Extracellular Vesicle-Based Communication May Contribute to the Co-Evolution of Cancer Stem Cells and Cancer-Associated Fibroblasts in Anti-Cancer Therapy

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Tumor-Stroma Co-Evolution Requires CAF Survival, Recruitment, and Differentiation
2.1. Therapy-Resistant Subpopulations of Carcinoma-Associated Fibroblasts
2.2. Treatment-Related Replenishment of CAFs
2.3. Differential Cellular Behavior of Resting and Chemotherapy-Treated CAFs
3. Extracellular Vesicle-Mediated Resistance at the Tumor Cell-CAF Interface
3.1. Small EV-Induced Resistance in the CAF-Cancer Cell/CSC Axis
3.2. Medium-Sized EV Communication in Resistance Development of Tumor Cells and CAFs
3.3. Apoptotic Cell-Derived EVs in the Development of Resistance, Relapse, CSC Phenotype and Metastasis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Easwaran, H.; Tsai, H.C.; Baylin, S.B. Cancer epigenetics: Tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell 2014, 54, 716–727. [Google Scholar] [CrossRef] [Green Version]
- Hinohara, K.; Polyak, K. Intratumoral Heterogeneity: More than Just Mutations. Trends Cell Biol. 2019, 29, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Luskin, M.R.; Murakami, M.A.; Manalis, S.R.; Weinstock, D.M. Targeting minimal residual disease: A path to cure? Nat. Rev. Cancer 2018, 18, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Ghiaur, G.; Gerber, J.; Jones, R.J. Concise review: Cancer stem cells and minimal residual disease. Stem Cells 2012, 30, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.Y.; Tang, J.N.; Xie, H.X.; Du, Y.A.; Huang, L.; Yu, P.F.; Cheng, X.D. 5-Fluorouracil chemotherapy of gastric cancer generates residual cells with properties of cancer stem cells. Int. J. Biol. Sci. 2015, 11, 284–294. [Google Scholar] [CrossRef] [Green Version]
- Kyrochristos, I.D.; Ziogas, D.E.; Roukos, D.H. Drug resistance: Origins, evolution and characterization of genomic clones and the tumor ecosystem to optimize precise individualized therapy. Drug Discov. Today 2019, 24, 1281–1294. [Google Scholar] [CrossRef]
- Tang, D.G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012, 22, 457–472. [Google Scholar] [CrossRef]
- Li, S.C.; Lee, K.L.; Luo, J.; Zhong, J.F.; Loudon, W.G. Convergence of normal stem cell and cancer stem cell developmental stage: Implication for differential therapies. World J. Stem Cells 2011, 3, 83–88. [Google Scholar] [CrossRef]
- Kim, W.T.; Ryu, C.J. Cancer stem cell surface markers on normal stem cells. BMB Rep. 2017, 50, 285–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells—Perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedmann-Morvinski, D.; Verma, I.M. Dedifferentiation and reprogramming: Origins of cancer stem cells. EMBO Rep. 2014, 15, 244–253. [Google Scholar] [CrossRef] [Green Version]
- Clarke, M.F.; Fuller, M. Stem cells and cancer: Two faces of eve. Cell 2006, 124, 1111–1115. [Google Scholar] [CrossRef] [Green Version]
- Turdo, A.; Veschi, V.; Gaggianesi, M.; Chinnici, A.; Bianca, P.; Todaro, M.; Stassi, G. Meeting the Challenge of Targeting Cancer Stem Cells. Front. Cell Dev. Biol. 2019, 7, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fábián, A.; Barok, M.; Vereb, G.; Szöllosi, J. Die hard: Are cancer stem cells the Bruce Willises of tumor biology? Cytom. Part A J. Int. Soc. Adv. Cytom. 2009, 75, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M. Cancer stem cells as ‘units of selection’. Evol. Appl. 2013, 6, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Boesch, M.; Sopper, S.; Zeimet, A.G.; Reimer, D.; Gastl, G.; Ludewig, B.; Wolf, D. Heterogeneity of Cancer Stem Cells: Rationale for Targeting the Stem Cell Niche. Biochim. Biophys. Acta Rev. Cancer 2016, 1866, 276–289. [Google Scholar] [CrossRef] [Green Version]
