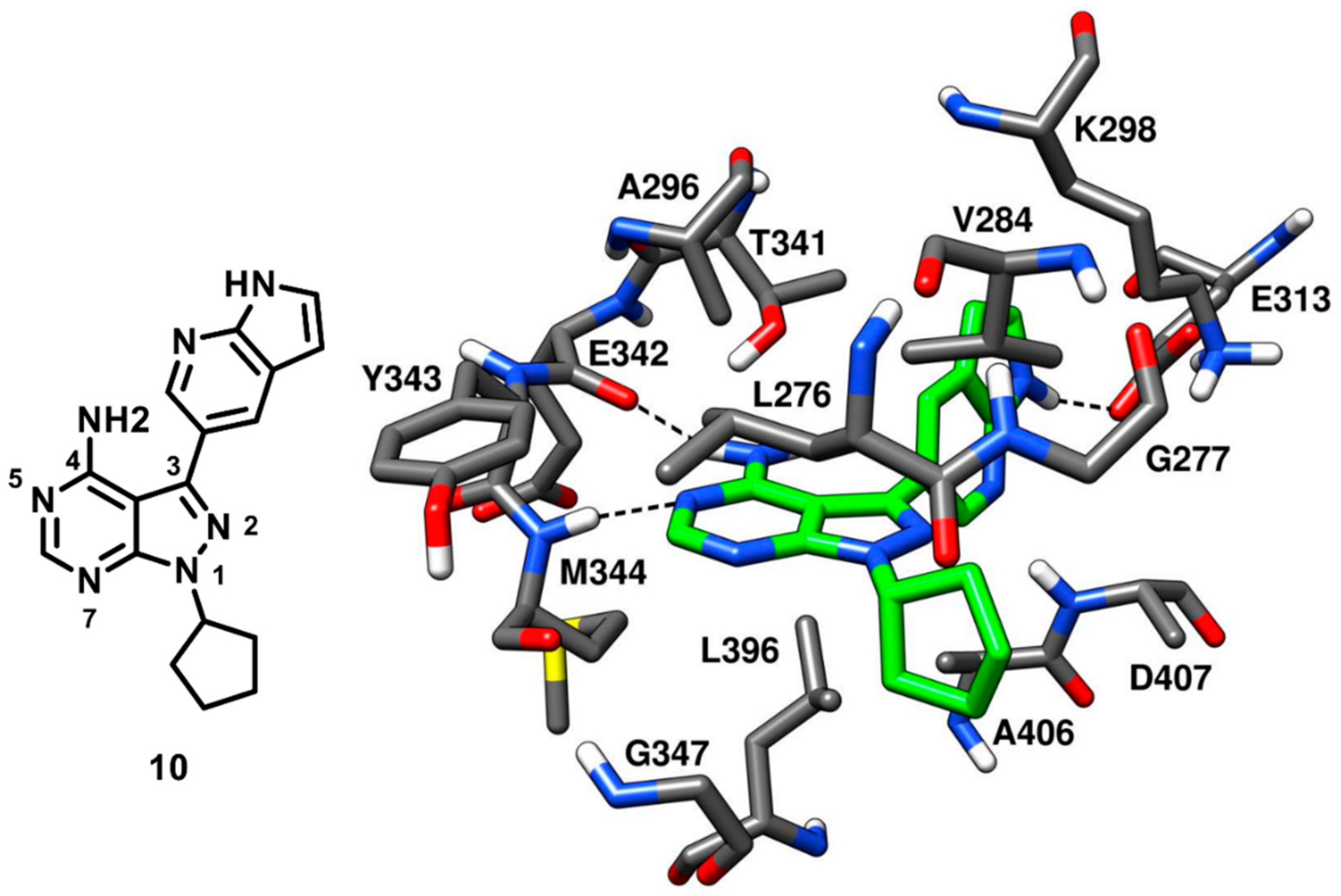
Apsel and coworkers in 2008 carried out an extensive research on the discovery of multitarget kinase inhibitors containing a pyrazolopyrimidine scaffold. An experimental screening of tyrosine kinase inhibitors for activity against the PI3-K p110α resulted in the identification of two pyrazolopyrimidines with nanomolar inhibitory potency that inspired the synthesis of more than 200 derivatives, which were tested against a panel of 14 tyrosine kinases and PI3-Ks. Four new pyrazolopyrimidine inhibitors that displayed interesting inhibition properties for c-Src kinase were identified from robust SAR studies. All four pyrazolopyrimidines were co-crystallized with c-Src [
57], which further revealed that one ligand (compound
10,
Figure 11), which showed an IC
50 of 14 nM against c-Src kinase, was a type-I inhibitor, while the other three were CHO type-II inhibitors (see next section for their description). The central pyrazolopyrimidine core of compound
10 occupied the adenine pocket of the ATP-binding site and formed two typical H-bonds in the hinge-region (PDB ID: 3EN4,
Figure 11). The 4-amino group of the pyrazolopyrimidine ring established an H-bond with the backbone oxygen of E342 while the 5-N of pyrazolopyrimidine ring formed another H-bond with the backbone nitrogen of M344. The C-3 pyrrolopyridine substituent of the inhibitor was projected towards the gatekeeper residue into hydrophobic pocket I, showing hydrophobic interactions with T341 and K298 and a crucial H-bond with the carboxylic group of E313, which is usually implicated in the salt bridge with K298 and in maintaining the αC-helix-in active kinase conformation. This particular H-bond interaction was not observed for other members of the series and is believed to contribute to the nanomolar potency of the inhibitor. The solvent-exposed cyclopentyl substituent of the inhibitor was well accommodated into the ribose pocket of the binding site showing van der Waals contacts with V284. Despite its high inhibitory potency against c-Src, compound
10 was found to have poor selectivity, as it showed a broad-spectrum inhibition profile with nanomolar potency against other kinases, such as Ret (IC
50 < 1 nM), VEGFR2 (IC
50 = 12 nM) and Abl kinase (IC
50 = 18 nM).
Recently, Tintori and coworkers have conducted a research focused on the development of novel c-Src/Abl inhibitors endowed with a pyrazolopyrimidine scaffold [
58,
59,
60,
61]. Several members of this series were found to induce apoptosis and to reduce cell proliferation in various solid tumor cell lines. In particular, compound
11a, characterized by a C-6 methylthio group on the pyrazolopyrimidine scaffold (
Figure S2), demonstrated a promising antiproliferative activity in SH-SY5Y cell cultures of human neuroblastoma [
62]. Compound 11a displayed an appreciable selectivity for c-Src when tested against a panel of tyrosine kinases (EGFR, KDR, Flt-3, IGF-IR, Tek and c-Src) [
63].
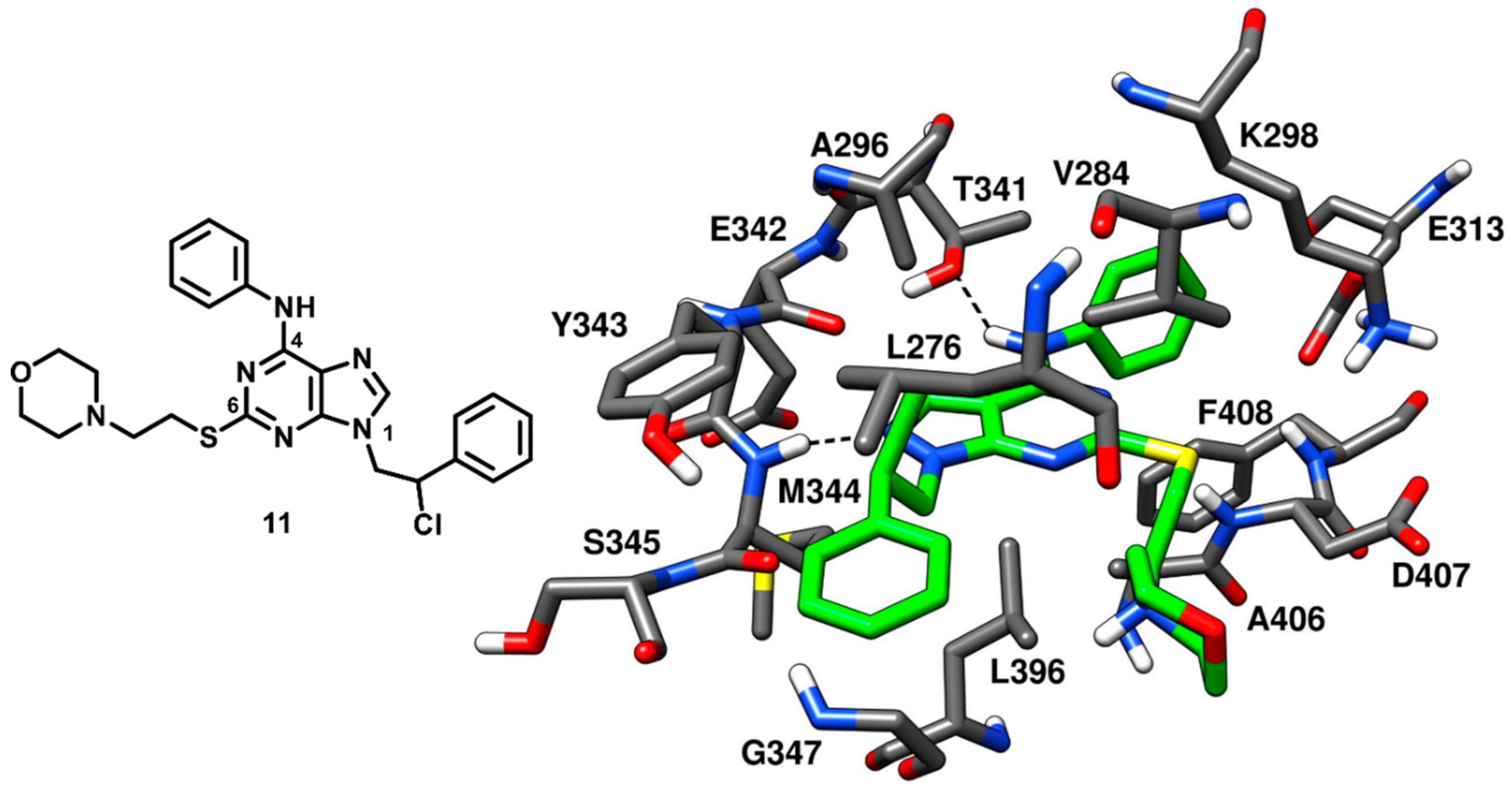
A small library of pyrazolopyrimidine analogues of compound
11a was designed and synthesized, aimed at better exploring N-1, C-4 and C-6 positions of the central scaffold with SAR studies. Molecular modeling suggested that the C-6 substituent of the pyrazolopyrimidine analogue was oriented toward the solvent-exposed region of c-Src binding site and SAR studies highlighted that the introduction of polar groups in C-6 position would be suitable for enhancing the water solubility of the compounds. This study led to the identification of a compound
11 (
Figure 12) that displayed a significantly improved profile in terms of both biological activity and ADME properties. Compound
11 was characterized by a high metabolic stability (95%), good water solubility (1.7 µg/mL) and a potent inhibitory activity against isolated c-Src (Ki = 0.21 µM). Compound
11 was co-crystallized with c-Src kinase (PDB ID: 4O2P,
Figure 12) to get deeper structural insights on the binding mode of this series of compounds and employ the X-ray complex for the structure-based design of more potent analogues. The central pyrazolopyrimidine moiety of the compound exhibited hydrophobic contacts with V284, A296 and L396, while its N-2 nitrogen showed a typical interaction with the backbone nitrogen of M344 located in the hinge region. The amino group of the C-4 aniline substituent of compound
11 formed a second H-bond with the gatekeeper residue T341 and the aromatic ring was placed between T341 and K298 in the hydrophobic pocket I, forming van der Waals interactions with these residues. The flexible ethylthio linker at C-6 position of the scaffold allowed the disposition of the morpholine ring in the ribose pocket, while the terminal phenyl ring of the N1 substituent was sandwiched between L276 and G347, thus occupying hydrophobic pocket II. Finally, the chlorine atom of the ligand was found to be in close proximity with L276 and G347 of hydrophobic pocket II and displayed strong van der Waals contacts with these residues. Compound
11 presented a chiral center and the racemic mixture was used for the preparation of the X-ray crystal structure; notably only the (
R)-enantiomer was able to bind the active conformation of the kinase, despite no differences in biological activity was observed for the two enantiomers [
64].
