Trabectedin and Lurbinectedin Extend Survival of Mice Bearing C26 Colon Adenocarcinoma, without Affecting Tumor Growth or Cachexia

 , , , ,
, , , ,  add
Show full author list
add
Show full author list
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
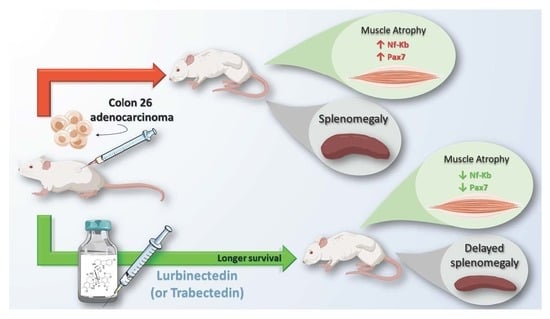
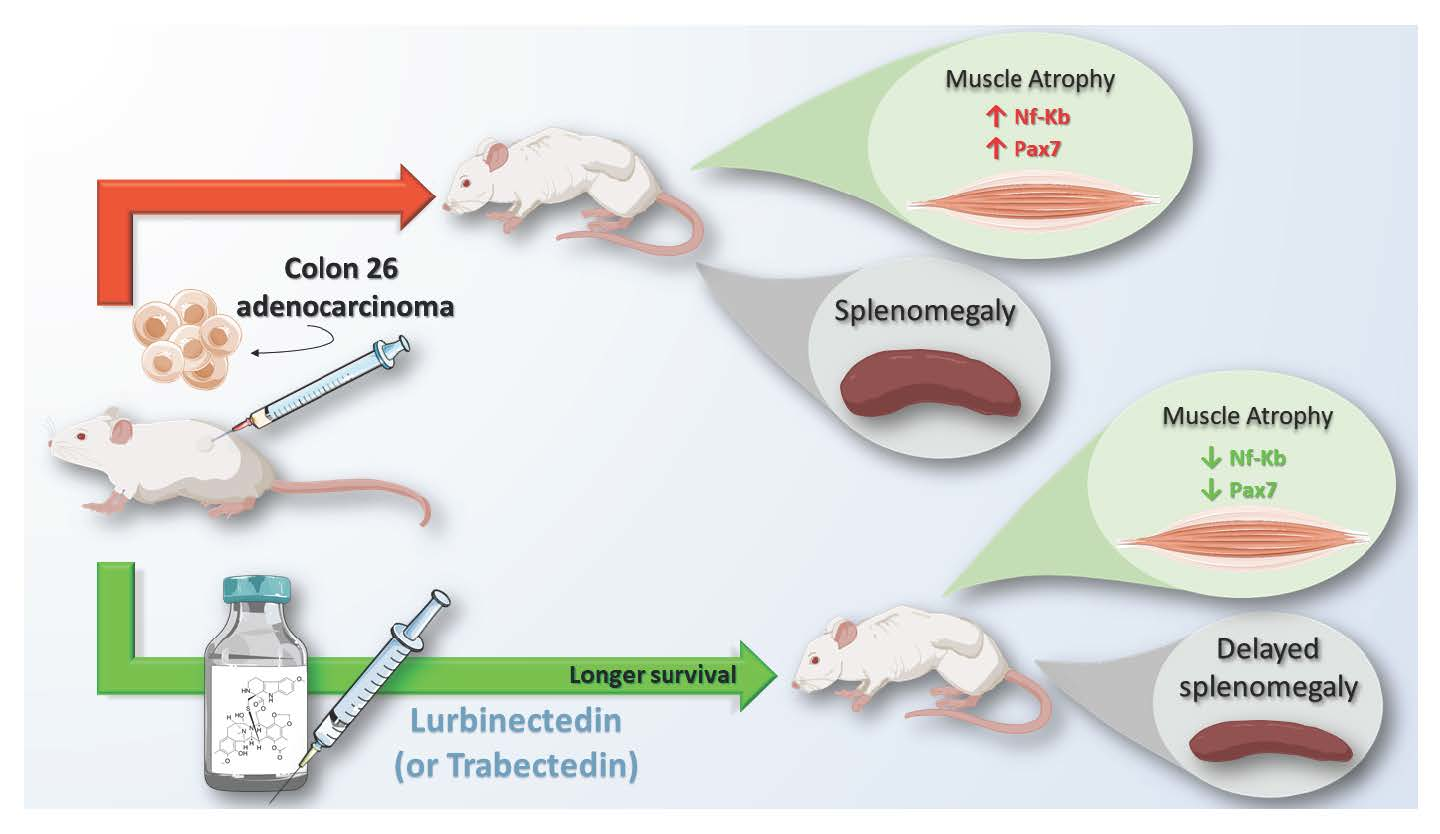
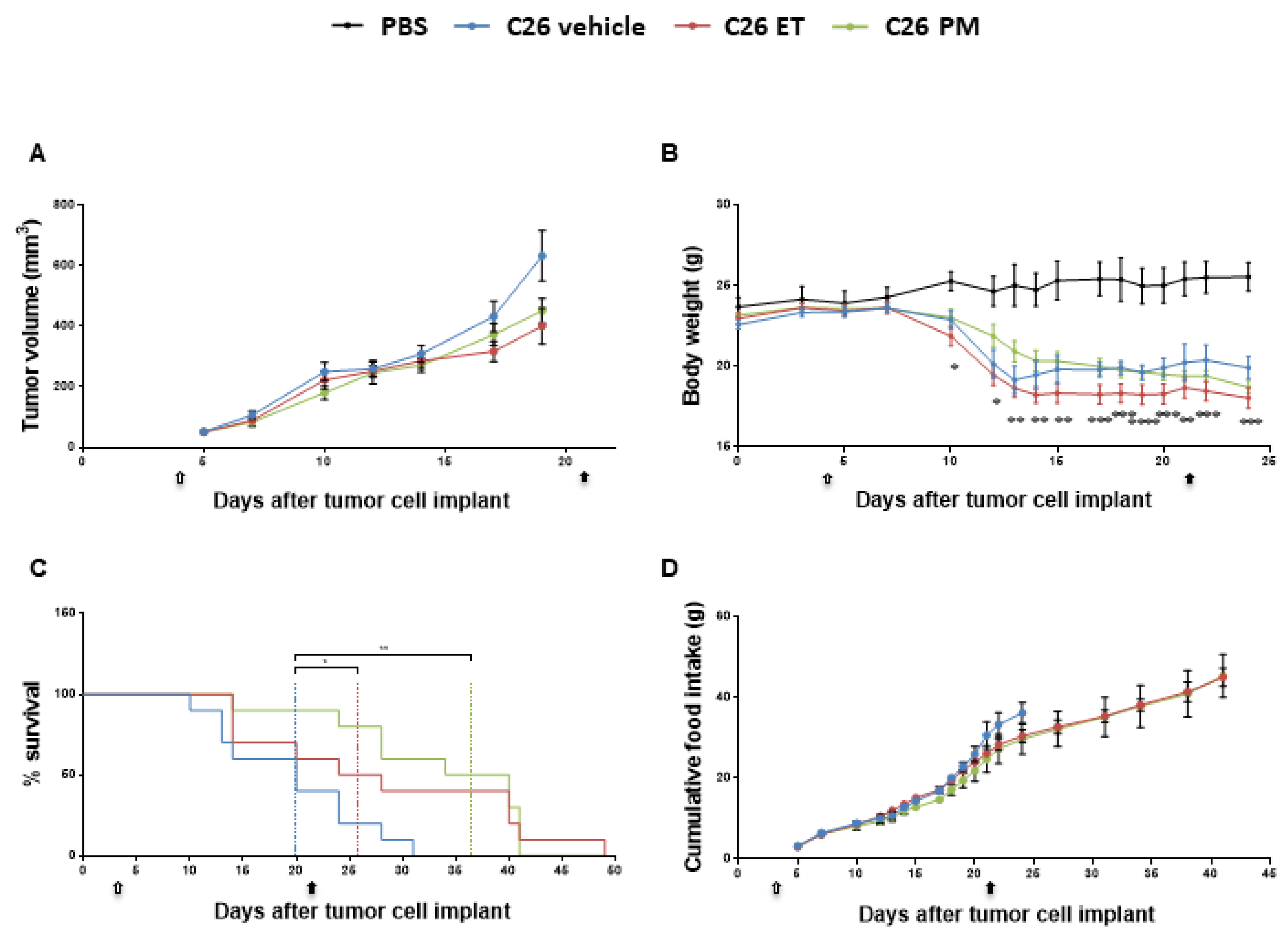
2.1. Trabectedin and Lurbinectedin Impressively Increase the Survival of C26 Tumor-Bearing Mice
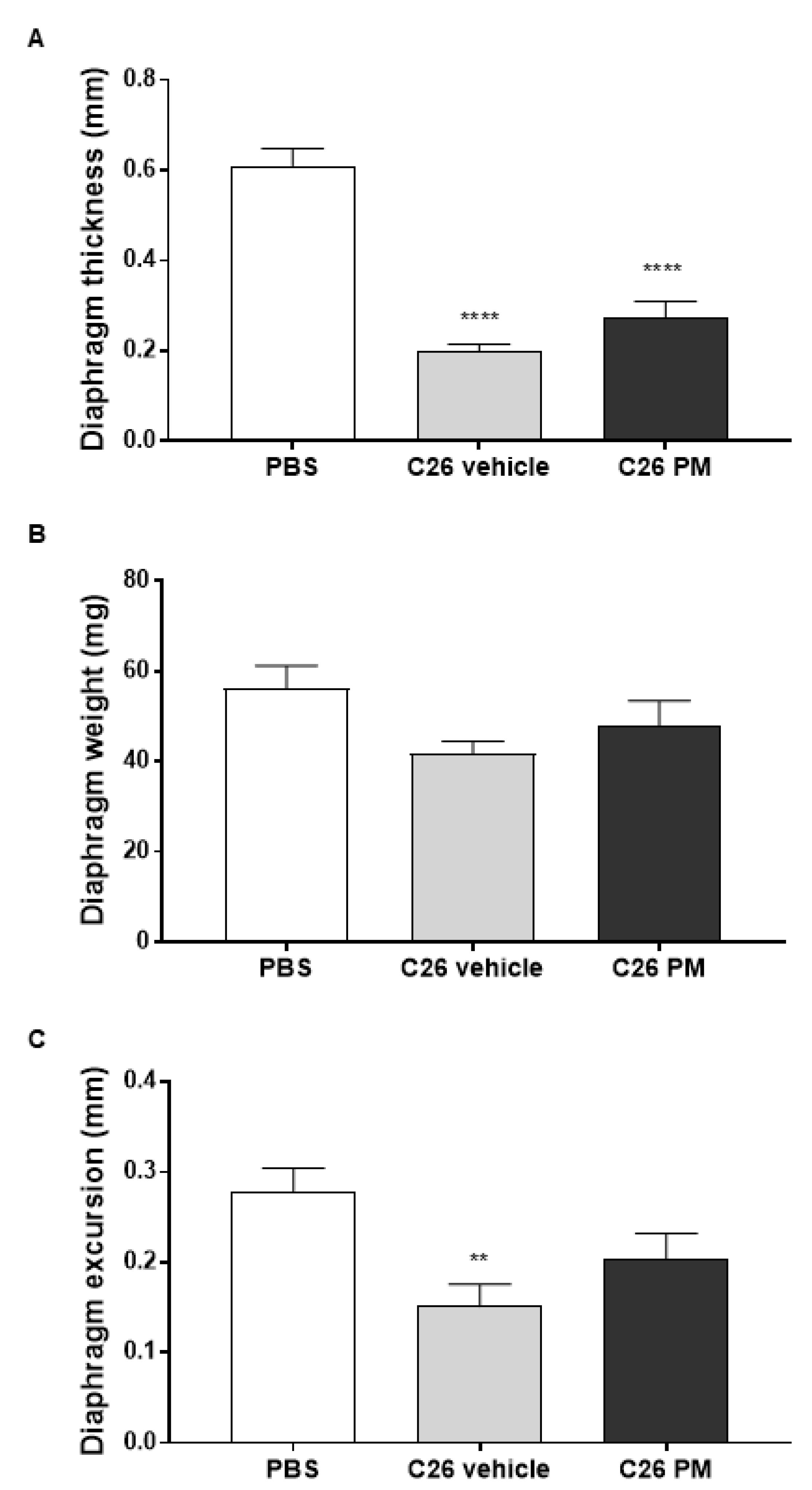
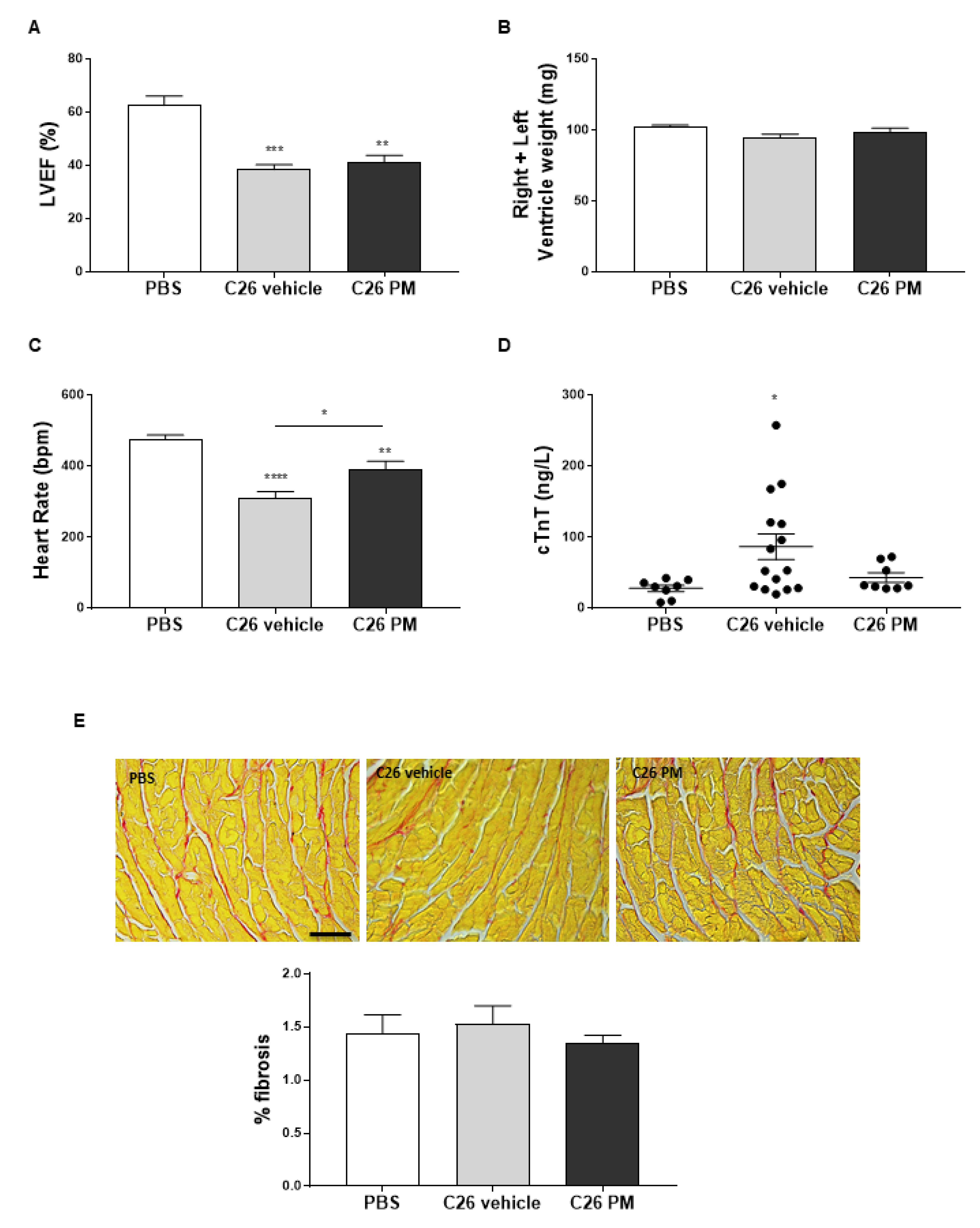
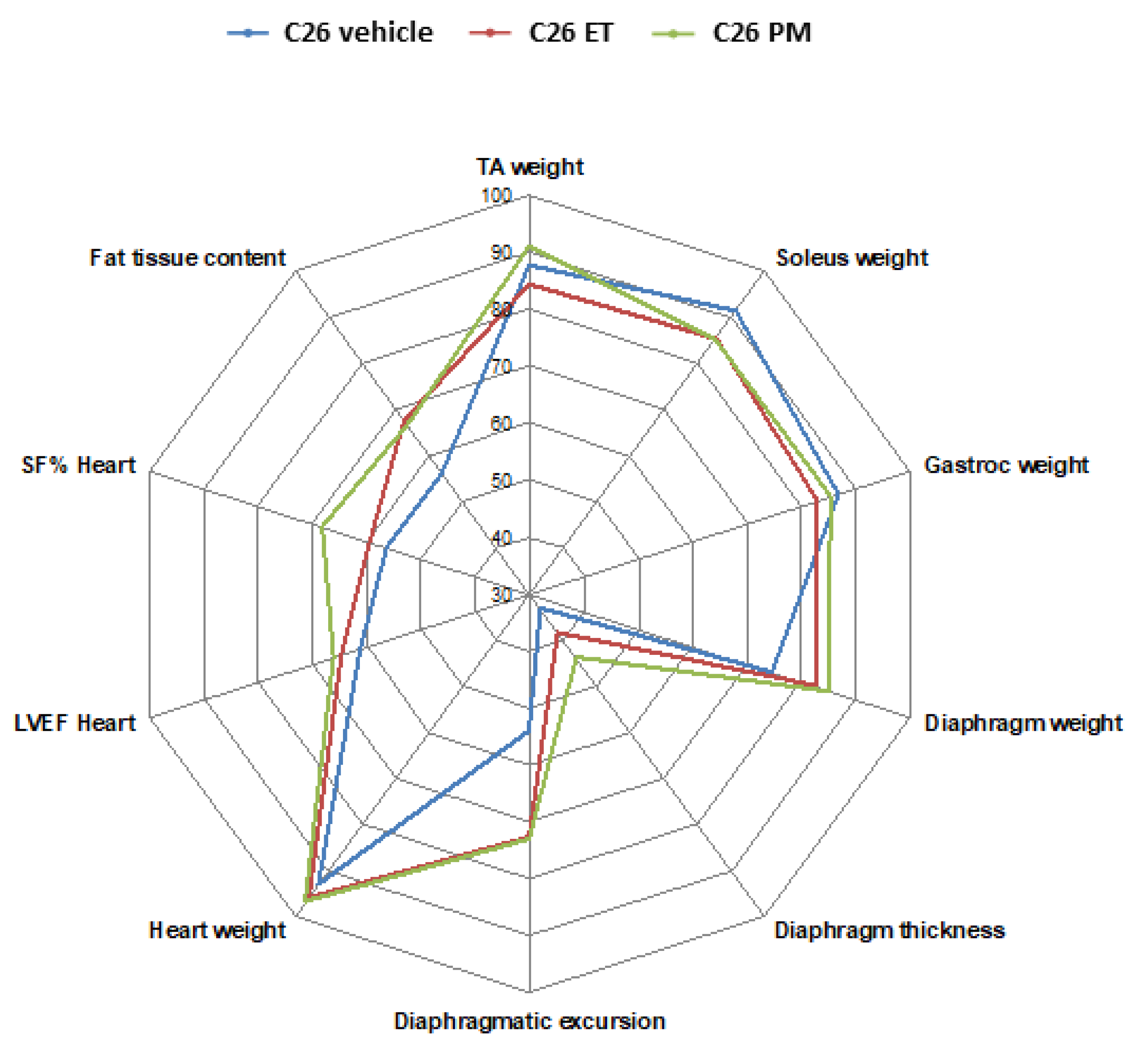
2.2. Lurbinectedin Does Not Protect C26-Carrying Mice from Cardiac and Diaphragm Dysfunctions during Cancer Cachexia
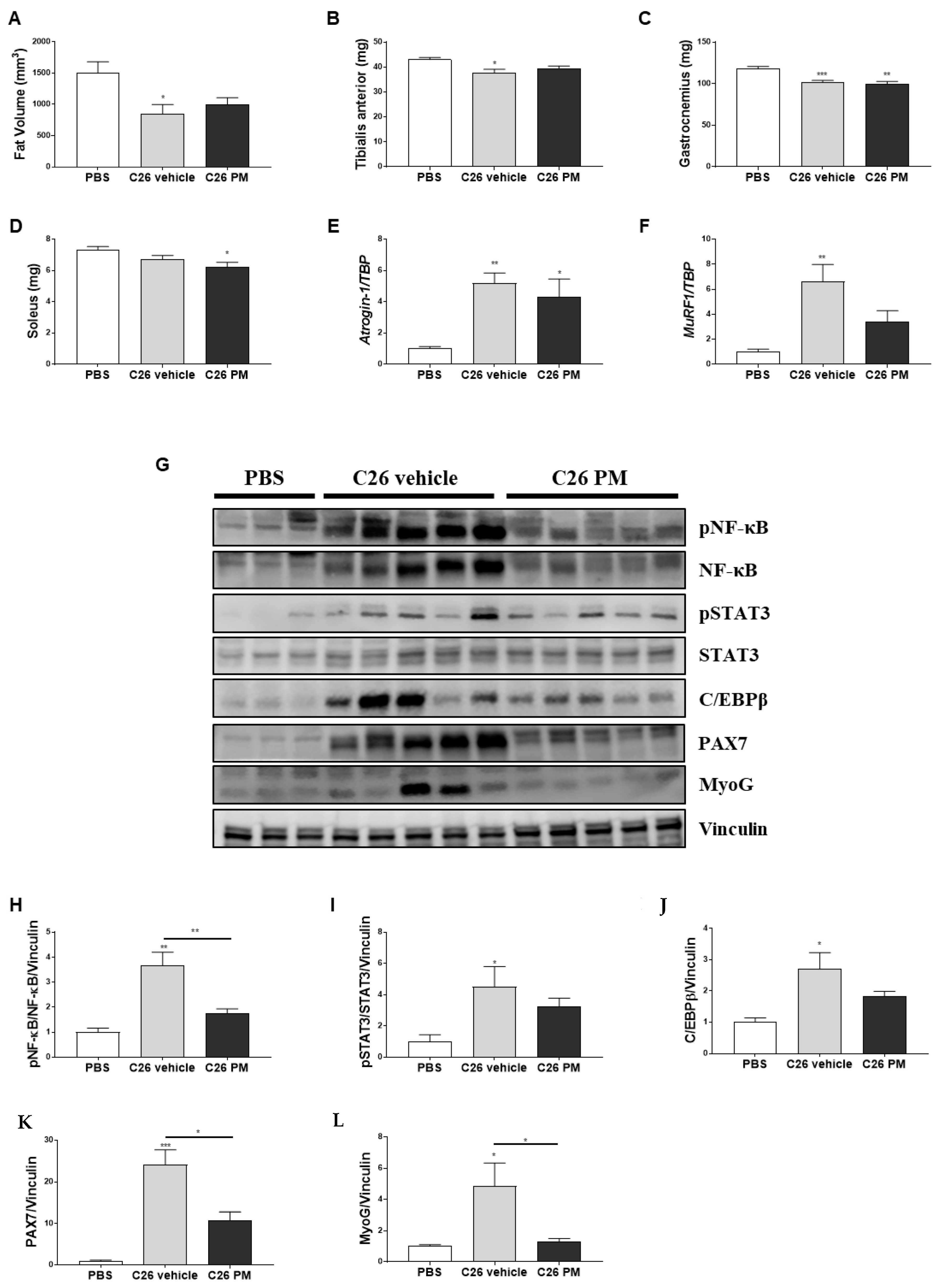
2.3. Lurbinectedin Does Not Protect either Fat or Hindlimb Muscles from Wasting but Restrains the Activation of the NF-κB/PAX7/Myogenin Axis in Tibialis Anterior of C26-Bearing Mice
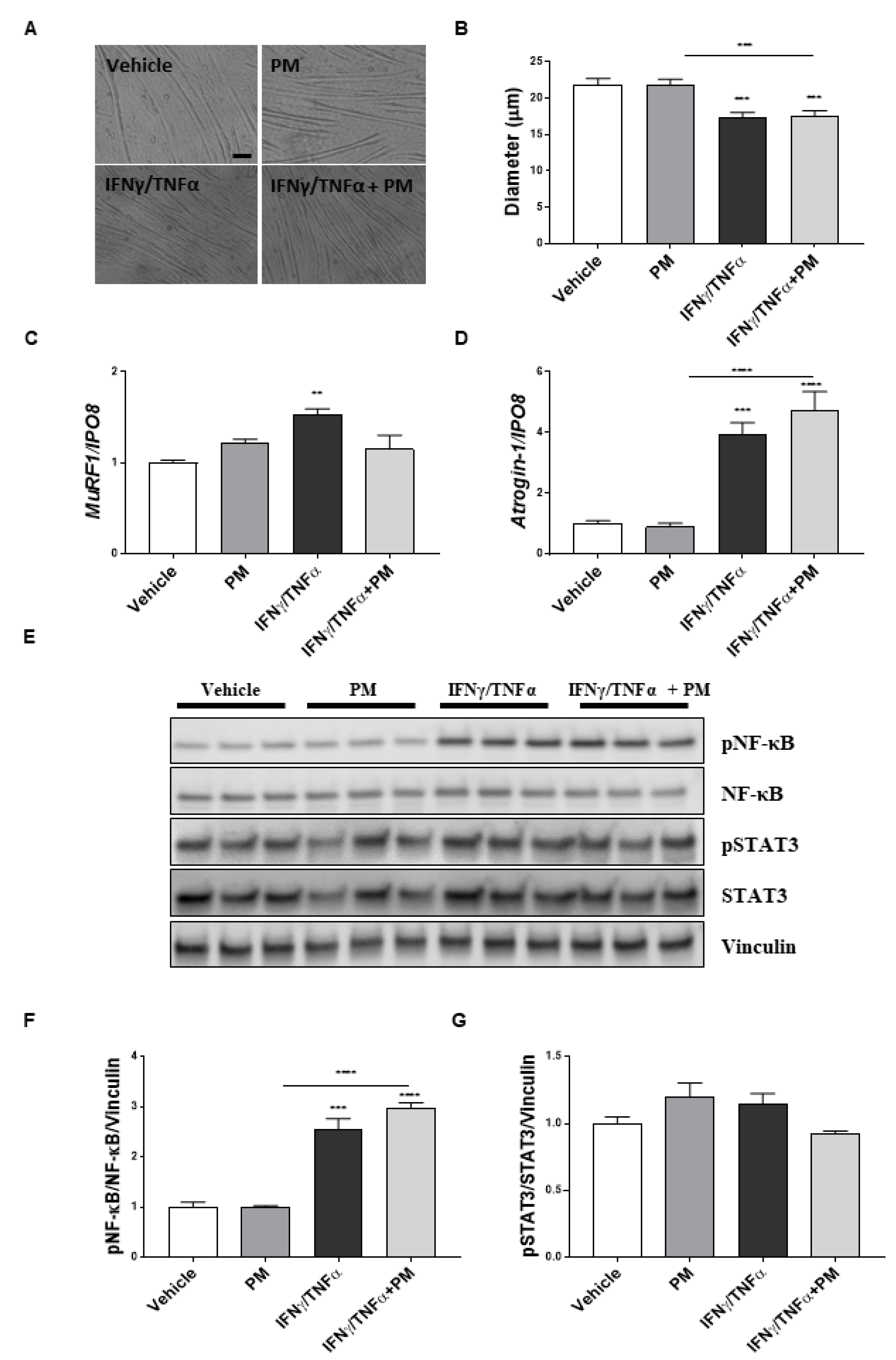
2.4. Lurbinectedin Abrogates Neither NF-κB Nor STAT3 Activation In Vitro in Atrophying Myotubes
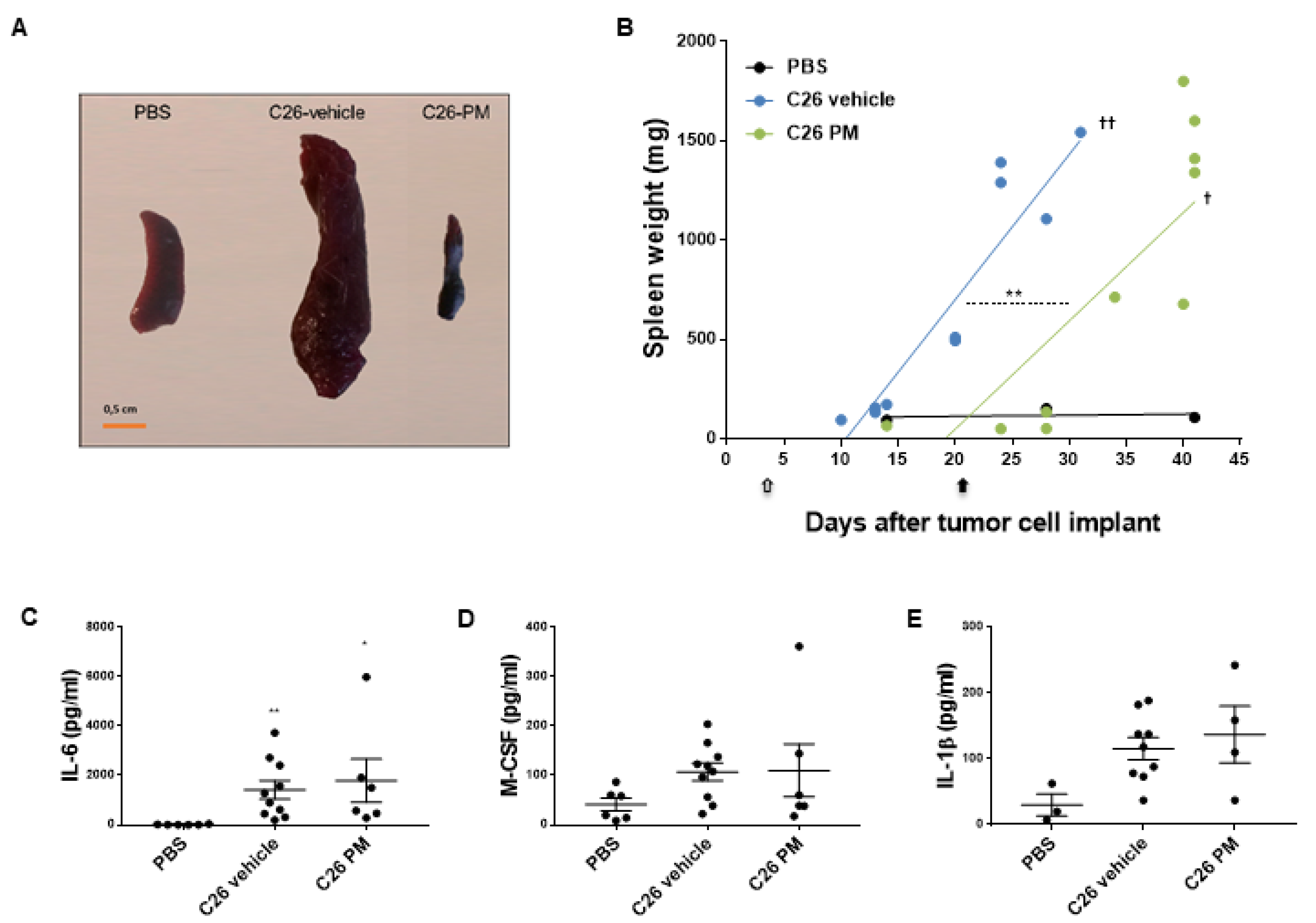
2.5. Lurbinectedin Inhibits C26-Induced Splenomegaly in Mice for as Long as the Treatment Lasts
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Drugs
4.3. Mice and Tumor Model
4.4. Diaphragmatic Ultrasonography
4.5. Echocardiography
4.5.1. Image Acquisition
4.5.2. LV Volumes and LVEF Calculation
4.6. Micro-CT Analysis
4.7. Collagen Detection in Cardiac Tissues by Picrosirius Red Staining
4.8. RNA Isolation from Cultured Cells or Muscles and Reverse Transcription
4.9. Quantitative Real-Time Polymerase Chain Reaction (PCR)
4.10. Protein Extraction and Western Blot
4.11. Cardiac Troponin and Cytokine Measurements
4.12. Myotube Diameter Measurements
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Roberts, B.M.; Ahn, B.; Smuder, A.J.; Al-Rajhi, M.; Gill, L.C.; Beharry, A.W.; Powers, S.K.; Fuller, D.D.; Ferreira, L.F.; Judge, A.R. Diaphragm and ventilatory dysfunction during cancer cachexia. FASEB J. 2013, 27, 2600–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houten, L.; Reilley, A.A. An investigation of the cause of death from cancer. J. Surg. Oncol. 1980, 13, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Nathan, J.A.; Goldberg, A.L. Muscle wasting in disease: Molecular mechanisms and promising therapies. Nat. Rev. Drug Discov. 2015, 14, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Yoneda, J.; Ohmori, H.; Sasaki, T.; Shimbo, K.; Eto, S.; Kato, Y.; Miyano, H.; Kobayashi, T.; Sasahira, T.; et al. Cancer usurps skeletal muscle as an energy repository. Cancer Res. 2014, 74, 330–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.; Laird, B.J.A.; Skipworth, R.J.E. The immunological regulation of cancer cachexia and its therapeutic implications. J. Cancer Metastasis Treat. 2019, 2019. [Google Scholar] [CrossRef]
- Cai, D.; Frantz, J.D.; Tawa, N.E.; Melendez, P.A.; Oh, B.-C.; Lidov, H.G.W.; Hasselgren, P.-O.; Frontera, W.R.; Lee, J.; Glass, D.J.; et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 2004, 119, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Todorov, P.; Cariuk, P.; McDevitt, T.; Coles, B.; Fearon, K.; Tisdale, M. Characterization of a cancer cachectic factor. Nature 1996, 379, 739–742. [Google Scholar] [CrossRef]