- Wicha, M.S.; Liu, S.; Dontu, G. Cancer stem cells: An old idea—A paradigm shift. Cancer Res. 2006, 66, 1883–1890. [Google Scholar] [CrossRef] [Green Version]
- Peitzsch, C.; Tyutyunnykova, A.; Pantel, K.; Dubrovska, A. Cancer stem cells: The root of tumor recurrence and metastases. Semin. Cancer Biol. 2017, 44, 10–24. [Google Scholar] [CrossRef]
- Lagasse, E. Cancer stem cells with genetic instability: The best vehicle with the best engine for cancer. Gene Ther. 2008, 15, 136–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlyukov, M.S.; Yu, H.; Bastola, S.; Minata, M.; Shender, V.O.; Lee, Y.; Zhang, S.; Wang, J.; Komarova, S.; Wang, J.; et al. Apoptotic Cell-Derived Extracellular Vesicles Promote Malignancy of Glioblastoma via Intercellular Transfer of Splicing Factors. Cancer Cell 2018, 34, 119–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwörer, S.; Vardhana, S.A.; Thompson, C.B. Cancer Metabolism Drives a Stromal Regenerative Response. Cell Metab. 2019, 29, 576–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1989, 133, 571–573. [Google Scholar] [CrossRef] [Green Version]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Ligorio, M.; Sil, S.; Malagon-Lopez, J.; Nieman, L.T.; Misale, S.; Di Pilato, M.; Ebright, R.Y.; Karabacak, M.N.; Kulkarni, A.S.; Liu, A.; et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019, 178, 160–175. [Google Scholar] [CrossRef]
- Kiskowski, M.A.; Jackson, R.S.; Banerjee, J.; Li, X.; Kang, M.; Iturregui, J.M.; Franco, O.E.; Hayward, S.W.; Bhowmick, N.A. Role for stromal heterogeneity in prostate tumorigenesis. Cancer Res. 2011, 71, 3459–3470. [Google Scholar] [CrossRef] [Green Version]
- Casazza, A.; Di Conza, G.; Wenes, M.; Finisguerra, V.; Deschoemaeker, S.; Mazzone, M. Tumor stroma: A complexity dictated by the hypoxic tumor microenvironment. Oncogene 2014, 33, 1743–1754. [Google Scholar] [CrossRef] [Green Version]
- Hira, V.V.V.; Van Noorden, C.J.F.; Molenaar, R.J. CXCR4 Antagonists as Stem Cell Mobilizers and Therapy Sensitizers for Acute Myeloid Leukemia and Glioblastoma? Biology 2020, 9, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Q.; Yun, Z. Impact of the hypoxic tumor microenvironment on the regulation of cancer stem cell characteristics. Cancer Biol. Ther. 2010, 9, 949–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morikawa, T.; Takubo, K. Hypoxia regulates the hematopoietic stem cell niche. Pflüg. Arch. Eur. J. Physiol. 2016, 468, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-Hoff, M.H.; Park, C.; Wright, E.G. Radiation and the microenvironment—Tumorigenesis and therapy. Nat. Rev. Cancer 2005, 5, 867–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856. [Google Scholar] [CrossRef] [PubMed]
- Hirata, E.; Sahai, E. Tumor Microenvironment and Differential Responses to Therapy. Cold Spring Harb. Perspect. Med. 2017, 7, a026781. [Google Scholar] [CrossRef] [Green Version]
- Vitale, I.; Manic, G.; Galassi, C.; Galluzzi, L. Stress responses in stromal cells and tumor homeostasis. Pharmacol. Ther. 2019, 200, 55–68. [Google Scholar] [CrossRef]