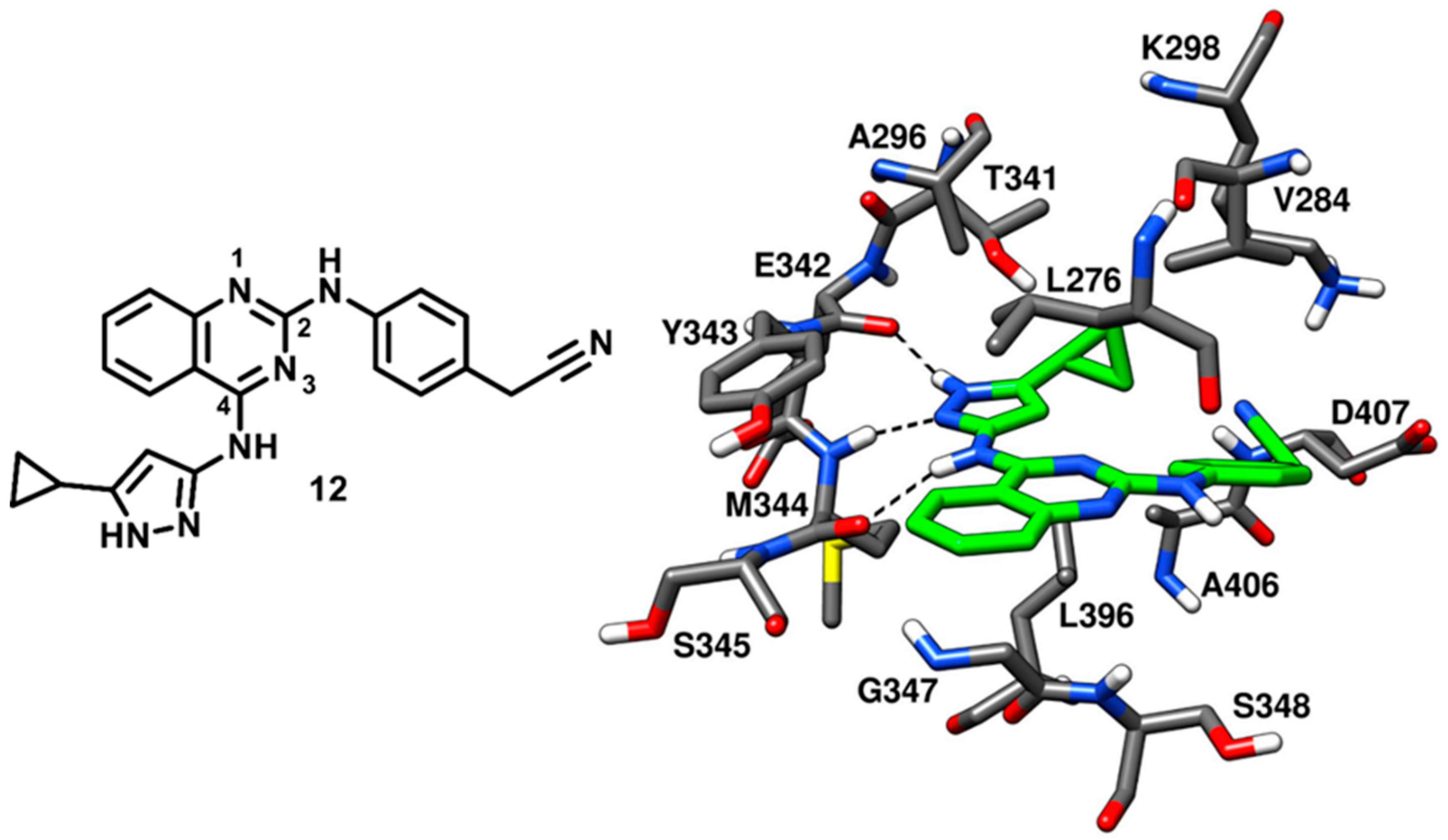
In 2008, Statsuk and coworkers focused on the discovery of multitarget kinase inhibitors and identified a moderately potent quinazoline-based inhibitor (
12) of c-Src, which served as a template in the optimization studies for the development of potent quinazoline-based c-Src ligands in the following years. Compound
12 was screened against a panel of 359 wild type kinases and found to bind most of them. The inhibitor displayed 0.1% residual c-Src kinase activity, as compared to control, in the enzyme assays and emerged as a mid-micromolar potent inhibitor of c-Src (
Kd~500 nM). Compound
12 was co-crystallized with c-Src (PDB ID: 3F6X,
Figure 13), showing to act as a type-I inhibitor, interacting with the active kinase conformation [
65]. The 4-amino pyrazole moiety of the inhibitor was accommodated into the adenine pocket and formed three H-bonds in the hinge-region of the binding site. In particular, the pyrazole ring displayed two H-bonds with the backbone oxygen of E342 and the backbone nitrogen of M344, while the 4-amino group showed an H-bond with the backbone oxygen of M344. Moreover, the 5-cyclopropyl group of the pyrazole ring laid in front of the gatekeeper residue, thus establishing van der Waals contacts with T341 and L396 of the C-terminal lobe. The central quinazoline core of compound
12 mainly showed van der Waals contacts with L276 and G347 in hydrophobic pocket II. The phenyl ring of the cyanobenzyl moiety occupied the ribose pocket and predominantly formed lipophilic interactions with V284, while the nitrile group showed van der Waals contacts with the side chains of D407 and K298.
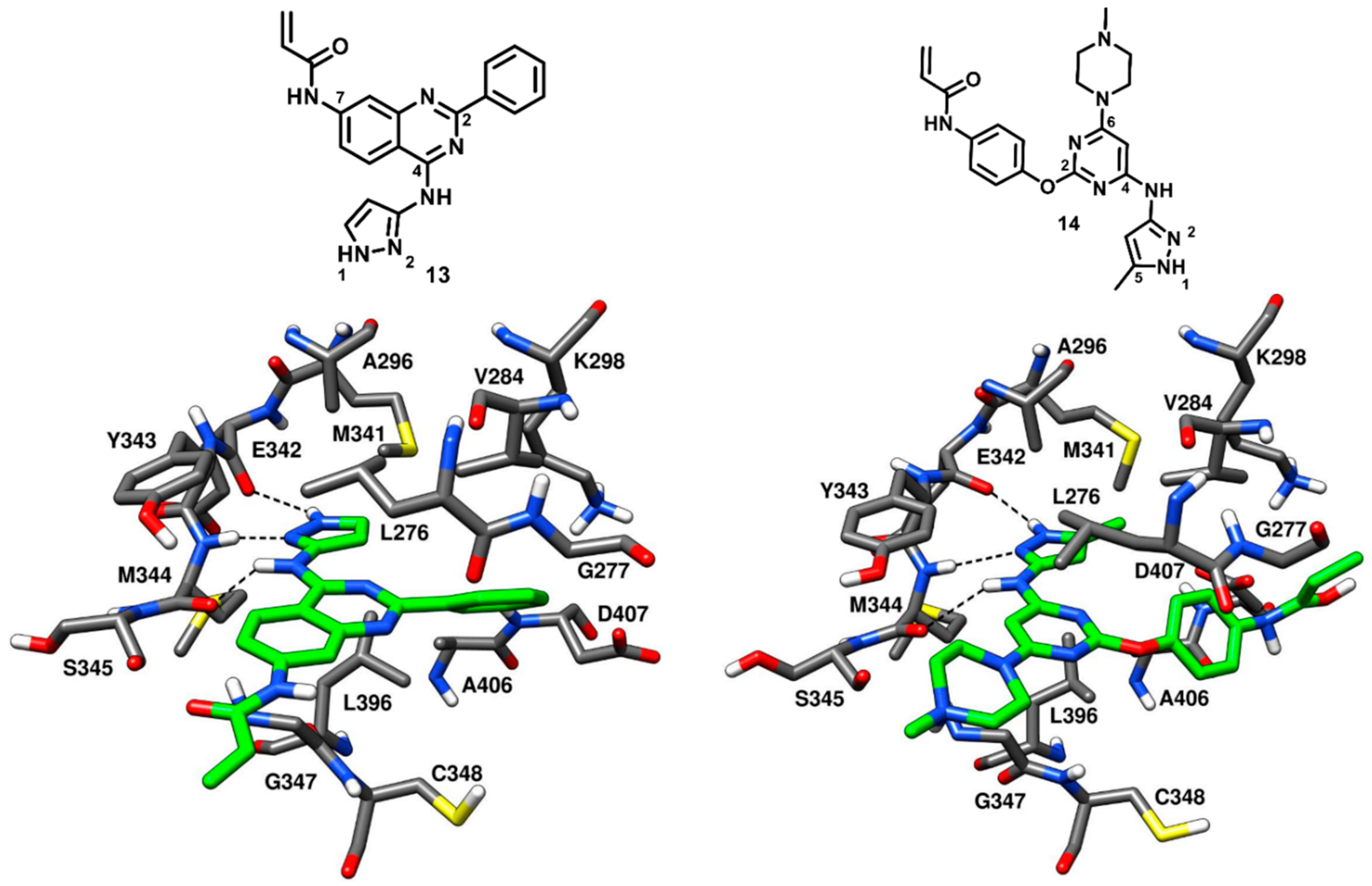
Engel and coworkers in 2015 designed and synthesized a series of irreversible quinazoline and pyrimidine-based EGFR inhibitors. The best compounds of the series were co-crystallized with engineered c-Src (T341M/S348C), which is a validated model system for EGFR and drug-resistant EGFR (T790M) [
36,
66]. However, two compounds of the series (
13 and
14,
Figure 14) were found binding to c-Src in a reversible manner. From the activity assays, the quinazoline-based inhibitor
13 demonstrated moderate inhibition of wild-type c-Src, with an IC
50 of 0.211 µM, but showed a 6- to 9-fold stronger potency against mutant c-Src, with IC
50 values of 65 nM and 43 nM against T341M c-Src and T341M/S348C c-Src, respectively. Similarly, the pyrimidine-based inhibitor
14 displayed IC
50 values of 0.218 µM, 0.107 µM and 72 nM against wild-type c-Src, T341M c-Src and T341M/S348C c-Src, respectively.
In order to gain structural insights into their binding mode, both
13 and
14 were co-crystallized with T341M/S348C mutated c-Src (PDB ID: 5D12 and 5D10, respectively,
Figure 14) [
67]. The 4-amino pyrazole moiety of both inhibitors occupied the adenine pocket, as observed for compound
12, and formed three H-bonds with the hinge-region residues. The 4-amino group of both
13 and
14 formed an H-bond with the backbone oxygen of M344, while the pyrazole ring established H-bonds with the backbone oxygen of E342 and the backbone nitrogen of M344. The orientation of the pyrazole ring of compound
13 was not affected by the mutated and larger gatekeeper residue M341; however, the presence of the 5-methyl group in the pyrazole ring of compound
14 displaced M341 away from the binding site due to steric repulsions and could be a reason of the slightly decreased potency of
14 with respect to
13. The central quinazoline ring of
13 and the pyrimidine ring of
14 established van der Waals interactions with L276 and G347 in the hydrophobic pocket II. The Michael acceptor (acrylamide) in the 7-position of the quinazoline inhibitor
13 projected outward from the ATP binding pocket, into the solvent-exposed region, thus preventing the covalent interaction with C348, whereas the 2-phenyl ring of the quinazoline core laid near the ribose pocket, showing van der Waals contacts mainly with V284. The methylpiperazine tail of
14 was solvent exposed, acting as a solubilizing group, while the 2-phenoxy ring linked to the pyrimidine core showed hydrophobic contacts with V284 similarly to phenyl ring of
13. The terminal acrylamide moiety of compound
14 was projected away from the binding site leaving no chance of covalent bonding with C348. The binding mode of compound
13 and
14 in complex with T341/S348C c-Src showed certain similarities to other reversible type-I inhibitors and due to the observed tolerance with larger mutated gatekeeper residues, the two compounds were further employed for the structure-based design of drug-resistant c-Src (T341M) inhibitors.