- Piccirillo, R.; Giavazzi, R. Inactivating STAT3: Bad for tumor, good for muscle. Cell Cycle 2015, 14, 939–940. [Google Scholar] [CrossRef] [Green Version]
- Gilabert, M.; Calvo, E.; Airoldi, A.; Hamidi, T.; Moutardier, V.; Turrini, O.; Iovanna, J. Pancreatic cancer-induced cachexia is Jak2-dependent in mice. J. Cell. Physiol. 2014, 229, 1437–1443. [Google Scholar] [CrossRef]
- He, W.A.; Berardi, E.; Cardillo, V.M.; Acharyya, S.; Aulino, P.; Thomas-Ahner, J.; Wang, J.; Bloomston, M.; Muscarella, P.; Nau, P.; et al. NF-κB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J. Clin. Investig. 2013, 123, 4821–4835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchildon, F.; Lamarche, É.; Lala-Tabbert, N.; St-Louis, C.; Wiper-Bergeron, N. Expression of CCAAT/Enhancer Binding Protein Beta in Muscle Satellite Cells Inhibits Myogenesis in Cancer Cachexia. PLoS ONE 2015, 10, e0145583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchildon, F.; Fu, D.; Lala-Tabbert, N.; Wiper-Bergeron, N. CCAAT/enhancer binding protein beta protects muscle satellite cells from apoptosis after injury and in cancer cachexia. Cell Death Dis. 2016, 7, e2109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Lecker, S.H.; Jagoe, R.T.; Gilbert, A.; Gomes, M.; Baracos, V.; Bailey, J.; Price, S.R.; Mitch, W.E.; Goldberg, A.L. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004, 18, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Bonetto, A.; Aydogdu, T.; Jin, X.; Zhang, Z.; Zhan, R.; Puzis, L.; Koniaris, L.G.; Zimmers, T.A. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E410–E421. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.; Zhang, S.; Hu, W.; Lu, X.; Lou, N.; Yang, Z.; Chen, S.; Zhang, X.; Yang, H. Valproic acid attenuates skeletal muscle wasting by inhibiting C/EBPβ-regulated atrogin1 expression in cancer cachexia. Am. J. Physiol. Cell Physiol. 2016, 311, C101–C115. [Google Scholar] [CrossRef] [Green Version]
- Gordon, E.M.; Sankhala, K.K.; Chawla, N.; Chawla, S.P. Trabectedin for Soft Tissue Sarcoma: Current Status and Future Perspectives. Adv. Ther. 2016, 33, 1055–1071. [Google Scholar] [CrossRef] [Green Version]
- Paz-Ares, L.G.; Trigo Perez, J.M.; Besse, B.; Moreno, V.; Lopez, R.; Sala, M.A.; Ponce Aix, S.; Fernandez, C.M.; Siguero, M.; Kahatt, C.M.; et al. Efficacy and safety profile of lurbinectedin in second-line SCLC patients: Results from a phase II single-agent trial. J. Clin. Oncol. 2019, 37, 8506. [Google Scholar] [CrossRef]
- Jimenez, P.C.; Wilke, D.V.; Branco, P.C.; Bauermeister, A.; Rezende-Teixeira, P.; Gaudêncio, S.P.; Costa-Lotufo, L.V. Enriching Cancer Pharmacology with Drugs of Marine Origin. Br. J. Pharmacol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Belgiovine, C.; Bello, E.; Liguori, M.; Craparotta, I.; Mannarino, L.; Paracchini, L.; Beltrame, L.; Marchini, S.; Galmarini, C.M.; Mantovani, A.; et al. Lurbinectedin reduces tumour-associated macrophages and the inflammatory tumour microenvironment in preclinical models. Br. J. Cancer 2017, 117, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Frapolli, R.; Zangarini, M.; Bello, E.; Porcu, L.; Galmarini, C.M.; García-Fernández, L.F.; Cuevas, C.; Allavena, P.; Erba, E.; et al. Comparison of in vitro and in vivo biological effects of trabectedin, lurbinectedin (PM01183) and Zalypsis® (PM00104). Int. J. Cancer 2013, 133, 2024–2033. [Google Scholar] [CrossRef] [PubMed]