- Klemm, F.; Joyce, J.A. Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 2015, 25, 198–213. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug. Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]
- D’Arcangelo, E.; Wu, N.C.; Cadavid, J.L.; McGuigan, A.P. The life cycle of cancer-associated fibroblasts within the tumour stroma and its importance in disease outcome. Br. J. Cancer 2020, 122, 931–942. [Google Scholar] [CrossRef]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef] [Green Version]
- Valcz, G.; Patai, A.V.; Kalmár, A.; Péterfia, B.; Fűri, I.; Wichmann, B.; Műzes, G.; Sipos, F.; Krenács, T.; Mihály, E.; et al. Myofibroblast-derived SFRP1 as potential inhibitor of colorectal carcinoma field effect. PLoS ONE 2014, 9, e106143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef] [Green Version]
- Dylla, S.J.; Beviglia, L.; Park, I.K.; Chartier, C.; Raval, J.; Ngan, L.; Pickell, K.; Aguilar, J.; Lazetic, S.; Smith-Berdan, S.; et al. Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS ONE 2008, 3, e2428. [Google Scholar] [CrossRef]
- Jia, D.; Tan, Y.; Liu, H.; Ooi, S.; Li, L.; Wright, K.; Bennett, S.; Addison, C.L.; Wang, L. Cardamonin reduces chemotherapy-enriched breast cancer stem-like cells in vitro and in vivo. Oncotarget 2016, 7, 771–785. [Google Scholar] [CrossRef] [Green Version]
- Peiris-Pagès, M.; Sotgia, F.; Lisanti, M.P. Chemotherapy induces the cancer-associated fibroblast phenotype, activating paracrine Hedgehog-GLI signalling in breast cancer cells. Oncotarget 2015, 10, 10728–10745. [Google Scholar] [CrossRef] [Green Version]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.; Takashi, S.; Baik, G.H.; Shibata, W.; Diprete, B.; Betz, K.S.; et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef] [Green Version]
- Winkler, F.; Wick, W. Harmful networks in the brain and beyond. Science 2018, 359, 1100–1101. [Google Scholar] [CrossRef]
- Roehlecke, C.; Schmidt, M.H.H. Tunneling Nanotubes and Tumor Microtubes in Cancer. Cancers 2020, 12, 857. [Google Scholar] [CrossRef] [Green Version]
- Resnik, N.; Erman, A.; Veranič, P.; Kreft, M.E. Triple labelling of actin filaments, intermediate filaments and microtubules for broad application in cell biology: Uncovering the cytoskeletal composition in tunneling nanotubes. Histochem. Cell Biol. 2019, 152, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Gleisner, M.A.; Navarrete, M.; Hofmann, F.; Salazar-Onfray, F.; Tittarelli, A. Mind the Gaps in Tumor Immunity: Impact of Connexin-Mediated Intercellular Connections. Front. Immunol. 2017, 8, 1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yáñez-Mó, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanada, M.; Bachmann, M.H.; Contag, C.H. Signaling by Extracellular Vesicles Advances Cancer Hallmarks. Trends Cancer 2016, 2, 84–94. [Google Scholar] [CrossRef]
- Samuel, P.; Fabbri, M.; Carter, D.R.F. Mechanisms of Drug Resistance in Cancer: The Role of Extracellular Vesicles. Proteomics 2017, 17, 23–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Li, M.; Yin, T.; Shi, H.; Wen, Y.; Zhang, B.; Chen, M.; Xu, G.; Ren, K.; Wei, Y. Targeting of cancer-associated fibroblasts enhances the efficacy of cancer chemotherapy by regulating the tumor microenvironment. Mol. Med. Rep. 2016, 13, 2476–2484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotti, F.; Jarrar, A.M.; Pai, R.K.; Hitomi, M.; Lathia, J.; Mace, A.; Gantt, G.A., Jr.; Sukhdeo, K.; DeVecchio, J.; Vasanji, A.; et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by IL-17A. J. Exp. Med. 2013, 210, 2851–2872. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Tang, Y.; Tan, Y.; Wei, Q.; Yu, W. Cancer-associated fibroblasts in radiotherapy: Challenges and new opportunities. Cell Commun. Signal. 2019, 17, 47. [Google Scholar] [CrossRef] [Green Version]