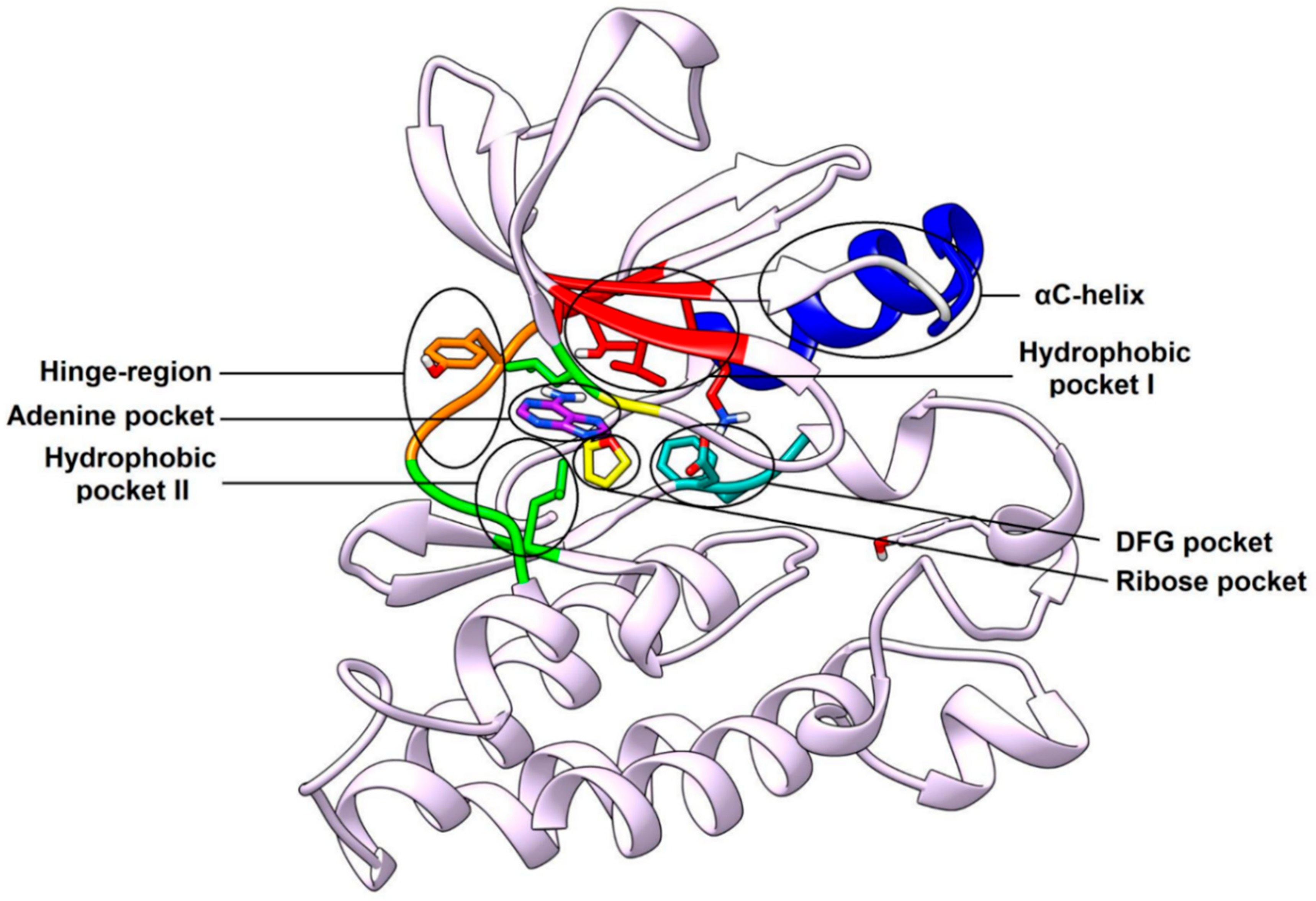
2.4. Type-II Inhibitors
Most type-I inhibitors are not able to fully occupy hydrophobic pocket I and to interact with the DFG pocket, leaving space within the kinase active site that could be targeted in order to improve their binding affinity. The design of type-II inhibitors has been mainly focused on extending the binding interactions of the ligands into these pockets. Successful strategies of type-II inhibitors design involved the structural optimization of known type-I inhibitors (e.g., design of type-II pyrazolopyrimidine inhibitors [
68]) and a hybrid design approach (e.g., design of dasatinib–imatinib hybrid compounds [
69]). Type II inhibitors bind inactive kinase conformations of c-Src and often show advanced pharmacological properties, such as slow dissociation rates, which increase drug-target residence time and result in significant enhancement in affinities over type I inhibitors. In the following paragraph, a description of the ligand–protein interactions of some important type-II inhibitors co-crystallized with c-Src kinase is reported (
Table S3).
In 2004, Ple and coworkers reported a series of 2-chloro-5-methoxyanilinoquinazolines substituted at the C-6 and C-7 positions of the quinazoline ring that demonstrated good inhibition potency against c-Src [
70]. However, molecular modeling studies suggested that the inhibitors were not able to occupy the ribose pocket of the kinase. Later on, the same research group investigated a new series of anilinoquinazolines substituted at the C-5 position of the quinazoline ring [
71]. Docking results confirmed that the C-5 position allowed the substituents to access the ribose pocket in terms of shape complementarity. The design of C-5 substitued anilinoquinazolines was derived from the discovery of CDK1-2 (cyclin-dependent kinase) inhibitors and from docking studies suggesting that the ribose pocket could be occupied by a broad range of substituents, including straight or branched alkyl chains, phenyl rings and cyclic amines [
72]. A total of 42 quinazoline compounds bearing either a 2-chloro-5-methoxyanilino or a chlorobenzodioxolylamino group were synthesized. An extensive SAR study was aimed at modifying the C-5 position of the quinazoline core to identify suitable substituents that could satisfactorily occupy the ribose pocket and improve the binding affinity for the target. Cyclic substituents, especially oxygen and nitrogen containing heterocycles, were found to be more favorable over acyclic and flexible substituents at C-5 position, leading to the identification of potent enzyme inhibitors. This class of compounds proved to be highly selective for c-Src over a variety of other kinases, such as CDK2, Aurora kinase, EGFR-TR, MEK and VEGFR-2. In particular, C-5 substituted anilinoquinazolines were reported to be 70-fold more potent towards c-Src over VEGFR-2 [
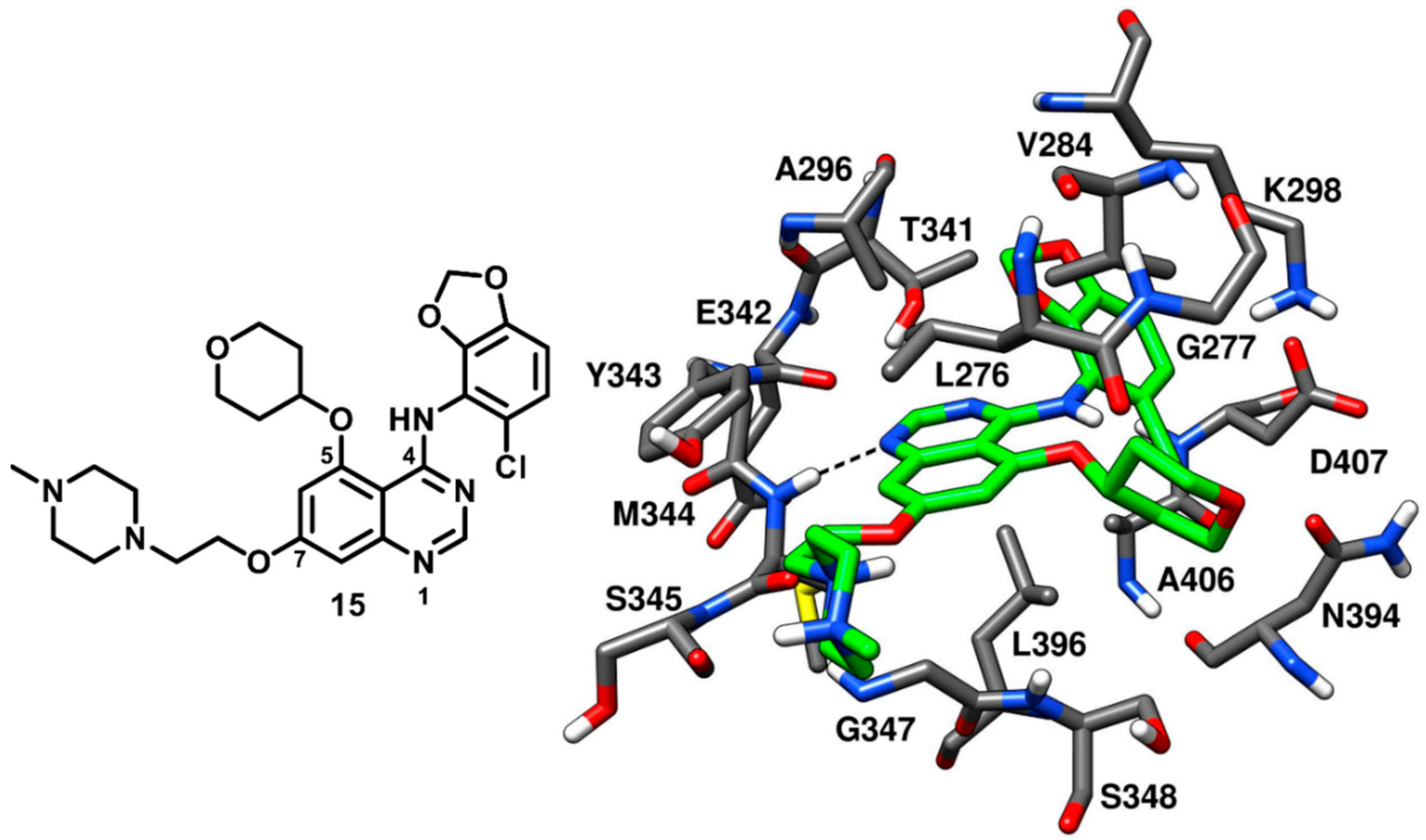
70]. The best compound of the series, compound
15 (AZD0530, saracatinib,
Figure 15), is a bicyclic [(chlorobenzodioxolyl)amino]-quinazoline with a tetrahydropyranyloxy group at the C-5 position, which demonstrated a remarkable IC
50 of 2.7 nM in c-Src kinase inhibition. Compound
15 was co-crystallized with c-Src kinase (
Figure 15, PDB ID: 2H8H) and showed binding to the inactive kinase conformation. The tetrahydropyran ring of the inhibitor occupied the ribose pocket, forming hydrophobic interactions with L276, V284 (N-terminal lobe) and L396 (C-terminal lobe). The binding of the inhibitor determined a slight distortion of the ATP pocket and allowed a little opening at the entrance of hydrophobic pocket I to accommodate the bulky benzodioxane ring. Due to the presence of the large benzodioxane group, the αC-helix of the catalytic domain was displaced outwards, causing the disruption of the salt bridge interaction between K298 and E313. The displacement of the αC-helix categorized compound
15 as a CHO (αC-helix-out) type-II inhibitor. The chlorine atom on the benzodioxane ring established lipophilic interactions with the methyl side chain of A406. Similarly to type-I inhibitors, compound
15 displayed a key H-bond interaction in the hinge-region of the ATP-binding site, since the N-1 of the quinazoline core formed an H-bond with the backbone nitrogen of M344. The piperazine tail of compound
15 laid predominantly on the solvent exposed region, whereas the ethyloxy linker established van der Waals contacts with L276 in hydrophobic pocket II. The discovery of type-II quinazoline inhibitors allowed a better exploration of hydrophobic pocket I and ribose pocket of the c-Src binding site, which could be utilized for the development of promising type-II analogues.