- Elez, M.E.; Tabernero, J.; Geary, D.; Macarulla, T.; Kang, S.P.; Kahatt, C.; Pita, A.S.-M.; Teruel, C.F.; Siguero, M.; Cullell-Young, M.; et al. First-in-human phase I study of Lurbinectedin (PM01183) in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 2205–2214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argilés, J.M.; López-Soriano, F.J.; Busquets, S. Mediators of cachexia in cancer patients. Nutrition 2019, 66, 11–15. [Google Scholar] [CrossRef]
- Germano, G.; Frapolli, R.; Simone, M.; Tavecchio, M.; Erba, E.; Pesce, S.; Pasqualini, F.; Grosso, F.; Sanfilippo, R.; Casali, P.G.; et al. Antitumor and anti-inflammatory effects of trabectedin on human myxoid liposarcoma cells. Cancer Res. 2010, 70, 2235–2244. [Google Scholar] [CrossRef] [Green Version]
- D’Incalci, M.; Galmarini, C.; de la Riba Alvarez, I.; Badri, N.; Schöffski, P. Association between body weight and efficacy outcomes during trabectedin therapy for recurrent advanced soft tissue sarcoma (STS). J. Clin. Oncol. 2012, 30, 10047. [Google Scholar] [CrossRef]
- Nakai, T.; Imura, Y.; Tamiya, H.; Yamada, S.; Nakai, S.; Yasuda, N.; Kaneko, K.; Outani, H.; Takenaka, S.; Hamada, K.; et al. Trabectedin is a promising antitumor agent potentially inducing melanocytic differentiation for clear cell sarcoma. Cancer Med. 2017, 6, 2121–2130. [Google Scholar] [CrossRef]
- Martinelli, G.B.; Olivari, D.; Re Cecconi, A.D.; Talamini, L.; Ottoboni, L.; Lecker, S.H.; Stretch, C.; Baracos, V.E.; Bathe, O.F.; Resovi, A.; et al. Activation of the SDF1/CXCR4 pathway retards muscle atrophy during cancer cachexia. Oncogene 2016, 35, 6212–6222. [Google Scholar] [CrossRef]
- Bonetto, A.; Rupert, J.E.; Barreto, R.; Zimmers, T.A. The Colon-26 Carcinoma Tumor-bearing Mouse as a Model for the Study of Cancer Cachexia. J. Vis. Exp. 2016. [Google Scholar] [CrossRef] [Green Version]
- Aulino, P.; Berardi, E.; Cardillo, V.M.; Rizzuto, E.; Perniconi, B.; Ramina, C.; Padula, F.; Spugnini, E.P.; Baldi, A.; Faiola, F.; et al. Molecular, cellular and physiological characterization of the cancer cachexia-inducing C26 colon carcinoma in mouse. BMC Cancer 2010, 10, 363. [Google Scholar] [CrossRef]
- Shukla, S.K.; Dasgupta, A.; Mehla, K.; Gunda, V.; Vernucci, E.; Souchek, J.; Goode, G.; King, R.; Mishra, A.; Rai, I.; et al. Silibinin-mediated metabolic reprogramming attenuates pancreatic cancer-induced cachexia and tumor growth. Oncotarget 2015, 6, 41146–41161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.K.; Gebregiworgis, T.; Purohit, V.; Chaika, N.V.; Gunda, V.; Radhakrishnan, P.; Mehla, K.; Pipinos, I.I.; Powers, R.; Yu, F.; et al. Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab. 2014, 2, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, M.; Nishijima, Y.; Asp, M.L.; Stout, M.B.; Reiser, P.J.; Belury, M.A. Cardiac alterations in cancer-induced cachexia in mice. Int. J. Oncol. 2010, 37, 347–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pretto, F.; Ghilardi, C.; Moschetta, M.; Bassi, A.; Rovida, A.; Scarlato, V.; Talamini, L.; Fiordaliso, F.; Bisighini, C.; Damia, G.; et al. Sunitinib prevents cachexia and prolongs survival of mice bearing renal cancer by restraining STAT3 and MuRF-1 activation in muscle. Oncotarget 2015, 6, 3043–3054. [Google Scholar] [CrossRef]
