Therapeutic Potential of Chemically Modified miR-489 in Triple-Negative Breast Cancers

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. MiR-489 Mediates Anti-Proliferative and DNA Damaging Response Blocking Function of ARRDC3 in TNBC Cells
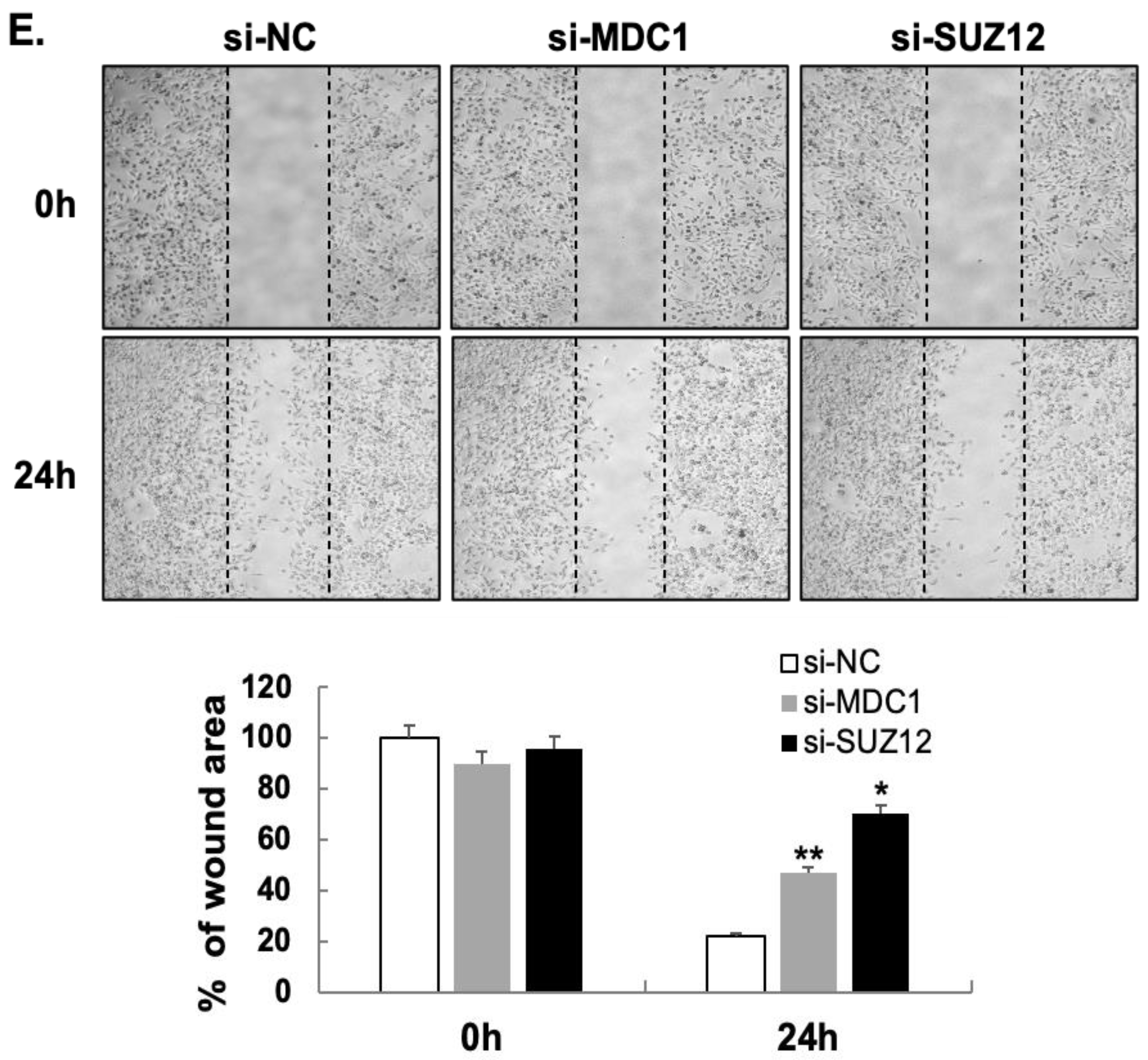
2.2. MDC1 and SUZ12 Are Novel Target Genes of MiR-489
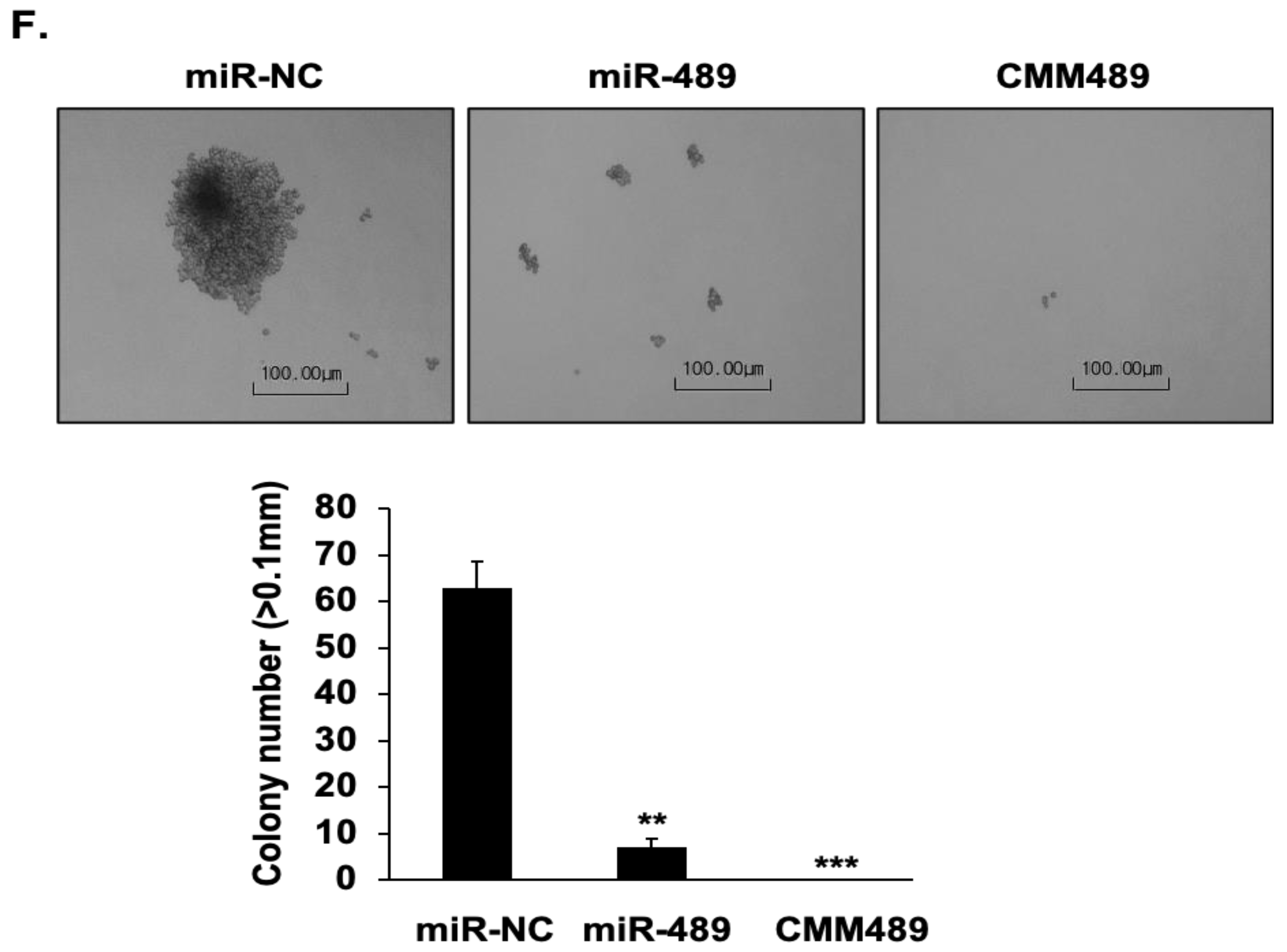