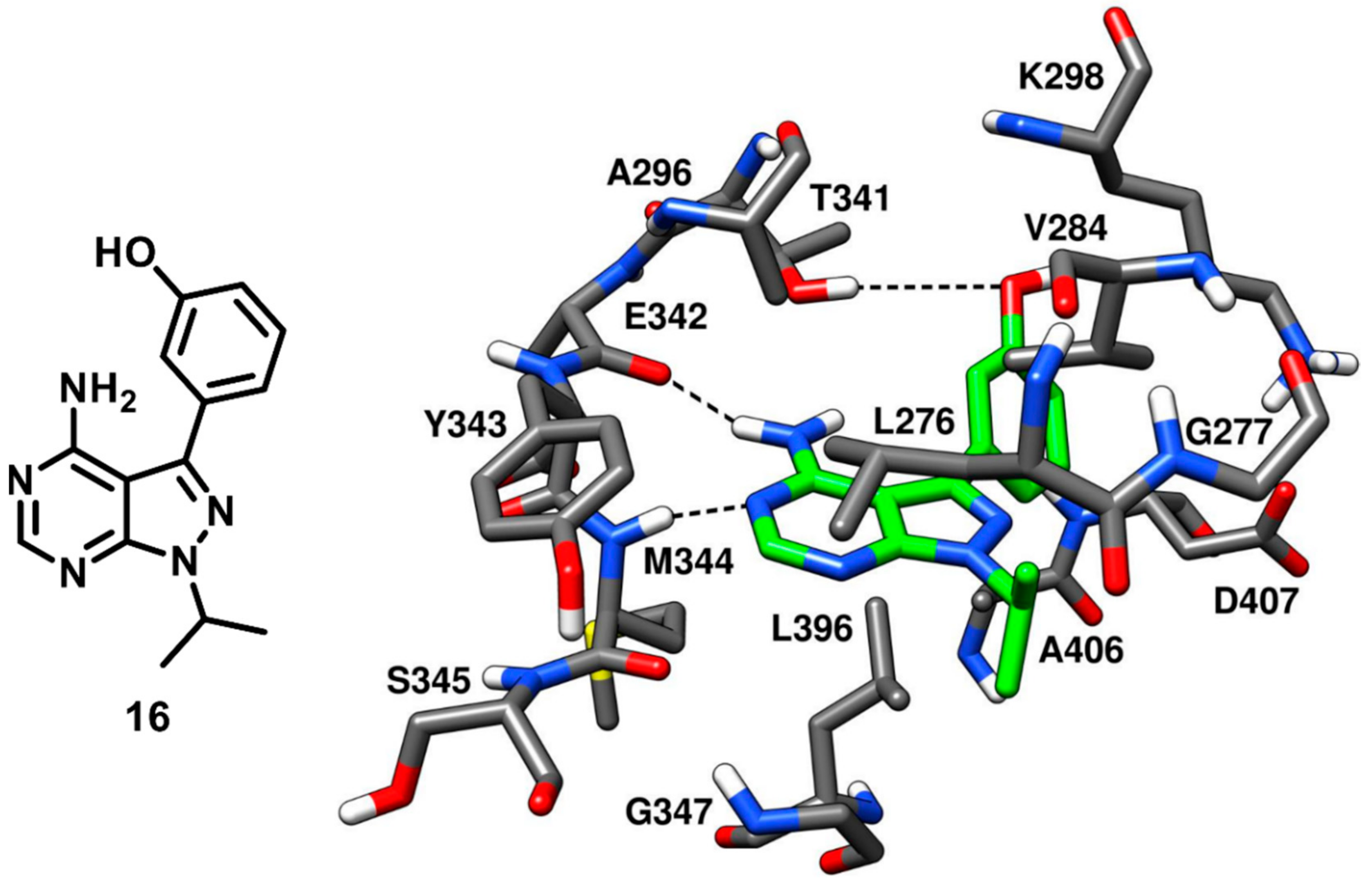
Compound
10 bearing a pyrazolopyrimidine scaffold, which was discussed earlier in type-I inhibitors’ section, was identified along with three more pyrazolopyrimidine inhibitors that were co-crystallized with c-Src. These ligands were able to bind the inactive kinase with CHO conformation, where the key salt bridge interaction between K298 and E313 was disrupted. Among the three inhibitors, compound
16 (PDB ID: 3EN7,
Figure 16) was the most potent, with an IC
50 of 15 nM, similarly to compound
10, while compounds
17 and
18 (PDB IDs 3EN5 and 3EN6 respectively,
Figure S3) showed IC
50 values of 360 nM and 235 nM, respectively [
57]. The central pyrazolopyrimidine core of all three inhibitors occupied the adenine pocket and showed two H-bond interactions with the hinge-region: as observed for compound
10, the 4-amino group of the pyrazolopyrimidine ring established an H-bond with the backbone oxygen of E342 and the 5-N nitrogen formed the second H-bond with the backbone nitrogen of M344. The C-3 aryl substituents of these inhibitors (the phenol, dimethoxyphenyl and quinoline groups of
16,
17 and
18, respectively) extended beyond the gatekeeper residue and were accommodated into the hydrophobic pocket I. The solvent exposed N-1 cyclic and acyclic substituents (isopropyl for
16,
18 and cyclobutyl for
17) occupied the ribose pocket and established van der Waals interactions with V284, L276 and G277 of the N-terminal lobe. The key feature that differentiated compound
16 from the other two ligands is that
16 was able to form a third additional H-bond with the gatekeeper residue, which significantly improved its inhibition potency over compound
17 and
18. Notably, the type-I analogue
10 that showed an IC
50 for c-Src of 14 nM was also able to form a third H-bond in addition to the H-bond interactions with the hinge region.
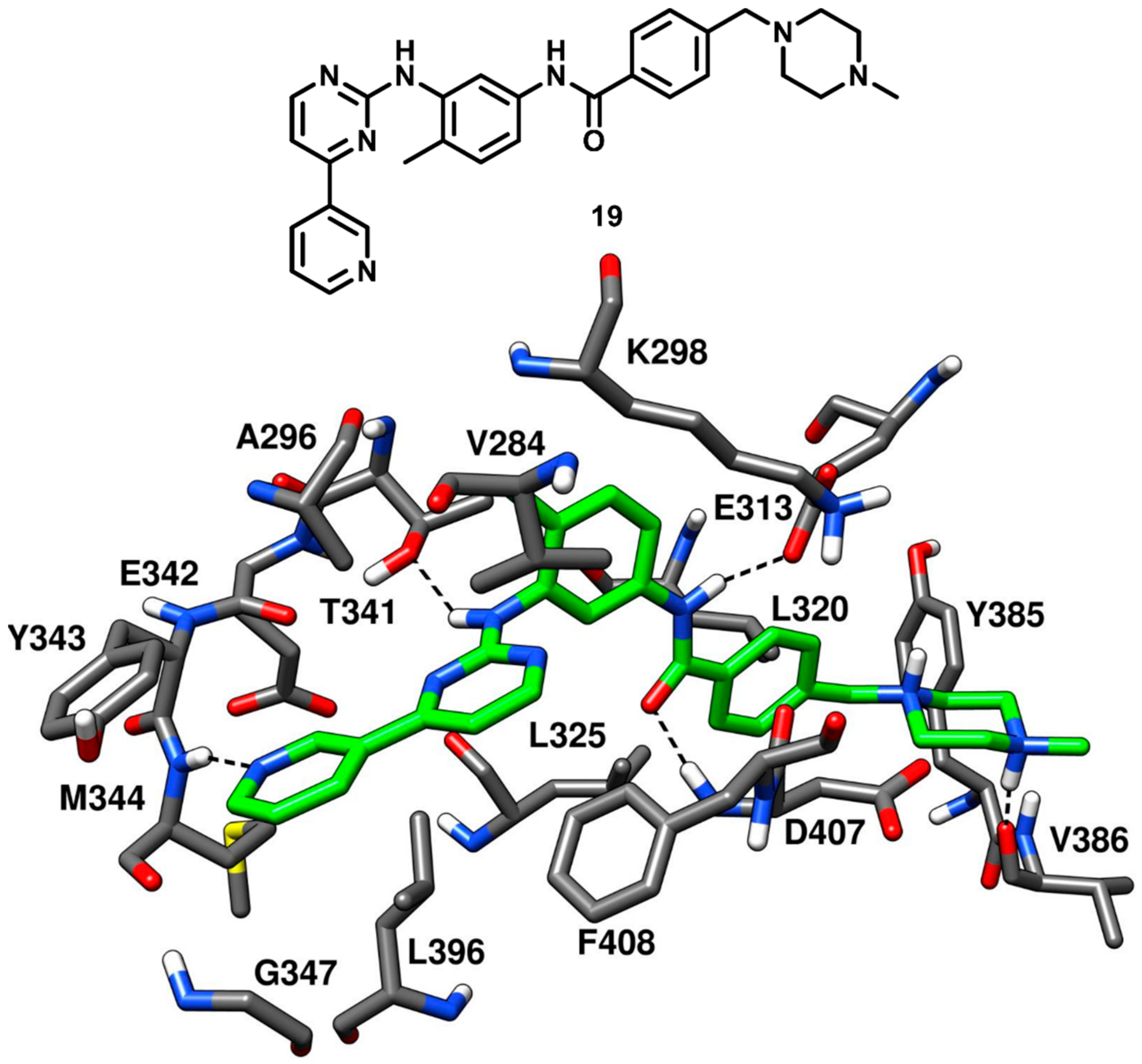
As discussed in type-I inhibitors’ category, imatinib (compound
19,
Figure 17) is a potent inhibitor of Abl kinase but a weak inhibitor of c-Src kinase; however, the des-methyl analogue of imatinib (compound
2, PDB ID 1Y57,
Figure 4) had shown an impressive IC
50 of 1.6 nM against c-Src kinase [
43]. Seeliger and coworkers in 2007 co-crystallized compound
19 with c-Src kinase (
Figure 17, PDB ID 2OIQ) and explored the structural basis of its affinity for c-Src, which could provide a useful rationale of its weak inhibitory activity [
42]. Compound
19 was bound to the DFG-out inactive kinase conformation where an outward flip of F408 in the DFG pocket was observed, which categorized compound
19 as a DFG-out type-II inhibitor of c-Src. Compound
19 adopted an inverted binding orientation in the active site of c-Src with respect to the binding mode of the demethylated analogue
2. The presence of a methyl group on the phenyl ring adjacent to the pyrimidine core of compound
19 determined steric repulsions with the hinge-region residues and the allocation of the methylated phenyl ring in the hydrophobic pocket I. The pyridine and pyrimidine rings of the inhibitor
19 occupied the adenine pocket of the ATP binding site. The nitrogen of the pyridine ring established one H-bond interaction with the backbone nitrogen of M344 whereas the pyrimidine ring was found to be in proximity with V284 of the N-terminal lobe and F408 of the DFG pocket, forming hydrophobic interactions with these residues. Moreover, the amino group of the pyrimidine moiety formed an H-bond with the gatekeeper residue T341. The benzamide group of the ligand occupied the DFG pocket and established key H-bonds with the E313 carboxylic group of the αC-helix and the backbone nitrogen of D407 from the DFG motif, while the adjacent phenyl ring showed van der Waals interactions mainly with the sidechains of L320 and L325. Finally, the piperazine tail of the inhibitor extended past the DFG pocket into the solvent exposed region, displayed an H-bond with the backbone oxygen of V386 and formed van der Waals contact primarily with Y385. The binding mode of compound
19 demonstrated that the presence of sterically bulky groups in the portion of the inhibitor occupying the adenine pocket prevented the formation of the classical H-bonding interactions in the hinge-region as compared to type-I inhibitors. This steric repulsion significantly changed the binding conformation of the inhibitor; however, the orientation of the benzamide moiety in the DFG pocket and the H-bonding interactions demonstrated by the amide linker with E313 and D407 were greatly exploited to develop novel DFG-out type-II ligands of c-Src.