- Petersen, O.W.; Nielsen, H.L.; Gudjonsson, T.; Villadsen, R.; Rank, F.; Niebuhr, E.; Bissell, M.J.; Ronnov-Jessen, L. Epithelial to mesenchymal transition in human breast cancer can provide a nonmalignant stroma. Am. J. Pathol. 2003, 162, 391–402. [Google Scholar] [CrossRef] [Green Version]
- Mink, S.R.; Vashistha, S.; Zhang, W.; Hodge, A.; Agus, D.B.; Jain, A. Cancer-associated fibroblasts derived from EGFR-TKI-resistant tumors reverse EGFR pathway inhibition by EGFR-TKIs. Mol. Cancer Res. 2010, 8, 809–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvořánková, B.; Smetana, K.J.; Říhová, B.; Kučera, J.; Mateu, R.; Szabo, P. Cancer-associated fibroblasts are not formed from cancer cells by epithelial-to-mesenchymal transition in nu/nu mice. Histochem. Cell Biol. 2015, 143, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Ok Lee, S.; Liang, L.; Huang, C.K.; Li, L.; Wen, S.; Chang, C. Infiltrating bone marrow mesenchymal stem cells increase prostate cancer stem cell population and metastatic ability via secreting cytokines to suppress androgen receptor signaling. Oncogene 2014, 33, 2768–2778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camorani, S.; Hill, B.S.; Fontanella, R.; Greco, A.; Gramanzini, M.; Auletta, L.; Gargiulo, S.; Albanese, S.; Lucarelli, E.; Cerchia, L.; et al. Inhibition of Bone Marrow-Derived Mesenchymal Stem Cells Homing Towards Triple-Negative Breast Cancer Microenvironment Using an Anti-PDGFRβ Aptamer. Theranostics 2017, 7, 3595–3607. [Google Scholar] [CrossRef]
- Bergfeld, S.A.; Blavier, L.; DeClerck, Y.A. Bone marrow-derived mesenchymal stromal cells promote survival and drug resistance in tumor cells. Mol. Cancer Ther. 2014, 13, 962–975. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.J.; Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008, 68, 4331–4339. [Google Scholar] [CrossRef] [Green Version]
- Paunescu, V.; Bojin, F.M.; Tatu, C.A.; Gavriliuc, O.I.; Rosca, A.; Gruia, A.T.; Tanasie, G.; Bunu, C.; Crisnic, D.; Gherghiceanu, M.; et al. Tumour-associated fibroblasts and mesenchymal stem cells: More similarities than differences. J. Cell Mol. Med. 2011, 15, 635–646. [Google Scholar] [CrossRef]
- Borriello, L.; Nakata, R.; Sheard, M.A.; Fernandez, G.E.; Sposto, R.; Malvar, J.; Blavier, L.; Shimada, H.; Asgharzadeh, S.; Seeger, R.C.; et al. Cancer-Associated Fibroblasts Share Characteristics and Protumorigenic Activity with Mesenchymal Stromal Cells. Cancer Res. 2017, 77, 5142–5157. [Google Scholar] [CrossRef] [Green Version]
- Shintani, Y.; Fujiwara, A.; Kimura, T.; Kawamura, T.; Funaki, S.; Minami, M.; Okumura, M. IL-6 Secreted from Cancer-Associated Fibroblasts Mediates Chemoresistance in NSCLC by Increasing Epithelial-Mesenchymal Transition Signaling. J. Thorac. Oncol. 2016, 11, 1482–1492. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Miyata, H.; Sugimura, K.; Fukuda, S.; Kanemura, T.; Yamashita, K.; Miyazaki, Y.; Takahashi, T.; Kurokawa, Y.; Yamasaki, M.; et al. miR-27 is associated with chemoresistance in esophageal cancer through transformation of normal fibroblasts to cancer-associated fibroblasts. Carcinogenesis 2015, 36, 894–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodningen, O.K.; Overgaard, J.; Alsner, J.; Hastie, T.; Borresen-Dale, A.L. Microarray analysis of the transcriptional response to single or multiple doses of ionizing radiation in human subcutaneous fibroblasts. Radiother. Oncol. 