2.3. Chemically Modified MiR-489 Mimics (CMM489) Have Enhanced Therapeutic Efficacy in TNBC Cells
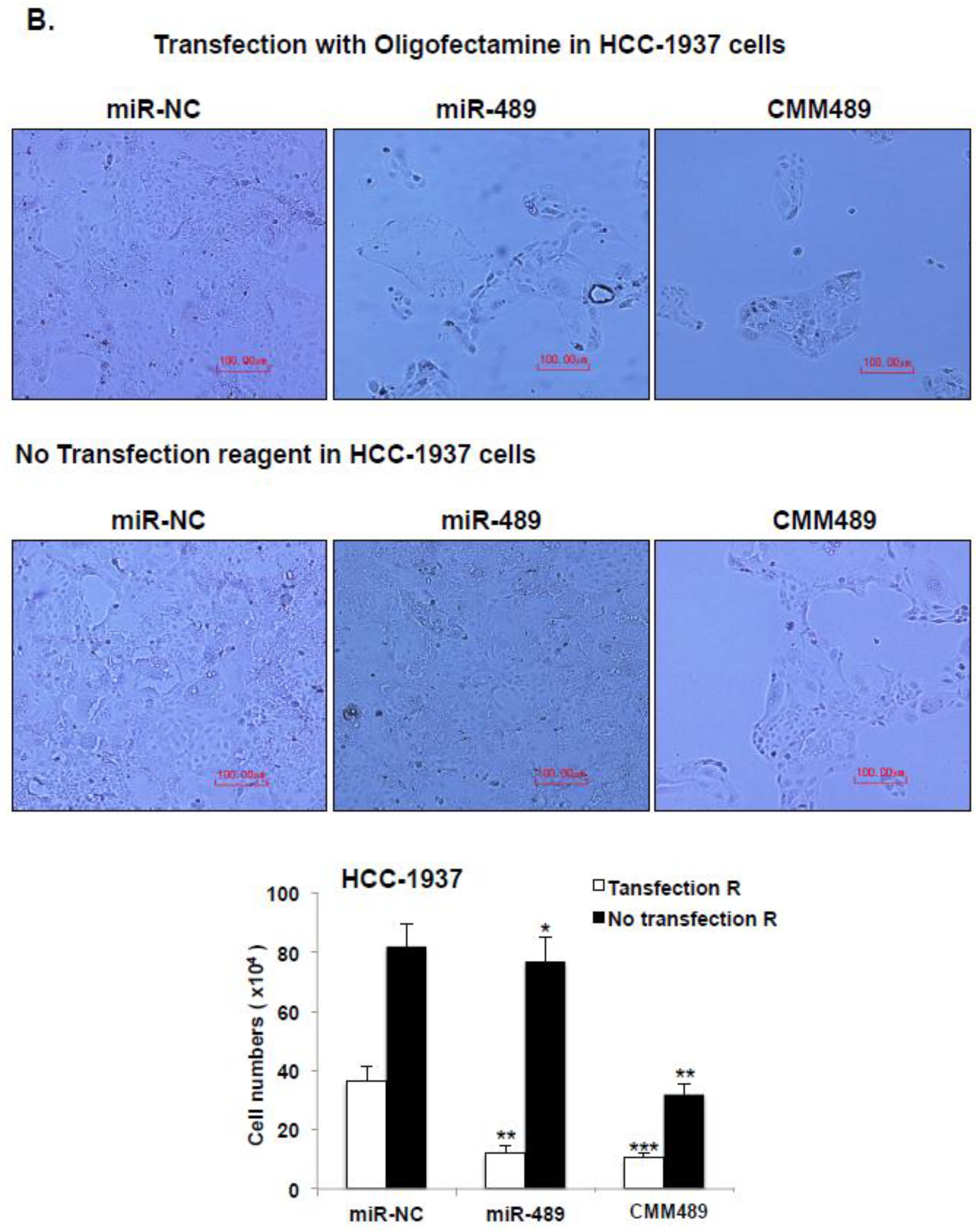
2.4. Vehicle Free Delivery of CMM489
2.5. CMM489 Inhibited Primary Tumor Growth and Metastasis in Metastatic Mouse Model
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. MiRNA Preparation and Transfection
4.3. Identification of MiR-489 Target Genes
4.4. SiRNA Transfection
4.5. Western Blot Analysis
4.6. Cell Viability by MTT Assay
4.7. Cell Cycle Assay
4.8. Cell Motility Assay
4.9. Colony Formation Assay
4.10. Wound-Healing Assay
4.11. Mouse Xenograft and Imaging
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Breast Cancer: Prevention and Control. Available online: http://www.who.int/cancer/detection/breastcancer/en/ (accessed on 10 June 2020).