- Re Cecconi, A.D.; Forti, M.; Chiappa, M.; Zhu, Z.; Zingman, L.V.; Cervo, L.; Beltrame, L.; Marchini, S.; Piccirillo, R. Musclin, a Myokine Induced by Aerobic Exercise, Retards Muscle Atrophy During Cancer Cachexia in Mice. Cancers 2019, 11, 1541. [Google Scholar] [CrossRef] [Green Version]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Gomes, M.D.; Lecker, S.H.; Jagoe, R.T.; Navon, A.; Goldberg, A.L. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. USA 2001, 98, 14440–14445. [Google Scholar] [CrossRef] [Green Version]
- Assi, M.; Derbré, F.; Lefeuvre-Orfila, L.; Rébillard, A. Antioxidant supplementation accelerates cachexia development by promoting tumor growth in C26 tumor-bearing mice. Free Radic. Biol. Med. 2016, 91, 204–214. [Google Scholar] [CrossRef]
- Moresi, V.; Williams, A.H.; Meadows, E.; Flynn, J.M.; Potthoff, M.J.; McAnally, J.; Shelton, J.M.; Backs, J.; Klein, W.H.; Richardson, J.A.; et al. Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell 2010, 143, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Costamagna, D.; Duelen, R.; Penna, F.; Neumann, D.; Costelli, P.; Sampaolesi, M. Interleukin-4 administration improves muscle function, adult myogenesis, and lifespan of colon carcinoma-bearing mice. J. Cachexia Sarcopenia Muscle 2020, 11, 783–801. [Google Scholar] [CrossRef] [Green Version]
- Bonetto, A.; Aydogdu, T.; Kunzevitzky, N.; Guttridge, D.C.; Khuri, S.; Koniaris, L.G.; Zimmers, T.A. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS ONE 2011, 6, e22538. [Google Scholar] [CrossRef] [Green Version]
- Jackman, R.W.; Floro, J.; Yoshimine, R.; Zitin, B.; Eiampikul, M.; El-Jack, K.; Seto, D.N.; Kandarian, S.C. Continuous Release of Tumor-Derived Factors Improves the Modeling of Cachexia in Muscle Cell Culture. Front. Physiol. 2017, 8, 738. [Google Scholar] [CrossRef]
- Ju, J.E.; Kim, M.-S.; Kang, J.H.; Lee, J.Y.; Lee, M.S.; Kim, E.H.; Chung, N.; Jeong, Y.K. Potential role of immunological factors in early diagnosis of cancer cachexia in C26 tumor-bearing mice. Appl. Biol. Chem. 2019, 62, 3. [Google Scholar] [CrossRef] [Green Version]
- Ramos, E.J.B.; Suzuki, S.; Marks, D.; Inui, A.; Asakawa, A.; Meguid, M.M. Cancer anorexia-cachexia syndrome: Cytokines and neuropeptides. Curr. Opin. Clin. Nutr. Metab. Care 2004, 7, 427–434. [Google Scholar] [CrossRef]
- Narsale, A.A.; Carson, J.A. Role of interleukin-6 in cachexia: Therapeutic implications. Curr. Opin. Support. Palliat. Care 2014, 8, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Valdés-Ferrer, S.I.; Rosas-Ballina, M.; Olofsson, P.S.; Lu, B.; Dancho, M.E.; Ochani, M.; Li, J.H.; Scheinerman, J.A.; Katz, D.A.; Levine, Y.A.; et al. Expression of Concern: HMGB 1 mediates splenomegaly and expansion of splenic CD 11b+ L y-6Chigh inflammatory monocytes in murine sepsis survivors. J. Intern. Med. 2013, 274, 381–390. [Google Scholar] [CrossRef]