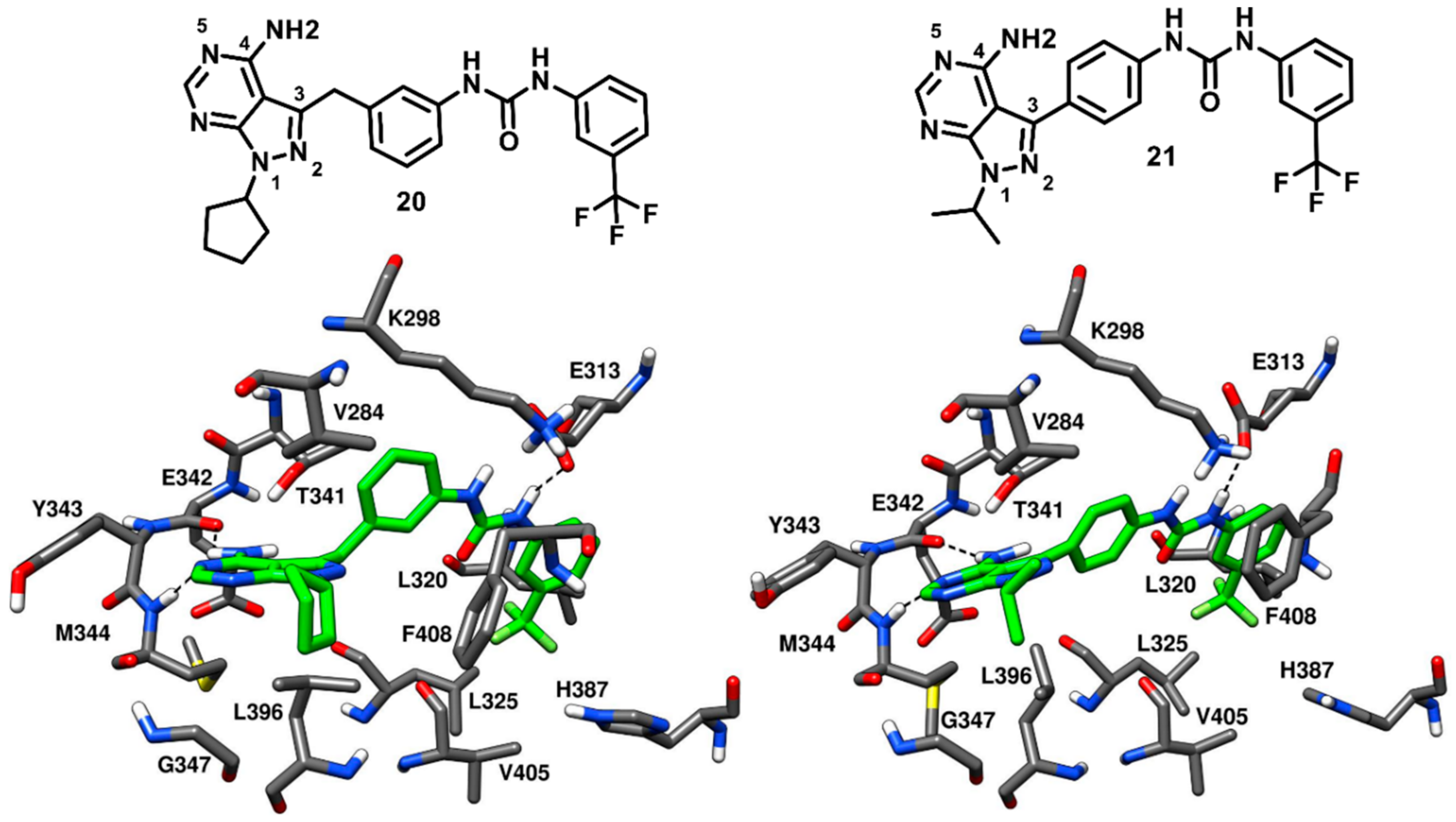
Dar and coworkers in 2008 designed and synthesized a panel of pyrazolopyrimidine-based inhibitors, with reference to the binding mode of imatinib, that would bind to the inactive DFG-out conformation of c-Src kinase. The synthesized ligands showed IC
50 values of c-Src inhibition from micromolar to low-nanomolar range. Two compounds of the series (
20 and
21,
Figure 18) were co-crystallized with c-Src (PDB ID: 3EL7 and 3EL8, respectively) and were both confirmed to act as type-II inhibitors, as they bound to the DFG-out inactive conformation of c-Src [
73]. Unlike imatinib, both inhibitors showed a dual inhibitory effect on c-Src and Abl. Compound
20 showed to be less potent, with IC
50 values of 0.480 µM and 2.087 µM against c-Src and Abl, respectively, whereas compound
21 demonstrated a higher activity, with IC
50 values of 25 nM and 41 nM against c-Src and Abl, respectively. The pyrazolopyrimidine core of both compounds occupied the adenine pocket, forming one H-bond with the backbone nitrogen of M344 in the hinge-region, while the 4-amino group established an H-bond with the backbone oxygen of E342. The C-3 substituents of both ligands were twisted out of plane relative to the pyrazolopyrimidine ring, with the benzyl ring of
20 showing to better fit in the hydrophobic pocket I, which could contribute to its higher potency compared to
21. The urea group of the ligands formed H-bonds with the carboxylic group of E313, whereas the trifluoromethyl phenyl ring of both inhibitors was well accommodated into the DFG pocket and displayed van der Waals contacts with the sidechains of L320, L325, H387 and V405, contributing to the binding affinity for the target. Hydrophobic interactions with V284 were also observed for the solvent exposed cyclopentyl and isopropyl groups of
20 and
21, respectively. In addition, a lipophilic interaction between the cyclopentyl of
20 and F408 from the DFG motif, not observed for
21, was noted and could further justify the higher activity of
20 compared to
21. The distinctive feature of these pyrazolopyrimidine ligands is the rotation of their benzyl/phenyl ring away from the gatekeeper residue (similarly to the pyrimidine ring of imatinib). The extra space from the gatekeeper residue, created as a result of this rotation suggested that inhibitors
20 and
21 could bind to the clinically relevant T341M/T341I mutant c-Src and show promising results, which could further provide a platform for optimization studies against drug-resistant c-Src kinase.
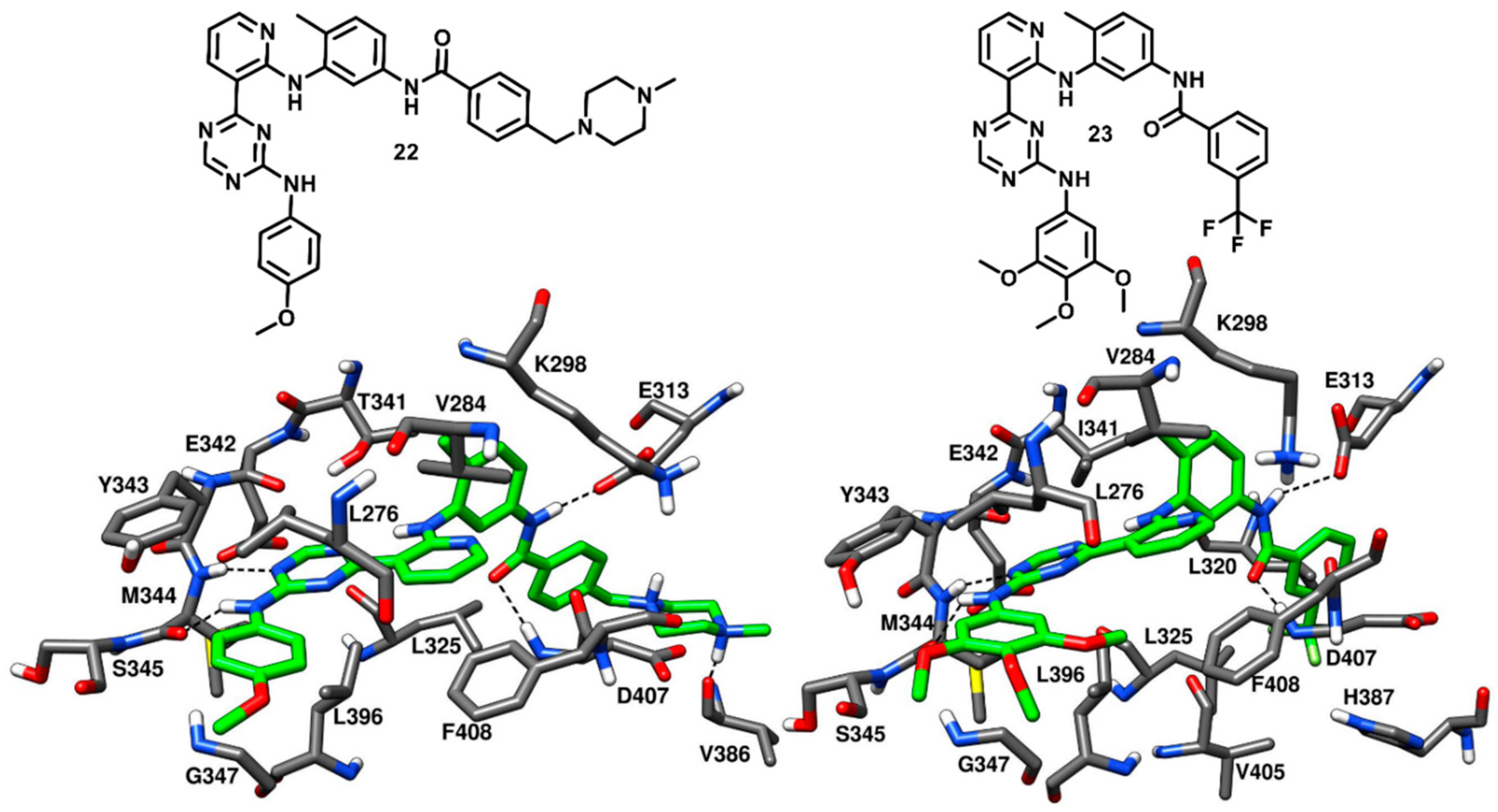
In 2009, Seeliger and coworkers identified a series pyridinyl-triazine inhibitors (also called as DSA compounds), that are based on the central scaffold of imatinib and were designed to bind c-Src with DFG-out kinase conformation. Unlike Imatinib, these compounds are nanomolar inhibitors of both c-Src and Abl with similar potencies. The most promising derivatives of the series, compounds
22 and
23 (
Figure 19), were co-crystallized with wild type (PDB ID: 3G6G) and T341I mutant c-Src (PDB ID: 3G6H), respectively, and showed binding to the DFG-out kinase conformation as type-II inhibitors (
Figure 19) [
74]. Compounds
22 and
23 displayed IC
50 values of 2.8 nM and 4.6 nM, respectively, against wild-type c-Src and IC
50 values of 18 nM and 6.4 nM, respectively, against T341I mutant c-Src, thus demonstrating the retention of their inhibition potencies against the mutant enzyme, with an only two- to eightfold increase in IC
50 values. The X-ray complexes of the ligands showed that the amino-group of their lateral aniline moieties formed an H-bond with the backbone oxygen of M344, while the triazine ring of both inhibitors formed another H-bond with the backbone nitrogen of M344. Moreover, the aniline moieties showed van der Waals contacts with hydrophobic pocket II residues, mainly with L276 and G347. Similarly to what observed for imatinib [
42], the pyridine ring of both inhibitors established van der Waals interactions with the sidechains of V284 of N-terminal lobe and F408 of the DFG motif, the adjacent aromatic phenyl ring occupied hydrophobic pocket I interacting with the gatekeeper residue, while the amide linker was placed at the entrance of the DFG pocket and established key H-bonds with the carboxylic group of E313 of the αC-helix and the backbone nitrogen of D407 from the DFG motif. By comparing the X-ray structures of wild type and mutant c-Src in complex with
22 and
23, respectively, it is possible to observe that the orientation of the central 4-methyl-
m-diaminophenyl ring of
23, which is placed in front of I341, is not perturbed by the larger side chain of the mutated gatekeeper residue, closely resembling the orientation observed for the same moiety of compound
22. In fact, the binding mode of
23 was found to be comparable with that observed for
22 and this rationalizes the retention of inhibitory activity of
23 against T341I mutant c-Src with respect to wild type c-Src. Finally, the phenyl ring of
22 and the trifluoromethylphenyl ring of
23 occupied the DFG pocket. The phenyl ring of
22 established van der Waals contacts mainly with L325, whereas the trifluoromethylphenyl ring of
23 was surrounded by the side chains of L320, L325, H387, V405 and displayed strong van der Waals interactions. However, the terminal methylpiperazine ring of
22 extended past the DFG pocket into the solvent-exposed region and established an H-bond interaction with V386 of the C-terminal lobe.
Although the binding mode of 22 and 23 resemble that of imatinib, some dissimilarities can be observed in their structural orientation, which may contribute to their nanomolar binding affinity, in contrast with the weak affinity of imatinib against c-Src. The triazine and pyridine rings of 22 and 23 occupied the adenine pocket like the pyridine and pyrimidine groups of imatinib but showed different interactions. The pyridine in both 22 and 23 is ortho-substituted, while the equivalent pyrimidine in imatinib is meta-substituted. The different orientation of the pyridine and triazine ring in 22 and 23 led to a better accommodation of the two ligands into the adenine pocket, favored by stronger hydrophobic contacts with V284 and F408. Moreover, although 22 and 23 lacked the H-bond with the sidechain of the gatekeeper residue T341, the two ligands formed an H-bond with the hinge region, precisely with the backbone oxygen of M344, through the amino group of their terminal aniline rings. These considerations may justify the higher inhibitory activity of 22 and 23 with respect to imatinib.