2005, 77, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Steer, A.; Cordes, N.; Jendrossek, V.; Klein, D. Impact of Cancer-Associated Fibroblast on the Radiation-Response of Solid Xenograft Tumors. Front. Mol. Biosci. 2019, 6, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, T.S.; Hsu, C.C.; Pai, V.C.; Liao, W.Y.; Huang, S.S.; Tan, K.T.; Yen, C.J.; Hsu, S.C.; Chen, W.Y.; Shan, Y.S.; et al. Metronomic chemotherapy prevents therapy-induced stromal activation and induction of tumor-initiating cells. J. Exp. Med. 2016, 213, 2967–2988. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Huang, G.; Wang, R.; Pan, Y.; He, Z.; Chu, X.; Song, H.; Chen, L. Cancer-associated fibroblasts treated with cisplatin facilitates chemoresistance of lung adenocarcinoma through IL-11/IL-11R/STAT3 signaling pathway. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Grinde, M.T.; Vik, J.; Camilio, K.A.; Martinez-Zubiaurre, I.; Hellevik, T. Ionizing radiation abrogates the pro-tumorigenic capacity of cancer-associated fibroblasts co-implanted in xenografts. Sci. Rep. 2017, 7, 46714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohuchida, K.; Mizumoto, K.; Murakami, M.; Qian, L.W.; Sato, N.; Nagai, E.; Matsumoto, K.; Nakamura, T.; Tanaka, M. Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor-stromal interactions. Cancer Res. 2004, 64, 3215–3222. [Google Scholar] [CrossRef] [Green Version]
- Tommelein, J.; De Vlieghere, E.; Verset, L.; Melsens, E.; Leenders, J.; Descamps, B.; Debucquoy, A.; Vanhove, C.; Pauwels, P.; Gespach, C.P.; et al. Radiotherapy-Activated Cancer-Associated Fibroblasts Promote Tumor Progression through Paracrine IGF1R Activation. Cancer Res. 2018, 78, 659–670. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Valcz, G.; Buzás, E.I.; Szállási, Z.; Kalmár, A.; Krenács, T.; Tulassay, Z.; Igaz, P.; Molnár, B. Perspective: Bidirectional exosomal transport between cancer stem cells and their fibroblast-rich microenvironment during metastasis formation. NPJ Breast Cancer 2018, 4, 1–7. [Google Scholar] [CrossRef]
- Suchorska, W.M.; Lach, M.S. The role of exosomes in tumor progression and metastasis. Oncol. Rep. 2016, 35, 1237–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowal, J.; Tkach, M.; Théry, C. Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 2014, 29, 116–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Paine, M.S.; Brooks, A.M.; McCubrey, J.A.; Renegar, R.H.; Wang, R.; Terrian, D.M. Senescence-associated exosome release from human prostate cancer cells. Cancer Res. 2008, 68, 7864–7871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jelonek, K.; Widlak, P.; Pietrowska, M. The Influence of Ionizing Radiation on Exosome Composition, Secretion and Intercellular Communication. Protein Pept. Lett. 2016, 23, 656–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Harris, S.L.; Levine, A.J. The regulation of exosome secretion: A novel function of the p53 protein. Cancer Res. 2006, 66, 4795–4801. [Google Scholar] [CrossRef] [Green Version]