- Gamba-Vitalo, C.; DiGiovanna, M.P.; Sartorelli, A.C. Effects of recombinant murine granulocyte-macrophage colony-stimulating factor in cyclophosphamide-treated mice. Blood Cells 1991, 17, 193–205; discussion 206–208. [Google Scholar]
- Paz-Ares, L.; Rivera-Herreros, F.; Díaz-Rubio, E.; García, M.; Casado, E.; Cubedo, R.; Gravalos, C.; Alfaro, V.; Gómez, J.; Izquierdo, M.A.; et al. Phase II study of trabectedin in pretreated patients with advanced colorectal cancer. Clin. Colorectal Cancer 2007, 6, 522–528. [Google Scholar] [CrossRef]
- Ciciliot, S.; Rossi, A.C.; Dyar, K.A.; Blaauw, B.; Schiaffino, S. Muscle type and fiber type specificity in muscle wasting. Int. J. Biochem. Cell Biol. 2013, 45, 2191–2199. [Google Scholar] [CrossRef]
- Devine, R.D.; Bicer, S.; Reiser, P.J.; Wold, L.E. Increased hypoxia-inducible factor-1α in striated muscle of tumor-bearing mice. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H1154–H1162. [Google Scholar] [CrossRef] [Green Version]
- Cosper, P.F.; Leinwand, L.A. Cancer causes cardiac atrophy and autophagy in a sexually dimorphic manner. Cancer Res. 2011, 71, 1710–1720. [Google Scholar] [CrossRef] [Green Version]
- Devine, R.D.; Bicer, S.; Reiser, P.J.; Velten, M.; Wold, L.E. Metalloproteinase expression is altered in cardiac and skeletal muscle in cancer cachexia. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H685–H691. [Google Scholar] [CrossRef] [Green Version]
- Seliger, S.L.; Hong, S.N.; Christenson, R.H.; Kronmal, R.; Daniels, L.B.; Lima, J.A.C.; de Lemos, J.A.; Bertoni, A.; de Filippi, C.R. High-Sensitive Cardiac Troponin T as an Early Biochemical Signature for Clinical and Subclinical Heart Failure: MESA (Multi-Ethnic Study of Atherosclerosis). Circulation 2017, 135, 1494–1505. [Google Scholar] [CrossRef] [Green Version]
- Braunwald, E. Biomarkers in heart failure. N. Engl. J. Med. 2008, 358, 2148–2159. [Google Scholar] [CrossRef] [Green Version]
- Ammann, P.; Maggiorini, M.; Bertel, O.; Haenseler, E.; Joller-Jemelka, H.I.; Oechslin, E.; Minder, E.I.; Rickli, H.; Fehr, T. Troponin as a risk factor for mortality in critically ill patients without acute coronary syndromes. J. Am. Coll. Cardiol. 2003, 41, 2004–2009. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Feng, X.; Dong, J.; Wang, Z.-M.; Lee, J.; Furdui, C.; Files, D.C.; Beavers, K.M.; Kritchevsky, S.; Milligan, C.; et al. Cardiac troponin T and fast skeletal muscle denervation in ageing. J. Cachexia Sarcopenia Muscle 2017, 8, 808–823. [Google Scholar] [CrossRef]
- Springer, J.; Tschirner, A.; Haghikia, A.; von Haehling, S.; Lal, H.; Grzesiak, A.; Kaschina, E.; Palus, S.; Pötsch, M.; von Websky, K.; et al. Prevention of liver cancer cachexia-induced cardiac wasting and heart failure. Eur. Heart J. 2014, 35, 932–941. [Google Scholar] [CrossRef]
- Kazemi-Bajestani, S.M.R.; Becher, H.; Butts, C.; Basappa, N.S.; Smylie, M.; Joy, A.A.; Sangha, R.; Gallivan, A.; Kavsak, P.; Chu, Q.; et al. Rapid atrophy of cardiac left ventricular mass in patients with non-small cell carcinoma of the lung. J. Cachexia Sarcopenia Muscle 2019, 10, 1070–1082. [Google Scholar] [CrossRef] [Green Version]