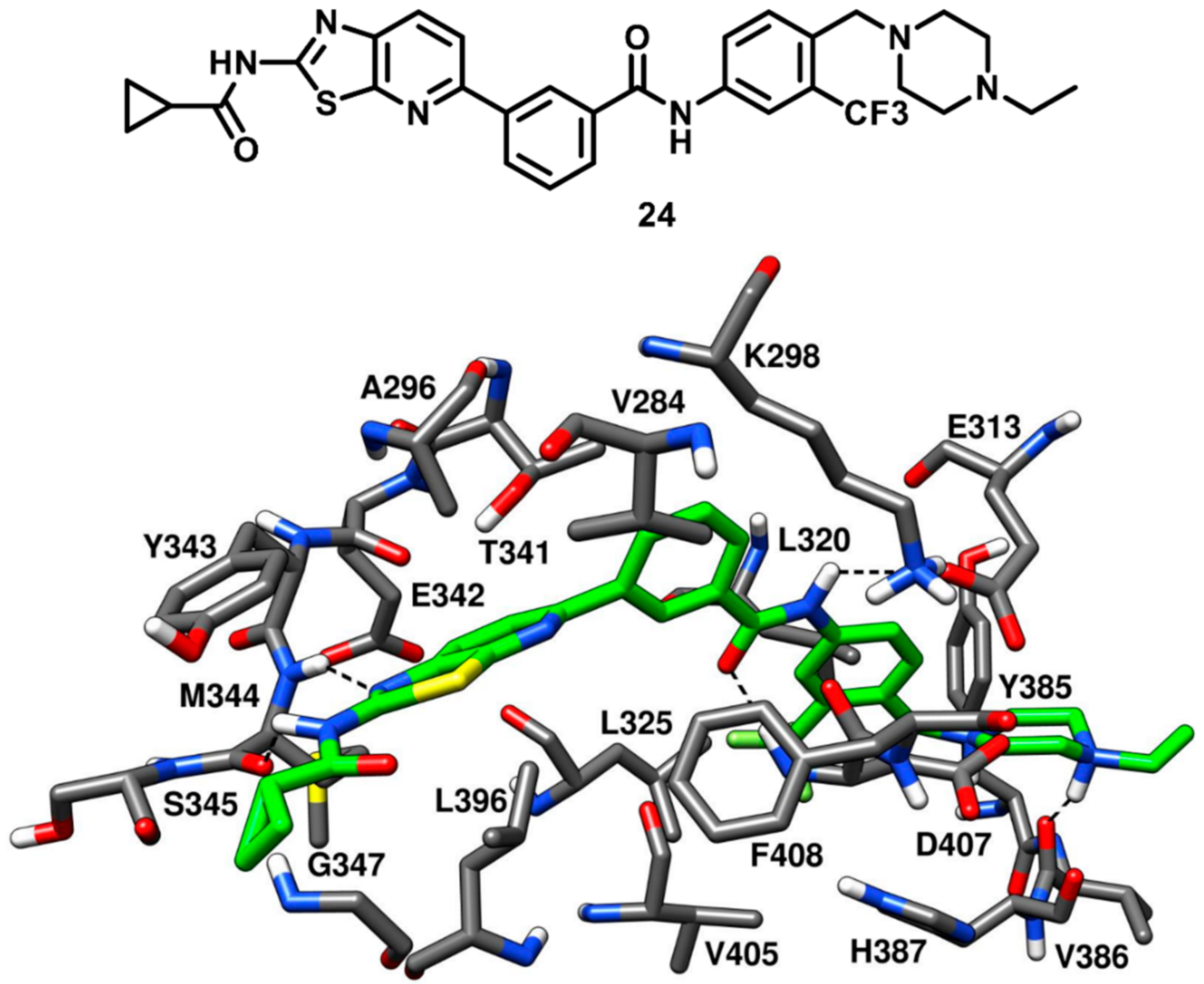
Weisberg and coworkers in 2010 developed a unique thiazolopyridine-based multitarget kinase inhibitor (
24,
Figure 20). The design of compound
24 involved a hybrid approach based on the type-I inhibitor dasatinib [
38] and the type-II inhibitor nilotinib [
75], which were both co-crystallized in complex with Abl kinase. The superimposition of the two ligand–protein co-crystal structures inspired the fusion of the hinge-interacting aminothiazole moiety of dasatinib and the N-(3-(trifluoromethylphenyl) benzamide moiety of nilotinib, which binds to the DFG-out conformation of the kinase. Compound
24 was thus designed as a type-II inhibitor, with the aim of developing a potent inhibitor of both wild-type and drug-resistant T341M/I mutant c-Src. Biochemical screening studies revealed that
24 was endowed with high inhibitory activity against a variety of wild-type and mutant forms of kinases. To confirm the proposed binding mode resulting from the hybrid design, compound
24 was co-crystallized with c-Src (PDB ID 4AGW,
Figure 20) [
76]. As expected, this inhibitor was bound to the inactive DFG-out kinase conformation and, interestingly, displayed five H-bonding interactions with the binding site residues of c-Src. The thiazolopyridine ring of compound
24 occupied the adenine pocket, the thiazole nitrogen formed one H-bond with the backbone nitrogen of M344 while the fused pyridine ring showed hydrophobic interactions with A296 and L396. The amino group of the cyclopropylamide of the inhibitor formed another H-bond with the backbone oxygen of M344, while the cyclopropyl moiety was solvent-exposed. The DFG-out conformation of c-Src kinase allowed the trifluoromethylphenyl ring of the inhibitor to occupy the DFG pocket, whereas the amide linker connecting phenyl and trifluoromethyl phenyl ring formed important H-bonds with the carboxylic group of E313 of the αC-helix and the backbone nitrogen of D407 from the DFG motif, which represented the prototypical feature of imatinib-based compounds. The phenyl of the benzamide moiety of the inhibitor displayed an edge-to-face π-π stacking with F408 of the DFG motif, while the trifluoromethylphenyl substituent demonstrated strong van der Waals contacts with L320, L325, H387 and V405. The ethylpiperazine tail of the inhibitor was allocated past the DFG pocket into the solvent-exposed area, displaying hydrophobic contacts with Y385 and forming an H-bond with the backbone oxygen of V386. Similarly to inhibitors
22 and
23, the loss of an H-bond with the sidechain of T341 observed for imatinib is compensated by an additional H-bond formation between the amino group of the cyclopropylamide and the backbone oxygen of M344. Molecular modeling studies performed with compound
24 suggested the possibility that mutant c-Src with larger gatekeeper residues (T341I/M) could accommodate the ligand without affecting its binding mode. In fact,
24 was reported to inhibit the proliferation of Ba/F3 cells transfected with wild type c-Src and both T341I and T341M mutant c-Src. The EC
50 values for compound
24 were determined to be 0.19 µM, 0.29 µM, and 0.15 µM for c-Src, T341I c-Src and T341M c-Src respectively. On the contrary, the nanomolar c-Src inhibitor dasatinib (
1) showed an EC
50 value < 10 nM for wild type c-Src but completely lost activity for both T341I and T341M mutant c-Src (EC
50 > 10 µM). This experimental data confirmed that the thiazolopyridine compound
24 is a potent inhibitor of both wild-type and drug-resistant c-Src kinase (T341I/M).
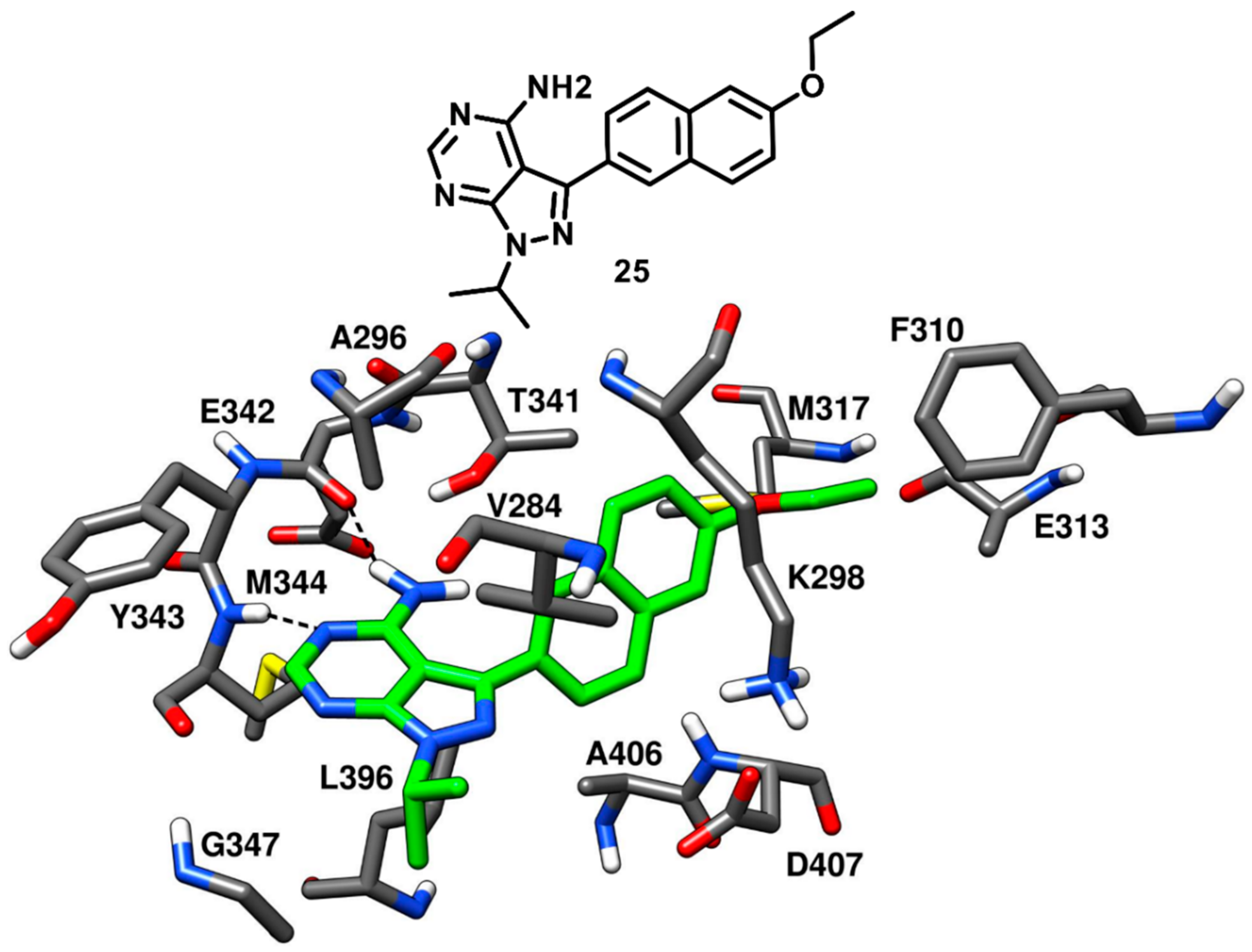
Larson and coworkers in 2012 developed a series of pyrazolopyrimidine-based apicomplexan CDPK1 inhibitors and reported a comparison analysis of the ligand–protein interactions observed in X-ray structures of CDPK1 and c-Src in complex with inhibitors. SAR studies were then performed with the aim to explore the pyrazolopyrimidine scaffold and a series of inhibitors comprising either isopropyl or 4-piperidinemethyl groups at 1-positon [
77] and various substituents at the 3-position of the pyrazolopyrimidine core was synthesized. One of the best compounds of the series (
25,
Figure 21) was co-crystallized with c-Src kinase (PDB ID: 3QUF) and demonstrated an IC
50 of 190 nM [
78]. Compound
25 was bound to inactive c-Src with CHO conformation and could be therefore categorized as a CHO-type-II inhibitor of c-Src. The 4-aminopyrazolopyrimidine core laid on the adenine pocket of ATP binding site, forming hydrophobic interactions with A296 and L396, and displayed classical H-bond interactions with the hinge-region residues: one H-bond with the backbone nitrogen of M344 and one with the backbone oxygen of E342. The highly hydrophobic and bulky ethoxynaphthalene substituent of the pyrazolopyrimidine ring well occupied hydrophobic pocket I, forming hydrophobic interactions with the sidechains of K298 and T341 residues and led to the disruption of the key salt bridge interaction between K298 of the N-terminal lobe and E313 of the αC-helix. Furthermore, the terminal ethoxy group was encircled by the sidechains of F310, E313 and M317, forming van der Waals contacts with these residues. Finally, the isopropyl group of the ligand was solvent-exposed but showed van der Waals contacts with V284. SAR studies highlighted that the naphthyl substituent, which was projected deeply towards the αC-helix, largely contributed to the activity of the ligand against c-Src. In fact, a synthesized analogue lacking the naphthyl substituent (
26,
Figure S4) showed a drastic loss of inhibition potency for c-Src, with an IC
50 > 10 µM. The X-ray structure of c-Src in complex with compound
26 (PDB ID: 3UQG) demonstrated that the absence of the large C-3 substituent prevented the ligand to interfere with the interaction between K298 and E313, and the compound bound to the αC-helix-in active conformation of c-Src as a type-I inhibitor. The pyrazolopyrimidine moiety of
26 displayed H-bonding interactions in the hinge-region similar to
25, while the solvent-exposed N-1 isopropyl substituent of
25 was replaced with 4-piperidinemethyl substituent, which did not make any contact with active site residues.