- Mayer, I.A.; Abramson, V.G.; Lehmann, B.D.; Pietenpol, J.A. New strategies for triple-negative breast cancer--deciphering the heterogeneity. Clin. Cancer Res. 2014, 20, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Xiang, L.; Li, T.; Bai, Z. Cancer hallmarks, biomarkers and breast cancer molecular subtypes. J. Cancer 2016, 7, 1281–1294. [Google Scholar] [CrossRef] [PubMed]
- Abramson, V.G.; Lehmann, B.D.; Ballinger, T.J.; Pietenpol, J.A. Subtyping of triple-negative breast cancer: Implications for therapy. Cancer 2015, 121, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, I.S. Molecular testing in breast cancer: A guide to current practices. Arch. Pathol. Lab. Med. 2016, 140, 815–824. [Google Scholar] [CrossRef]
- Collignon, J.; Lousberg, L.; Schroeder, H.; Jerusalem, G. Triple-negative breast cancer: Treatment challenges and solutions. Breast Cancer Targets Ther. 2016, 8, 93–107. [Google Scholar] [CrossRef]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Chartron, E.; Theillet, C.; Guiu, S.; Jacot, W. Targeting homologous repair deficiency in breast and ovarian cancers: Biological pathways, preclinical and clinical data. Crit. Rev. Oncol. Hematol. 2019, 133, 58–73. [Google Scholar] [CrossRef]
- Brandsma, I.; Fleuren, E.D.G.; Williamson, C.T.; Lord, C.J. Directing the use of DDR kinase inhibitors in cancer treatment. Expert Opin. Investig. Drugs 2017, 26, 1341–1355. [Google Scholar] [CrossRef]
- Soung, Y.H.; Pruitt, K.; Chung, J. Epigenetic silencing of ARRDC3 expression in basal-like breast cancer cells. Sci. Rep. 2014, 4, 3846. [Google Scholar] [CrossRef]
- Draheim, K.M.; Chen, H.-B.; Tao, Q.; Moore, N.; Roche, M.; Lyle, S. ARRDC3 suppresses breast cancer progression by negatively regulating integrin beta4. Oncogene 2010, 29, 5032–5047. [Google Scholar] [CrossRef]
- Soung, Y.H.; Kashyap, T.; Nguyen, T.; Yadav, G.; Chang, H.; Landesman, Y.; Chung, J. Selective Inhibitors of Nuclear Export (SINE) compounds block proliferation and migration of triple negative breast cancer cells by restoring expression of ARRDC3. Oncotarget 2017, 8, 52935–52947. [Google Scholar] [CrossRef] [PubMed]
- Karatas, O.F.; Oner, M.; Abay, A.; Diyapoglu, A. MicroRNAs in human tongue squamous cell carcinoma: From pathogenesis to therapeutic implications. Oral Oncol. 2017, 67, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Manasa, V.G.; Kannan, S. Impact of microRNA dynamics on cancer hallmarks: An oral cancer scenario. Tumour Biol. 2017, 39, 1010428317695920. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-F.; Hannafon, B.N.; Ding, W.-Q. microRNA regulation of human pancreatic cancer stem cells. Stem Cell Investig. 2017, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Soung, Y.H.; Chung, H.; Yan, C.; Ju, J.; Chung, J. Arrestin domain containing 3 reverses epithelial to mesenchymal transition and chemo-resistance of TNBC cells by up-regulating expression of miR-200b. Cells 2019, 8, 692. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; He, D.; Yang, D.; Chen, Z.; Pan, Q.; Mao, A.; Cai, Y.; Li, X.; Xing, H.; Shi, M.; et al. MiR-489 regulates chemoresistance in breast cancer via epithelial mesenchymal transition pathway. FEBS Lett. 2014, 588, 2009–2015. [Google Scholar] [CrossRef]
- Zhao, H.; Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 2001, 21, 4129–4139. [Google Scholar] [CrossRef]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef]
- Xu, M.; Wang, S.; Wang, Y.; Wu, H.; Frank, J.A.; Zhang, Z.; Luo, J. Role of p38γ MAPK in regulation of EMT and cancer stem cells. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3605–3617. [Google Scholar] [CrossRef]