- Kleveland, O.; Kunszt, G.; Bratlie, M.; Ueland, T.; Broch, K.; Holte, E.; Michelsen, A.E.; Bendz, B.; Amundsen, B.H.; Espevik, T.; et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: A double-blind, randomized, placebo-controlled phase 2 trial. Eur. Heart J. 2016, 37, 2406–2413. [Google Scholar] [CrossRef]
- Chen, Z.-W.; Qian, J.-Y.; Ma, J.-Y.; Chang, S.-F.; Yun, H.; Jin, H.; Sun, A.-J.; Zou, Y.-Z.; Ge, J.-B. TNF-α-induced cardiomyocyte apoptosis contributes to cardiac dysfunction after coronary microembolization in mini-pigs. J. Cell. Mol. Med. 2014, 18, 1953–1963. [Google Scholar] [CrossRef]
- Seale, P.; Sabourin, L.A.; Girgis-Gabardo, A.; Mansouri, A.; Gruss, P.; Rudnicki, M.A. Pax7 is required for the specification of myogenic satellite cells. Cell 2000, 102, 777–786. [Google Scholar] [CrossRef] [Green Version]
- Coletti, D.; Aulino, P.; Pigna, E.; Barteri, F.; Moresi, V.; Annibali, D.; Adamo, S.; Berardi, E. Spontaneous Physical Activity Downregulates Pax7 in Cancer Cachexia. Stem Cells Int. 2016, 2016, 6729268. [Google Scholar] [CrossRef] [Green Version]
- Dubois, V.; Laurent, M.R.; Sinnesael, M.; Cielen, N.; Helsen, C.; Clinckemalie, L.; Spans, L.; Gayan-Ramirez, G.; Deldicque, L.; Hespel, P.; et al. A satellite cell-specific knockout of the androgen receptor reveals myostatin as a direct androgen target in skeletal muscle. FASEB J. 2014, 28, 2979–2994. [Google Scholar] [CrossRef]
- Rana, K.; Lee, N.K.L.; Zajac, J.D.; MacLean, H.E. Expression of androgen receptor target genes in skeletal muscle. Asian J. Androl. 2014, 16, 675–683. [Google Scholar] [CrossRef]
- Penna, F.; Costamagna, D.; Fanzani, A.; Bonelli, G.; Baccino, F.M.; Costelli, P. Muscle wasting and impaired myogenesis in tumor bearing mice are prevented by ERK inhibition. PLoS ONE 2010, 5, e13604. [Google Scholar] [CrossRef]
- Levolger, S.; van den Engel, S.; Ambagtsheer, G.; IJzermans, J.N.M.; de Bruin, R.W.F. Caloric restriction is associated with preservation of muscle strength in experimental cancer cachexia. Aging 2018, 10, 4213–4223. [Google Scholar] [CrossRef]
- Op den Kamp, C.M.; Langen, R.C.; Snepvangers, F.J.; de Theije, C.C.; Schellekens, J.M.; Laugs, F.; Dingemans, A.-M.C.; Schols, A.M. Nuclear transcription factor κ B activation and protein turnover adaptations in skeletal muscle of patients with progressive stages of lung cancer cachexia. Am. J. Clin. Nutr. 2013, 98, 738–748. [Google Scholar] [CrossRef] [Green Version]
- Loria, R.; Laquintana, V.; Bon, G.; Trisciuoglio, D.; Frapolli, R.; Covello, R.; Amoreo, C.A.; Ferraresi, V.; Zoccali, C.; Novello, M.; et al. HMGA1/E2F1 axis and NFkB pathways regulate LPS progression and trabectedin resistance. Oncogene 2018, 37, 5926–5938. [Google Scholar] [CrossRef] [Green Version]
- Wysong, A.; Asher, S.A.; Yin, X.R.; Gore, M.; Weinstein, L. Selective Inhibition of NF-kappa-B with NBD Peptide Reduces Tumor-Induced Wasting in a Murine Model of Cancer Cachexia In vivo. J. Cancer Sci. Ther. 2011, 3. [Google Scholar] [CrossRef]
- Allavena, P.; Signorelli, M.; Chieppa, M.; Erba, E.; Bianchi, G.; Marchesi, F.; Olimpio, C.O.; Bonardi, C.; Garbi, A.; Lissoni, A.; et al. Anti-inflammatory properties of the novel antitumor agent yondelis (trabectedin): Inhibition of macrophage differentiation and cytokine production. Cancer Res. 2005, 65, 2964–2971. [Google Scholar] [CrossRef] [Green Version]