- Au Yeung, C.L.; Co, N.N.; Tsuruga, T.; Yeung, T.L.; Kwan, S.Y.; Leung, C.S.; Li, Y.; Lu, E.S.; Kwan, K.; Wong, K.K.; et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat. Commun. 2016, 7, 11150. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Ruan, H.; Zhang, X.; Xu, X.; Zhu, Y.; Peng, H.; Zhang, X.; Kong, F.; Guan, M. Long noncoding RNA CCAL transferred from fibroblasts by exosomes promotes chemoresistance of colorectal cancer cells. Int. J. Cancer 2019, 146, 1700–1716. [Google Scholar] [CrossRef]
- Kreger, B.T.; Johansen, E.R.; Cerione, R.A.; Antonyak, M.A. The Enrichment of Survivin in Exosomes from Breast Cancer Cells Treated with Paclitaxel Promotes Cell Survival and Chemoresistance. Cancers 2016, 8, 111. [Google Scholar] [CrossRef] [Green Version]
- Boelens, M.C.; Wu, T.J.; Nabet, B.Y.; Xu, B.; Qiu, Y.; Yoon, T.; Azzam, D.J.; Twyman-Saint Victor, C.; Wiemann, B.Z.; Ishwaran, H.; et al. Exosome Transfer from Stromal to Breast Cancer Cells Regulates Therapy Resistance Pathways. Cell 2014, 159, 499–513. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Yan, C.; Mu, L.; Huang, K.; Li, X.; Tao, D.; Wu, Y.; Qin, J. Fibroblast-Derived Exosomes Contribute to Chemoresistance through Priming Cancer Stem Cells in Colorectal Cancer. PLoS ONE 2015, 10, e0125625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.B.; Yan, C.; Mu, L.; Mi, Y.L.; Zhao, H.; Hu, H.; Li, X.L.; Tao, D.D.; Wu, Y.Q.; Gong, J.P.; et al. Exosomal Wnt-induced dedifferentiation of colorectal cancer cells contributes to chemotherapy resistance. Oncogene 2019, 38, 1951–1965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Göktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeilstra, J.; Joosten, S.P.; Dokter, M.; Verwiel, E.; Spaargaren, M.; Pals, S.T. Deletion of the WNT target and cancer stem cell marker CD44 in Apc(Min/+) mice attenuates intestinal tumorigenesis. Cancer Res. 2008, 68, 3655–3661. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.L.; Wang, W.; Lan, X.L.; Zeng, Z.C.; Liang, Y.S.; Yan, Y.R.; Song, F.Y.; Wang, F.F.; Zhu, X.H.; Liao, W.J.; et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol. Cancer 2019, 8, 91. [Google Scholar] [CrossRef] [Green Version]
- Ab Razak, N.S.; Ab Mutalib, N.S.; Mohtar, M.A.; Abu, N. Impact of Chemotherapy on Extracellular Vesicles: Understanding the Chemo-EVs. Front. Oncol. 2019, 9, 1113. [Google Scholar] [CrossRef]
- Ren, J.; Ding, L.; Zhang, D.; Shi, G.; Xu, Q.; Shen, S.; Wang, Y.; Wang, T.; Hou, Y. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics 2018, 8, 3932–3948. [Google Scholar] [CrossRef]
- Guo, H.; Ha, C.; Dong, H.; Yang, Z.; Ma, Y.; Ding, Y. Cancer-associated fibroblast-derived exosomal microRNA-98-5p promotes cisplatin resistance in ovarian cancer by targeting CDKN1A. Cancer Cell Int. 2019, 19, 1–15. [Google Scholar] [CrossRef]
- Qin, X.; Guo, H.; Wang, X.; Zhu, X.; Yan, M.; Wang, X.; Xu, Q.; Shi, J.; Lu, E.; Chen, W.; et al. Exosomal miR-196a derived from cancer-associated fibroblasts confers cisplatin resistance in head and neck cancer through targeting CDKN1B and ING5. Genome Biol. 2019, 20, 12. [Google Scholar] [CrossRef]