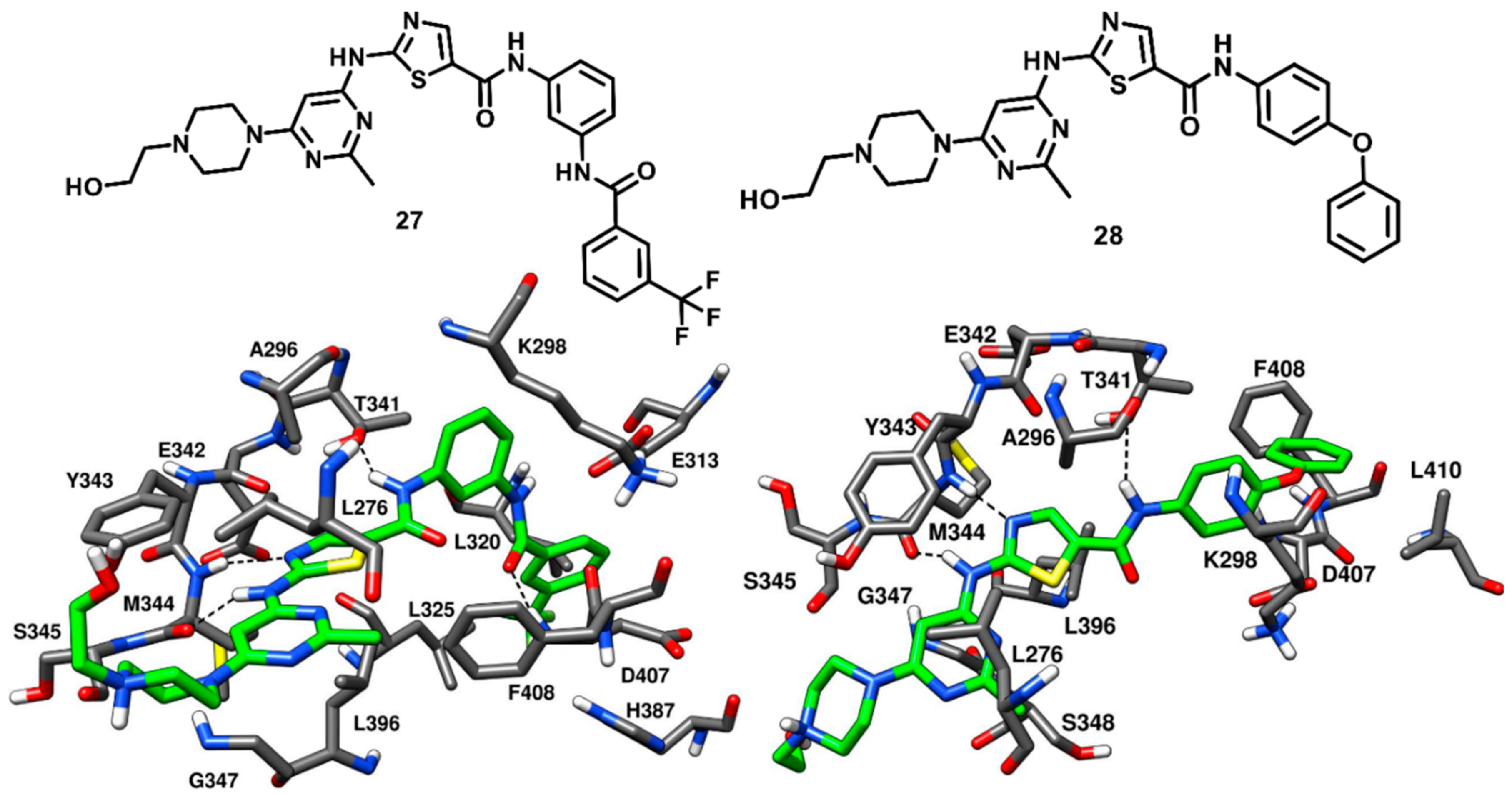
Kwarcinski and coworkers in 2015 adopted a hybrid design strategy to develop dasatinib analogues, that could bind to either CHO or DFG-out inactive kinase conformation. An overlay of the X-ray structures of c-Src bound to datasinib (compound
1, PDB ID: 3G5D,
Figure 3) and imatinib (compound
19, PDB ID: 2OIQ,
Figure 17) suggested that the addition of a benzamide group to the terminal phenyl ring of dasatinib could result in a potent type-II derivative. Two analogues of dasatinib were synthesized, and the most active compound
27, which demonstrated an impressive binding affinity, with a K
d value of 0.52 nM, was co-crystallized with c-Src (PDB ID: 4YBJ,
Figure 22) [
69]. Compound
27 demonstrated binding to the inactive kinase conformation, with the portion belonging from dasatinib accommodated into the ATP binding site and that derived from imatinib located into the DFG pocket of the activation loop. An outward flip of F408 of the DFG motif was observed in the inactive kinase conformation, which classified compound
27 as a DFG-out type-II inhibitor. Compound
27 showed the two classical H-bond interactions in the hinge-region of the ATP-binding site; the amino group of the 2-aminothiazole moiety formed an H-bond with the backbone oxygen of M344, while the nitrogen of the thiazole ring formed another H-bond with the backbone nitrogen of M344. The amide nitrogen of the thiazole-5-carboxamide established a crucial H-bond with T341, which could provide selectivity for c-Src. The 2-methyl pyrimidine ring of
27 showed van der Waals contacts with L276, G347 and L396, while the thiazole ring of the inhibitor displayed van der Waals interactions with A296 and L396. The carbonyl oxygen of the benzamide moiety of the inhibitor displayed an H-bond with the backbone nitrogen of D407 in the DFG pocket, whereas the trifluoromethyl ring of the inhibitor occupied the DFG pocket and formed van der Waals contacts with L320, L325, H387 and V405.
A similar design strategy was applied for the development of few more dasatinib analogues as type-II CHO inhibitors of c-Src. An overlay of dasatinib-c-Src complex with a group of known CHO inhibitors of other kinases (such as EGFR, HER2, MET and BTK) [
79,
80] suggested that addition of a
para-phenoxy group to dasatinib could lead to a potent type-II CHO analogue. Two dasatinib-CHO analogues were synthesized and the most active compound (
28) showed high binding affinity for the kinase, with a K
d value of 15 nM, and was co-crystallized with c-Src (PDB ID: 4YBK,
Figure 22). Compound
28 was bound to the inactive kinase conformation, with type-II CHO mode of inhibition. Compound
28 displayed H-bonding interactions with the backbone of M344 in the hinge-region and with the gatekeeper residue in the hydrophobic pocket I, similarly to compound
27. The inhibitor penetrated very deeply into hydrophobic pocket I and the αC-helix was projected outwards, undergoing a big rotation of around 5 Å from its original position, with the consequent disruption of the key salt bridge interaction between K298 and E313. The phenoxy group of compound
28 was not able to access the DFG pocket of the activation loop, but occupied an empty space emerged from the outward movement of the αC-helix and formed a T-shaped π-π stacking with the phenyl ring of F408. More van der Waals contacts were noted for the phenoxy group of the inhibitor with the sidechains of K298 and L410. Both compounds
27 and
28, together with dasatinib, were subjected to competition binding assays and tested on a panel of 124 non-mutant kinase constructs at the concentration of 500 nM, in order to assess the selectivity of the two ligands. While compound
27 showed a selectivity profile comparable to dasatinib, compound
28 showed to be much more selective, demonstrating affinity for a reduced number of kinases (with an S
35 score of 0.05 compared to the S
35 score of 0.21 obtained for dasatinib).