- Patel, A.N.; Goyal, S.; Wu, H.; Schiff, D.; Moran, M.S.; Haffty, B.G. Mediator of DNA damage checkpoint protein 1 (MDC1) expression as a prognostic marker for nodal recurrence in early-stage breast cancer patients treated with breast-conserving surgery and radiation therapy. Breast Cancer Res. Treat. 2011, 126, 601–607. [Google Scholar] [CrossRef]
- Wang, S.; Zou, Z.; Luo, X.; Mi, Y.; Chang, H.; Xing, D. LRH1 enhances cell resistance to chemotherapy by transcriptionally activating MDC1 expression and attenuating DNA damage in human breast cancer. Oncogene 2018, 37, 3243–3259. [Google Scholar] [CrossRef] [PubMed]
- Nomura, H.; Kataoka, F.; Aoki, D.; Jinno, H.; Kitagawa, Y.; Sato, Y.; Womack, C.; Wombwell, H.; Hodgson, D.; O’Connor, M.; et al. Expression of potential biomarkers associated with homologous recombination repair in patients with ovarian or triple-negative breast cancer. Cancer Biomark. Sect. Dis. Markers 2016, 16, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Su, P.; Jiao, M.; Bai, X.; Qi, M.; Liu, H.; Wu, Z.; Sun, J.; Zhou, G.; Han, B. TRPS1 suppresses breast cancer epithelial-mesenchymal transition program as a negative regulator of SUZ12. Transl. Oncol. 2018, 11, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Fesler, A.; Liu, H.; Ju, J. Development of novel miR-129 mimics with enhanced efficacy to eliminate chemoresistant colon cancer stem cells. Oncotarget 2018, 9, 8887–8897. [Google Scholar] [CrossRef] [PubMed]
- Aguilera-Aguirre, L.; Bacsi, A.; Radak, Z.; Hazra, T.K.; Mitra, S.; Sur, S.; Brasier, A.R.; Ba, X.; Boldogh, I. Innate inflammation induced by the 8-oxoguanine DNA glycosylase-1-KRAS-NF-κB pathway. J. Immunol. 2014, 193, 4643–4653. [Google Scholar] [CrossRef]
- Nicolas, E.; Bertucci, F.; Sabatier, R.; Gonçalves, A. Targeting Deficiency in Breast Cancer: What are the Clinical Evidences and the Next Perspectives? Cancers 2018, 10, 506. [Google Scholar] [CrossRef]
- Norquist, B.; Wurz, K.A.; Pennil, C.C.; Garcia, R.; Gross, J.; Sakai, W.; Karlan, B.Y.; Taniguchi, T.; Swisher, E.M. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J. Clin. Oncol. 2011, 29, 3008–3015. [Google Scholar] [CrossRef]
- Lingor, P.; Michel, U.; Schöll, U.; Bähr, M.; Kügler, S. Transfection of “naked” siRNA results in endosomal uptake and metabolic impairment in cultured neurons. Biochem. Biophys. Res. Commun. 2004, 315, 1126–1133. [Google Scholar] [CrossRef]
- Stein, C.A.; Hansen, J.B.; Lai, J.; Wu, S.; Voskresenskiy, A.; Høg, A.; Worm, J.; Hedtjärn, M.; Souleimanian, N.; Miller, P.; et al. Efficient gene silencing by delivery of locked nucleic acid antisense oligonucleotides, unassisted by transfection reagents. Nucleic Acids Res. 2010, 38, e3. [Google Scholar] [CrossRef]
- Buscail, L.; Bournet, B.; Vernejoul, F.; Cambois, G.; Lulka, H.; Hanoun, N.; Dufresne, M.; Meulle, A.; Vignolle-Vidoni, A.; Ligat, L.; et al. First-in-man phase 1 clinical trial of gene therapy for advanced pancreatic cancer: Safety, biodistribution, and preliminary clinical findings. Mol. Ther. 2015, 23, 779–789. [Google Scholar] [CrossRef]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soung, Y.H.; Chung, H.; Yan, C.; Fesler, A.; Kim, H.; Oh, E.-S.; Ju, J.; Chung, J. Therapeutic Potential of Chemically Modified miR-489 in Triple-Negative Breast Cancers. Cancers 2020, 12, 2209. https://doi.org/10.3390/cancers12082209
Soung YH, Chung H, Yan C, Fesler A, Kim H, Oh E-S, Ju J, Chung J. Therapeutic Potential of Chemically Modified miR-489 in Triple-Negative Breast Cancers. Cancers. 2020; 12(8):2209. https://doi.org/10.3390/cancers12082209
Chicago/Turabian StyleSoung, Young Hwa, Heesung Chung, Cecilia Yan, Andrew Fesler, Hyungjin Kim, Eok-Soo Oh, Jingfang Ju, and Jun Chung. 2020. "Therapeutic Potential of Chemically Modified miR-489 in Triple-Negative Breast Cancers" Cancers 12, no. 8: 2209. https://doi.org/10.3390/cancers12082209