- Castellana, D.; Zobairi, F.; Martinez, M.C.; Panaro, M.A.; Mitolo, V.; Freyssinet, J.M.; Kunzelmann, C. Membrane microvesicles as actors in the establishment of a favorable prostatic tumoral niche: A role for activated fibroblasts and CX3CL1-CX3CR1 axis. Cancer Res. 2009, 69, 785–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wysoczynski, M.; Ratajczak, M.Z. Lung cancer secreted microvesicles: Underappreciated modulators of microenvironment in expanding tumors. Int. J. Cancer 2009, 25, 1595–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minciacchi, V.R.; Freeman, M.R.; Di Vizio, D. Extracellular vesicles in cancer: Exosomes, microvesicles and the emerging role of large oncosomes. Semin. Cell Dev. Biol. 2015, 40, 41–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santi, A.; Caselli, A.; Ranaldi, F.; Paoli, P.; Mugnaioni, C.; Michelucci, E.; Cirri, P. Cancer associated fibroblasts transfer lipids and proteins to cancer cells through cargo vesicles supporting tumor growth. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 3211–3223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, E.; Xu, Z.; Wang, M.; Yan, T.; Huang, C.; Zhou, X.; Liu, Q.; Wang, L.; Chen, Y.; Wang, H.; et al. Tumoral microvesicle-activated glycometabolic reprogramming in fibroblasts promotes the progression of oral squamous cell carcinoma. FASEB J. 2019, 33, 5690–5703. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Lin, Z.; Ko, Y.H.; Goldberg, A.F.; Flomenberg, N.; Wang, C.; Pavlides, S.; Pestell, R.G.; Howell, A.; Sotgia, F.; et al. Understanding the metabolic basis of drug resistance: Therapeutic induction of the Warburg effect kills cancer cells. Cell Cycle 2011, 10, 2521–2528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Q.; Peng, J.; Liu, W.; He, X.; Cui, L.; Chen, X.; Yang, M.; Liu, H.; Liu, S.; Wang, H. Elevated cleaved caspase-3 is associated with shortened overall survival in several cancer types. Int. J. Clin. Exp. Pathol. 2014, 7, 5057–5070. [Google Scholar]
- Huang, Q.; Li, F.; Liu, X.; Li, W.; Shi, W.; Liu, F.F.; O’Sullivan, B.; He, Z.; Peng, Y.; Tan, A.C.; et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 2011, 17, 860–866. [Google Scholar] [CrossRef]
- Lynch, C.; Panagopoulou, M.; Gregory, C.D. Extracellular Vesicles Arising from Apoptotic Cells in Tumors: Roles in Cancer Pathogenesis and Potential Clinical Applications. Front. Immunol. 2017, 8, 1174. [Google Scholar] [CrossRef] [Green Version]
- Gregory, C.D.; Dransfield, I. Apoptotic Tumor Cell-Derived Extracellular Vesicles as Important Regulators of the Onco-Regenerative Niche. Front. Immunol. 2018, 9, 1111. [Google Scholar] [CrossRef] [Green Version]
- Bergsmedh, A.; Szeles, A.; Henriksson, M.; Bratt, A.; Folkman, M.J.; Spetz, A.L.; Holmgren, L. Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proc. Natl. Acad. Sci. USA 2001, 98, 6407–6411. [Google Scholar] [CrossRef] [Green Version]
- Ehnfors, J.; Kost-Alimova, M.; Persson, N.L.; Bergsmedh, A.; Castro, J.; Levchenko-Tegnebratt, T.; Yang, L.; Panaretakis, T.; Holmgren, L. Horizontal transfer of tumor DNA to endothelial cells in vivo. Cell Death Differ. 2009, 16, 749–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, G.; Chida, S.; Yanagihara, K.; Yashiro, M.; Aiba, N.; Tanaka, M. Cancer-associated fibroblasts induce cancer cell apoptosis that regulates invasion mode of tumours. Oncogene 2017, 36, 4434–4444. [Google Scholar] [CrossRef] [PubMed]



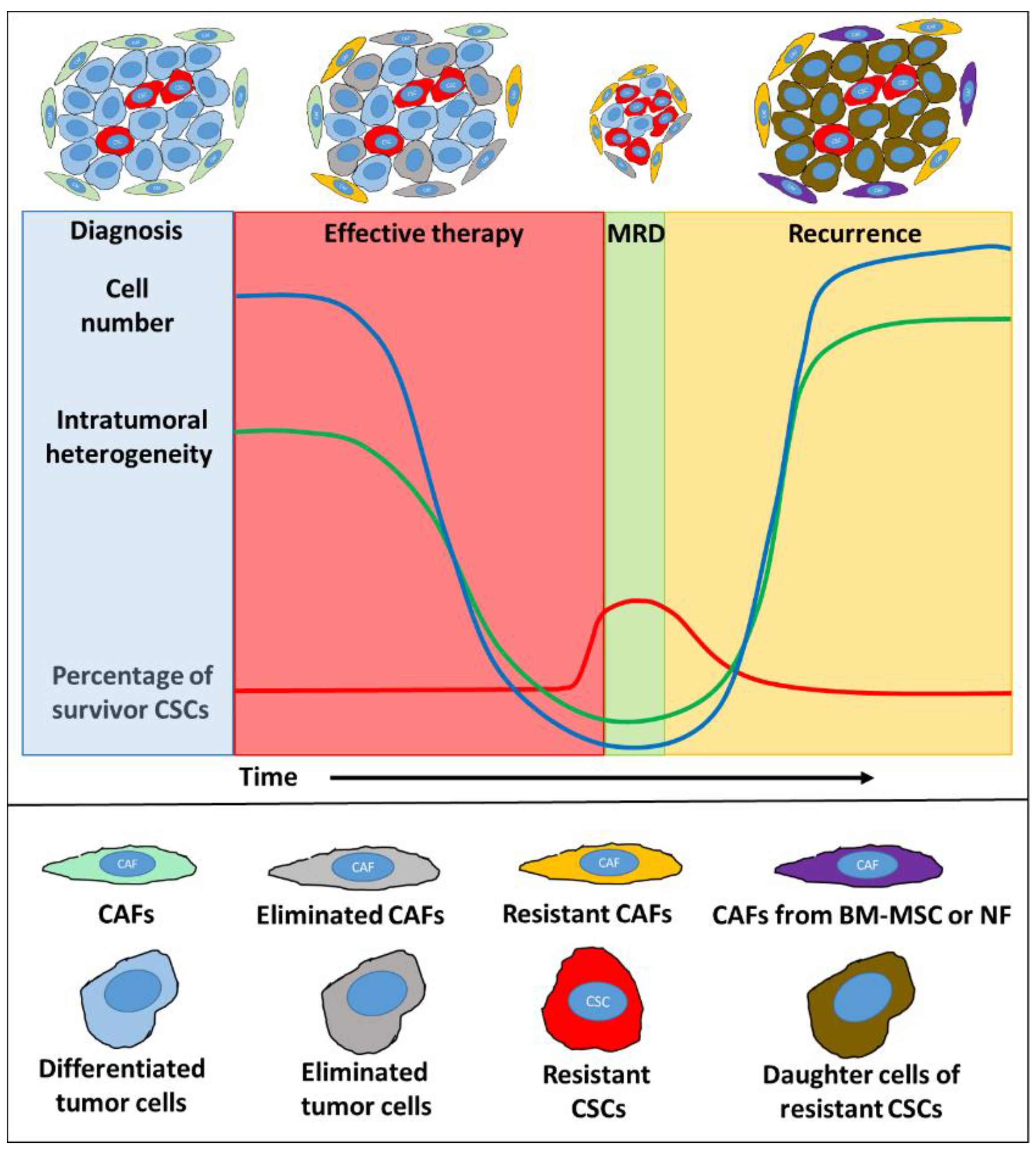{kind=link}
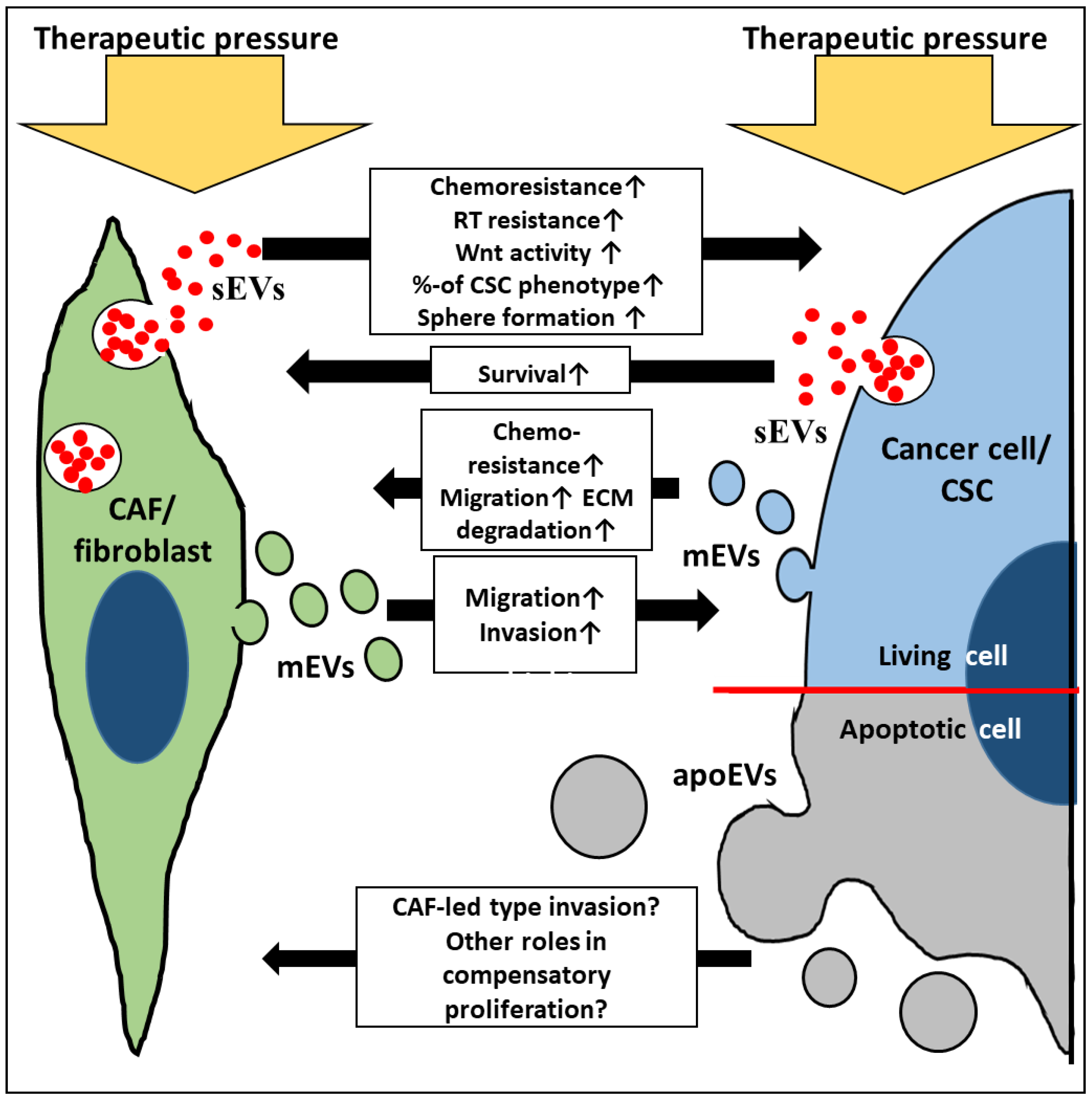{kind=link}
| Tumor Models (Cell Lines) | Origin of Extracellular Vesicles (and Used Terms) | Changes (↑↓) of Resistance- and CSC Properties as Well as Expression of Relevant Molecules and Activation of Pathways in the Recipient Cell Population. | Ref |
|---|---|---|---|
| Colorectal cancer: (SW620), xenograft | CAF-derived sEV(exosome) | Resistance to oxaliplatin and 5-FU↑. percentage of CD133+ CSCs↑, Wnt activity↑, in cancer cells | [91] |