- DuPre’, S.A.; Hunter, K.W. Murine mammary carcinoma 4T1 induces a leukemoid reaction with splenomegaly: Association with tumor-derived growth factors. Exp. Mol. Pathol. 2007, 82, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Michaelides, M.C.; Wallack, M.K. Characterization of tumorigenicity, mortality, metastasis, and splenomegaly of two cultured murine colon lines. Cancer Res. 1981, 41, 2267–2272. [Google Scholar] [PubMed]
- Wang, Z.; Zhao, C.; Moya, R.; Davies, J.D. A novel role for CD4+ T cells in the control of cachexia. J. Immunol. 2008, 181, 4676–4684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winfield, R.D.; Delano, M.J.; Pande, K.; Scumpia, P.O.; Laface, D.; Moldawer, L.L. Myeloid-derived suppressor cells in cancer cachexia syndrome: A new explanation for an old problem. J. Parenter. Enter. Nutr. 2008, 32, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Cuenca, A.G.; Cuenca, A.L.; Winfield, R.D.; Joiner, D.N.; Gentile, L.; Delano, M.J.; Kelly-Scumpia, K.M.; Scumpia, P.O.; Matheny, M.K.; Scarpace, P.J.; et al. Novel Role for Tumor-Induced Expansion of Myeloid-Derived Cells in Cancer Cachexia. J. Immunol. 2014, 192, 6111–6119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soda, K.; Kawakami, M.; Takagi, S.; Kashii, A.; Miyata, M. Splenectomy before tumor inoculation prolongs the survival time of cachectic mice. Cancer Immunol. Immunother. 1995, 41, 203–209. [Google Scholar] [CrossRef]
- Sato, N.; Michaelides, M.C.; Wallack, M.K. Effect of splenectomy on the growth of murine colon tumors. J. Surg. Oncol. 1983, 22, 73–76. [Google Scholar] [CrossRef]
- Kuroda, H.; Mabuchi, S.; Kozasa, K.; Yokoi, E.; Matsumoto, Y.; Komura, N.; Kawano, M.; Hashimoto, K.; Sawada, K.; Kimura, T. PM01183 inhibits myeloid-derived suppressor cells in vitro and in vivo. Immunotherapy 2017, 9, 805–817. [Google Scholar] [CrossRef]
- Larsen, A.K.; Galmarini, C.M.; D’Incalci, M. Unique features of trabectedin mechanism of action. Cancer Chemother. Pharmacol. 2016, 77, 663–671. [Google Scholar] [CrossRef]
- Allavena, P.; Liguori, M.; Belgiovine, C. Trabectedin, a Drug Acting on Both Cancer Cells and the Tumor Microenvironment. In Approaching Complex Diseases: Network-Based Pharmacology and Systems Approach in Bio-Medicine; Bizzarri, M., Ed.; Springer International Publishing: Cham, Switzerland, 2020; pp. 287–300. ISBN 978-3-030-32857-3. [Google Scholar]
- Mesa, R.A.; Passamonti, F. Individualizing Care for Patients with Myeloproliferative Neoplasms: Integrating Genetics, Evolving Therapies, and Patient-Specific Disease Burden. Am. Soc. Clin. Oncol. Educ. Book 2016, e324–e335. [Google Scholar] [CrossRef]
- Nissinen, T.A.; Hentilä, J.; Penna, F.; Lampinen, A.; Lautaoja, J.H.; Fachada, V.; Holopainen, T.; Ritvos, O.; Kivelä, R.; Hulmi, J.J. Treating cachexia using soluble ACVR2B improves survival, alters mTOR localization, and attenuates liver and spleen responses: Treating cachexia using soluble ACVR2B. J. Cachexia Sarcopenia Muscle 2018, 9, 514–529. [Google Scholar] [CrossRef] [PubMed]