2.5. Type-III and Type-III-Derived Inhibitors
Emergence of drug resistance is a fundamental challenge in the development of kinase inhibitors for prolonged treatments. As discussed before, the most common mutation occurs at the gatekeeper position in the hinge region, where the small amino acid T341 is exchanged with a larger hydrophobic residue (I or M) [
35,
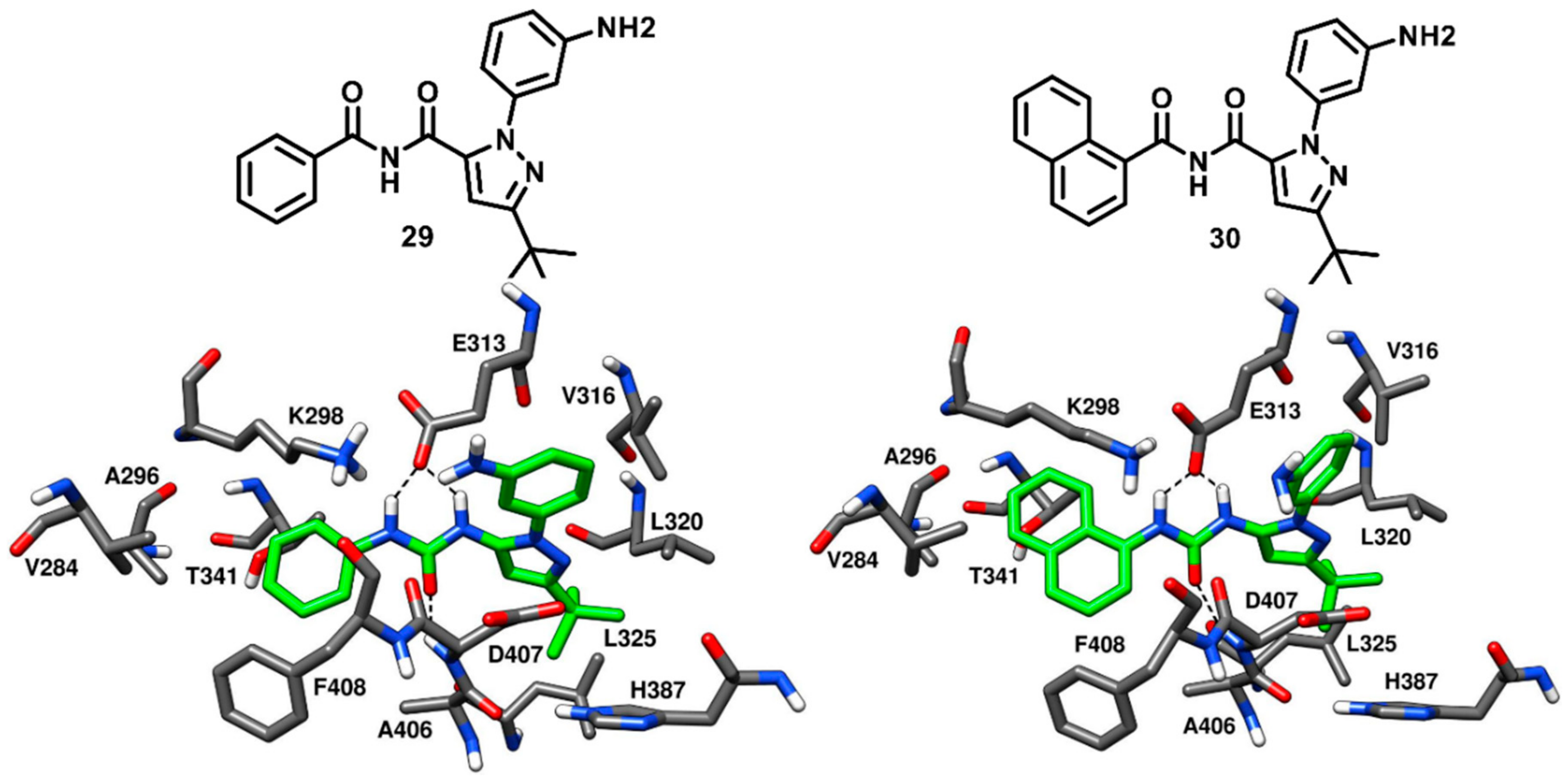
36]. Some of the type-I and type-II inhibitors demonstrated drug resistance due to the steric clashes with the mutated gatekeeper residues. In this context, another important class of c-Src ligands is represented by type-III inhibitors, which exclusively bind to the less-conserved allosteric site in the DFG pocket of the inactive c-Src conformation and do not interact with hinge-region and ATP binding site. For this reason, type-III inhibitors form either no or limited interactions with the gatekeeper residue. Though allosteric type-III inhibitors often show mid-micromolar inhibitory potency, they have been found to overcome drug resistance caused by mutations at the gatekeeper position that induce a steric clashes and prevents inhibitors from effectively binding to the hinge region. Moreover, the binding mode of type-III inhibitors have been employed for the development of potent type-I and type-III hybrid inhibitors that are capable of circumventing drug resistance. Several pyrazoloureas were known to be potent type-III binders of p38α kinase with affinities in the low nanomolar range [
81,
82]. Inspired by these compounds, Simard and coworkers in 2009 identified four pyrazolourea derivatives as the first type-III inhibitors of c-Src by screening a compound library. Enzyme activity assays confirmed that two out of these four hit compounds (
29 and
30,
Figure 23) inhibited c-Src kinase activity with IC
50 values in the mid-micromolar range (
Table S4). Compounds
29 and
30 demonstrated an IC
50 of 32.1 µM and 64.1 µM against c-Src, respectively, and were co-crystallized with c-Src kinase (PDB IDs: 3F3U and 3F3T, respectively,
Figure 23) to confirm the type-III allosteric binding mode. To our knowledge, these are the first and only reported crystal structures of c-Src kinase in complex with type-III inhibitors. Both ligands were bound to the inactive kinase conformation and were located outside the ATP binding pocket, which is a prototypical character of type-III kinase inhibitors. An outward flip of F408 was observed in the DFG pocket, thus characterizing the two ligands as DFG-out type-III inhibitors of c-Src. Both the phenyl and naphthyl ring of
29 and
30, respectively, occupied hydrophobic pocket I, with the naphthyl ring of
30 being closer to the gatekeeper residue. Furthermore, both substituents were found to be in the proximity of F408 of the DFG motif, forming hydrophobic interactions with this residue. The urea moiety of the ligands formed H-bonds with the carboxylic group of E313 of the αC-helix and the backbone nitrogen of D407 in the DFG pocket. The C-3 tert-butyl substituted pyrazole ring of both inhibitors was accommodated into the DFG pocket, with the tert-butyl group displaying van der Waals contacts with L320, L325 and H387. The terminal 3-aminophenyl ring exhibited van der Waals interactions predominantly with E313 and V316 [
37].
Despite the micromolar potency of compound
29 and
30, the identification of type-III c-Src inhibitors encouraged the development of potent analogues active against drug-resistant c-Src (T341M/I). Similar to the hybrid design approach employing type-I and type-II inhibitors discussed before, type-I/II and type-III hybrid design demonstrated to offer a profitable opportunity for scaffold development to circumvent drug resistance. In this context, Getlik and coworkers in 2009 employed a fragment-based design strategy to develop hybrid molecules from type-III pyrazolourea inhibitors and type-I quinazoline inhibitors. Activity assays using drug resistant c-Src variant (T341M) [
83] performed for compound
29 and
30 showed that the presence of the bulkier gatekeeper residue had no effect on the potency of compound
29, while compound
30 dramatically lost its activity. The X-ray structures suggested that compound
30 was likely to be impeded by steric repulsions with the larger gatekeeper residue due to the presence of its bulky naphthalene moiety, which was placed in close proximity to T341, while the phenyl moiety of compound
29, which was more distant from the gatekeeper residue, was unaffected by its mutation. The pyrazolourea ligand
29 was thus used as a reference scaffold for hybrid-inhibitor development.
The covalent 4-(phenylamino)quinazoline inhibitor (compound
31a,
Figure S5), previously developed by the same research group was used as the second scaffold. The quinazoline inhibitor showed IC
50 values of 0.23 µM and 5.71 µM for wild-type and drug-resistant (T341M) c-Src, respectively [
36]. The X-ray structure of c-Src in complex with compound
29 (PDB ID: 3F3U) was superimposed with the c-Src-
31a co-crystal structure (PDB ID: 2QLQ), which showed that the aniline moieties of both inhibitors were well aligned to each other in the vicinity of the gatekeeper residue. The alignment suggested that more potent inhibitors could be generated by fusing the scaffolds of the two compounds via 1,3 or 1,4-disubstituted aryl linkage. Docking studies suggested that 1,4-para hybrid compounds would be better accommodated into the c-Src active site, provided by optimal geometry to avoid steric clashes with the larger gatekeeper residue. Thus, the 1,4-substitution pattern was principally exploited to design and synthesize a small library of compounds with a proposed type-II inhibitor binding mode. However, few 1,3-disubstituted hybrids were also designed and synthesized for comparison studies. The synthesized 1,4-substituted hybrids showed at least a fourfold improvement in activity compared to the inhibition potencies of both parent ligands. Most importantly, 1,4-substituted hybrids showed no loss of potency against the drug-resistant T341M c-Src while 1,3-substituted hybrids demonstrated weak inhibition of wild-type c-Src kinase and complete loss of activity against drug-resistant T341M c-Src.
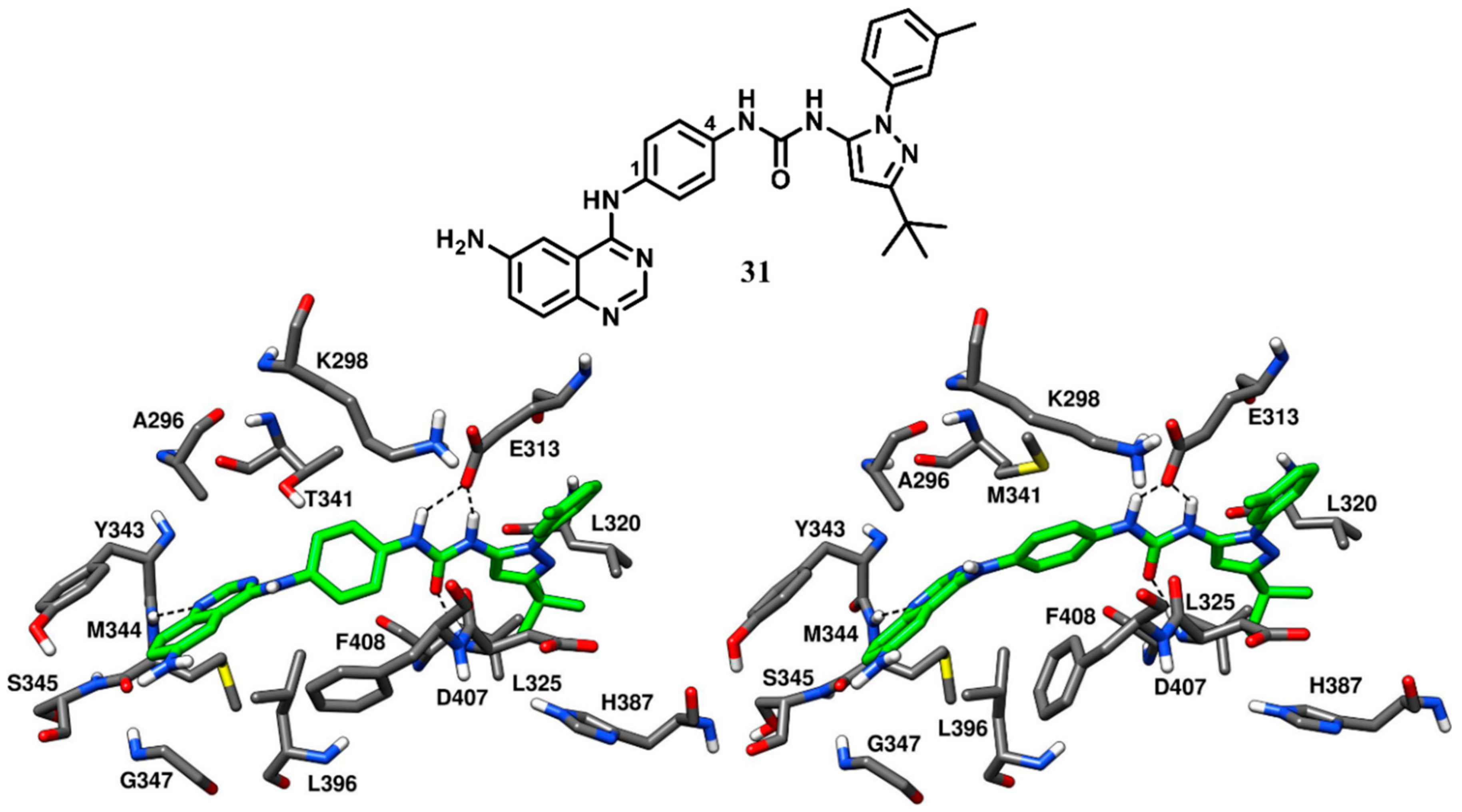
One of the 1,4-substitued hybrid inhibitors (compound
31,
Figure 24) was co-crystallized with both wild-type and drug-resistant T341M c-Src (PDB ID: 3F3V and 3F3W, respectively) to get deeper insights into its binding mode. The IC
50 values of compound
31 were found to be 21 nM and 34 nM against wild-type c-Src and drug-resistant T341M c-Src, respectively [
71]. Compound
31 was bound to the DFG-out inactive conformation of the kinase, where an outward flip of F408 of the DFG motif was observed. The quinazoline core of compound
31 occupied the ATP-binding site of both wild-type and drug-resistant c-Src, showing classical H-bond interactions with the backbone nitrogen of M344 in the hinge-region. The central phenyl ring that links the quinazoline and pyrazolourea scaffolds of the parent compounds was sandwiched between the gatekeeper residue and the side chain of F408 of the DFG motif, forming π-π staking interactions with this latter residue. The presence of the larger gatekeeper residue in the T341M c-Src forced the central phenyl ring of the inhibitor to rotate by 90 degrees in order avoid steric clashes with the side chain of M341. The side of chain of F408 was also rotated by 90 degrees to conserve the edge-to-face π-π stacking with the phenyl ring of the inhibitor [
84]. The urea group of the inhibitors displayed H-bond interactions with the carboxylic group of E313 and the backbone nitrogen of D407, while the adjacent pyrazole ring was placed within the DFG pocket in both wild-type and T34IM mutant c-Src, as observed for the parent compounds
29 and
30. The C-3 tert-butyl group of the inhibitor showed van der Waals interactions with L320, L325 and H387, while the 3-amino phenyl ring displayed van der Waals contacts with the sidechains of E313 and V316.
Docking results suggested that the 1,3-substituted hybrid (compound
32,
Figure S5) was tolerated in the wild-type c-Src. However, in the drug-resistant T341M c-Src, the rotation of the central phenyl core in compound
32 was not possible without disrupting the binding orientation of either the quinazoline or the pyrazolourea moiety. The decreased inhibitor flexibility could result in the loss of H-bonds or the displacement of the pyrazolourea from the allosteric site, in order to adapt the ligand conformation to the larger gatekeeper residue. This explains why compound
32 was not able to bind the mutated c-Src. Therefore, the 1,4-substitution pattern employed in the development of quinazoline-pyrazolourea hybrid inhibitors proved to be a robust rational to overcome T341M c-Src drug resistance with significantly improved potency.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}