| Colorectal cancer: (HT29, SW620), xenograft | Fibroblast (18Co) and CAF-derived sEVs (exosome) | Resistance to oxaliplatin and 5-FU↑. Wnt activity↑, cancer cell dedifferentiation to CSC↑, sphere formation ability↑ | [92] |
| Colorectal cancer: (HCT116, SW480), xenograft | CAF-derived sEV (exosome) | Resistance to oxaliplatin↑, percentage of CSC marker positive cells↑, sphere formation ability↑ | [97] |
| Colorectal cancer: (SW480, SW620, LOVO), xenograft | CAF-derived sEV (exosome) | Resistance to oxaliplatin and 5-FU↑. EMT and CSC markers↑, Wnt activity↑, mitochondrial apoptosis↓ | [95] |
| Colorectal cancer: (HCT116, SW480), xenograft | CAF-derived sEV (exosome) | Resistance to oxaliplatin and 5-FU↑. colorectal cancer-associated lncRNA↑, Wnt activity↑ | [88] |
| Pancreatic cancer: (PANC1, AsPC1), xenograft | CAF-derived sEV (exosome) | Gemcitabine resistance↑, Snail↑, miR-146a↑, proliferation↑, survival↑ in PaC cells | [83] |
| Ovarian cancer: (OVCA432, SKOV), xenograft | CAF-derived sEV (exosome) | Resistance to paclitaxel↑, miR-21↑, APAF1↓ in ovarian cancer cells | [87] |
| Ovarian cancer: (A2780, SKOV3), xenograft | CAF-derived sEV (exosome) | Resistance to cisplatin↑, CDKN1A↓ in ovarian cancer cells | [98] |
| Breast cancer: (MDA-MB-231), xenograft | CAF-derived sEV (exosome) | Resistance to RT and cisplatin↑, IRDS activation↑, RIG-I signaling↑, CD44+/CD24low CSC subpopulation↑ in basal-like subtypes breast cancer cells | [90] |
| Head and neck cancer: (HN4, CAL27), xenograft | CAF-derived sEV (exosome) | Resistance to cisplatin↑, miR-196a↑, CDKN1B↓, ING5↓ proliferation↑, in recipient cancer cells | [99] |
| Breast cancer cells: (MDAMB23) | Tumor-derived sEV (exosome) | Paclitaxel treated tumor-derived sEVs promotes survival of fibroblasts | [89] |
| Prostate cancer: (PC3, LnCaP) | Tumor-derived mEV, fibroblast-derived mEV (microvesicle) | Tumor-derived mEV treated fibroblasts: chemosensitivity against actinomycin D↓, ERK1/2 phosphorylation↑, MMP9↑, migration↑. Fibroblast-derived mEV treated PC3 cells: migration↑, invasion ↑ | [100] |
| Lung cancer: (LL-2, A549, HTB177), xenograft | Tumor-derived mEV (microvesicle) | Release of IL-1, -6, -8, -11↑, VEGF↑, LIF↑, OSM↑ and MMP9↑ in fibroblasts. Conditioned media of tumor-mEV treated fibroblasts: adhesion between LL-2 and HUVECs cells↑, metastatic potential of lung cancer cells↑ | [101] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valcz, G.; Buzás, E.I.; Sebestyén, A.; Krenács, T.; Szállási, Z.; Igaz, P.; Molnár, B. Extracellular Vesicle-Based Communication May Contribute to the Co-Evolution of Cancer Stem Cells and Cancer-Associated Fibroblasts in Anti-Cancer Therapy. Cancers 2020, 12, 2324. https://doi.org/10.3390/cancers12082324
Valcz G, Buzás EI, Sebestyén A, Krenács T, Szállási Z, Igaz P, Molnár B. Extracellular Vesicle-Based Communication May Contribute to the Co-Evolution of Cancer Stem Cells and Cancer-Associated Fibroblasts in Anti-Cancer Therapy. Cancers. 2020; 12(8):2324. https://doi.org/10.3390/cancers12082324
Chicago/Turabian StyleValcz, Gábor, Edit I. Buzás, Anna Sebestyén, Tibor Krenács, Zoltán Szállási, Péter Igaz, and Béla Molnár. 2020. "Extracellular Vesicle-Based Communication May Contribute to the Co-Evolution of Cancer Stem Cells and Cancer-Associated Fibroblasts in Anti-Cancer Therapy" Cancers 12, no. 8: 2324. https://doi.org/10.3390/cancers12082324