- Wiegant, J.; van Hall, T.; van der Burg, M.; Colombo, M.; Tanke, H.J.; Offringa, R.; Rosenberg, C. Application of multicolor fluorescence in situ hybridization analysis for detection of cross-contamination and in vitro progression in commonly used murine tumor cell lines. Cancer Genet. Cytogenet. 2002, 139, 126–132. [Google Scholar] [CrossRef]
- Wang, W.; Pan, Y.; Zhou, H.; Wang, L.; Chen, X.; Song, G.; Liu, J.; Li, A. Ferulic acid suppresses obesity and obesity-related metabolic syndromes in high fat diet-induced obese C57BL/6J mice. Food Agric. Immunol. 2018, 29, 1116–1125. [Google Scholar] [CrossRef] [Green Version]
- Mosa, R.; Huang, L.; Wu, Y.; Fung, C.; Mallawakankanamalage, O.; LeRoith, D.; Chen, C. Hexarelin, a Growth Hormone Secretagogue, Improves Lipid Metabolic Aberrations in Nonobese Insulin-Resistant Male MKR Mice. Endocrinology 2017, 158, 3174–3187. [Google Scholar] [CrossRef] [Green Version]
- Moschetta, M.; Pretto, F.; Berndt, A.; Galler, K.; Richter, P.; Bassi, A.; Oliva, P.; Micotti, E.; Valbusa, G.; Schwager, K.; et al. Paclitaxel enhances therapeutic efficacy of the F8-IL2 immunocytokine to EDA-fibronectin-positive metastatic human melanoma xenografts. Cancer Res. 2012, 72, 1814–1824. [Google Scholar] [CrossRef] [Green Version]
- Goligher, E.C.; Fan, E.; Herridge, M.S.; Murray, A.; Vorona, S.; Brace, D.; Rittayamai, N.; Lanys, A.; Tomlinson, G.; Singh, J.M.; et al. Evolution of Diaphragm Thickness during Mechanical Ventilation. Impact of Inspiratory Effort. Am. J. Respir. Crit. Care Med. 2015, 192, 1080–1088. [Google Scholar] [CrossRef]
- Russo, I.; Micotti, E.; Fumagalli, F.; Magnoli, M.; Ristagno, G.; Latini, R.; Staszewsky, L. A novel echocardiographic method closely agrees with cardiac magnetic resonance in the assessment of left ventricular function in infarcted mice. Sci. Rep. 2019, 9, 3580. [Google Scholar] [CrossRef]
- Luu, Y.K.; Lublinsky, S.; Ozcivici, E.; Capilla, E.; Pessin, J.E.; Rubin, C.T.; Judex, S. In vivo quantification of subcutaneous and visceral adiposity by micro-computed tomography in a small animal model. Med. Eng. Phys. 2009, 31, 34–41. [Google Scholar] [CrossRef] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aquila, G.; Re Cecconi, A.D.; Forti, M.; Frapolli, R.; Bello, E.; Novelli, D.; Russo, I.; Licandro, S.A.; Staszewsky, L.; Martinelli, G.B.; et al. Trabectedin and Lurbinectedin Extend Survival of Mice Bearing C26 Colon Adenocarcinoma, without Affecting Tumor Growth or Cachexia. Cancers 2020, 12, 2312. https://doi.org/10.3390/cancers12082312
Aquila G, Re Cecconi AD, Forti M, Frapolli R, Bello E, Novelli D, Russo I, Licandro SA, Staszewsky L, Martinelli GB, et al. Trabectedin and Lurbinectedin Extend Survival of Mice Bearing C26 Colon Adenocarcinoma, without Affecting Tumor Growth or Cachexia. Cancers. 2020; 12(8):2312. https://doi.org/10.3390/cancers12082312
Chicago/Turabian StyleAquila, Giorgio, Andrea David Re Cecconi, Mara Forti, Roberta Frapolli, Ezia Bello, Deborah Novelli, Ilaria Russo, Simonetta Andrea Licandro, Lidia Staszewsky, Giulia Benedetta Martinelli, and et al. 2020. "Trabectedin and Lurbinectedin Extend Survival of Mice Bearing C26 Colon Adenocarcinoma, without Affecting Tumor Growth or Cachexia" Cancers 12, no. 8: 2312. https://doi.org/10.3390/cancers12082312