Multiple Myeloma-Derived Extracellular Vesicles Induce Osteoclastogenesis through the Activation of the XBP1/IRE1α Axis

 , ,
, ,  , , , ,
, , , ,  ,
,
Abstract
:1. Introduction
2. Results
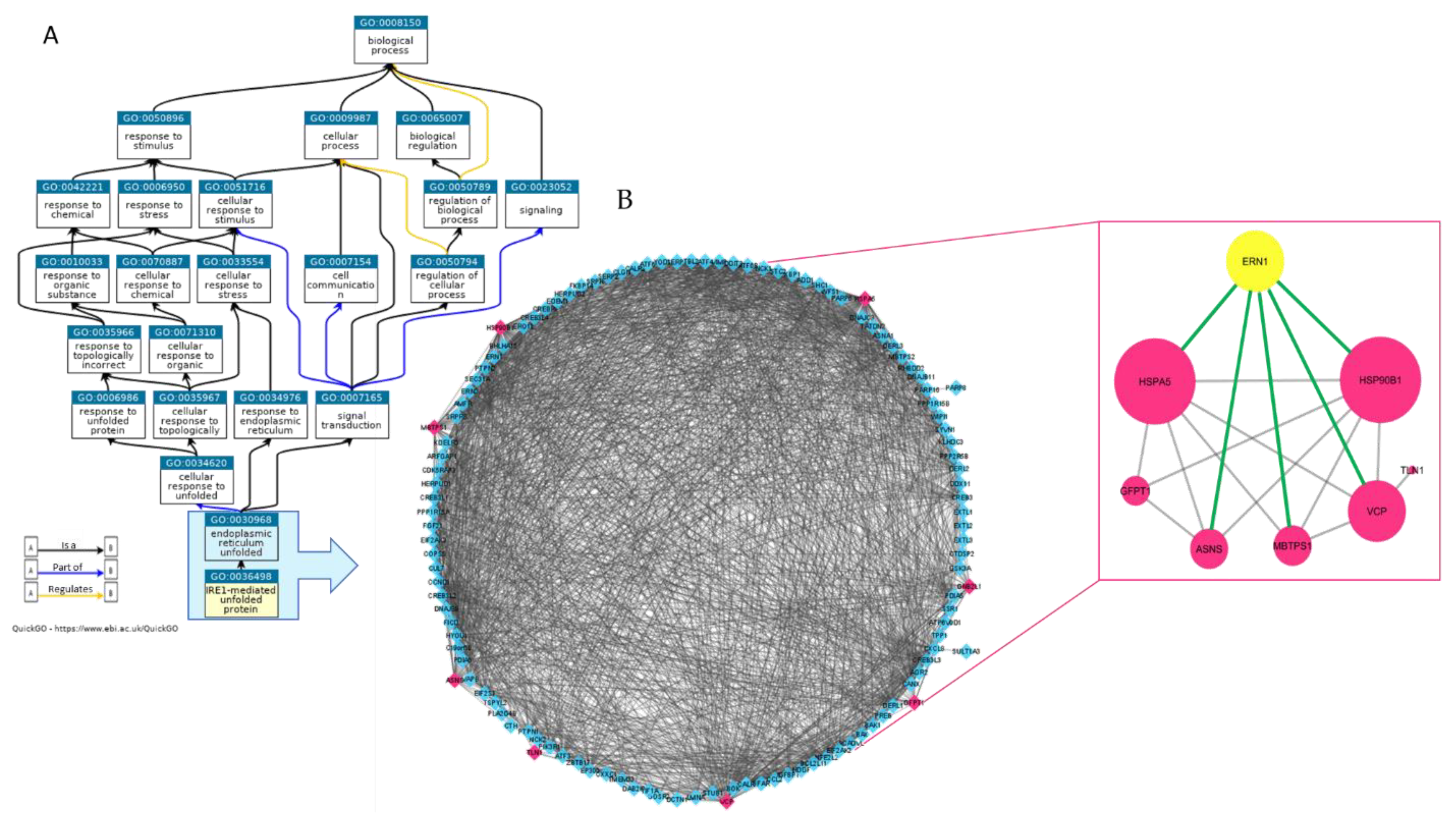
2.1. Proteomic Analysis of MM1.s-EVS Identifies a Cargo of UPR-Related Signaling Molecules
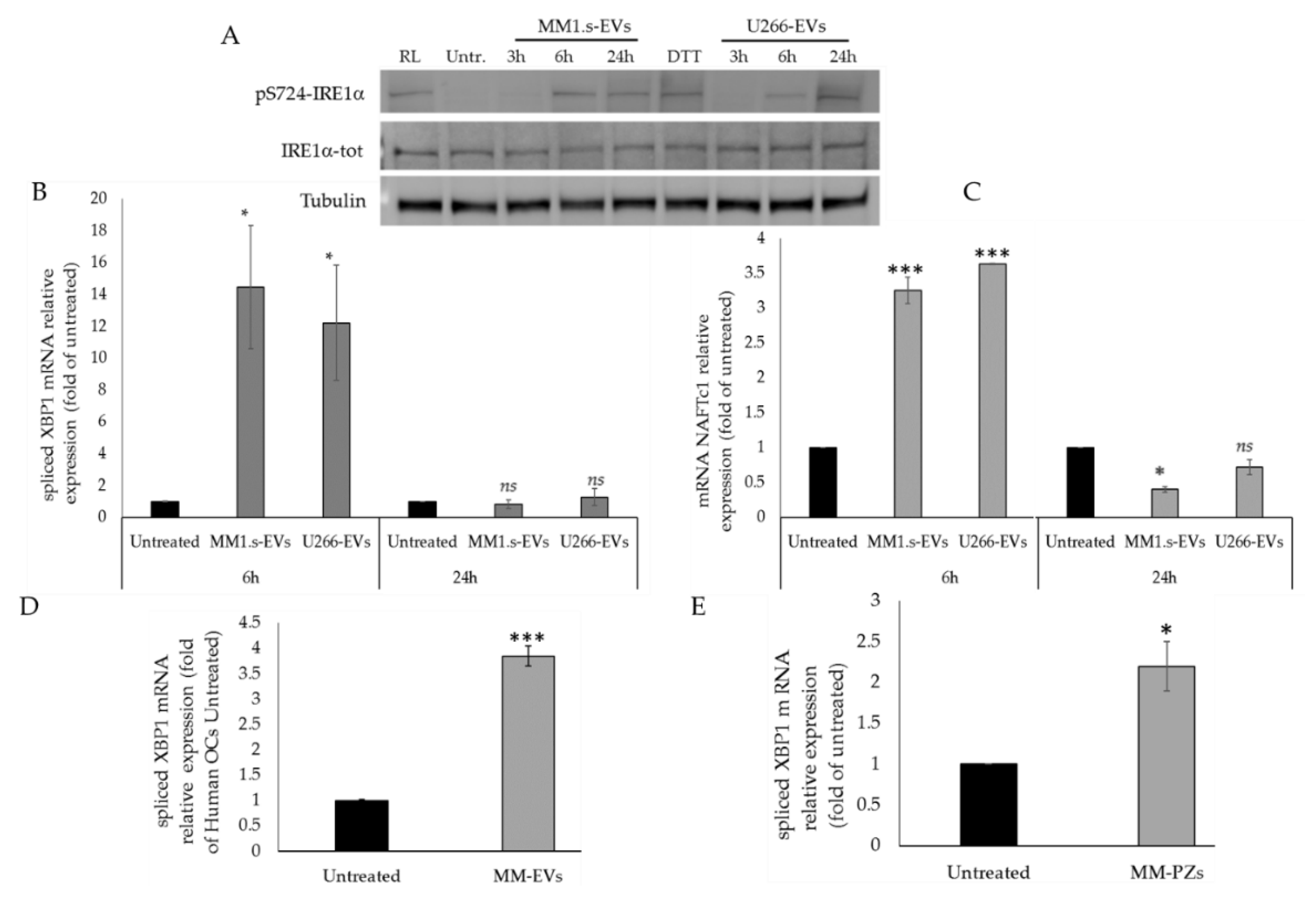
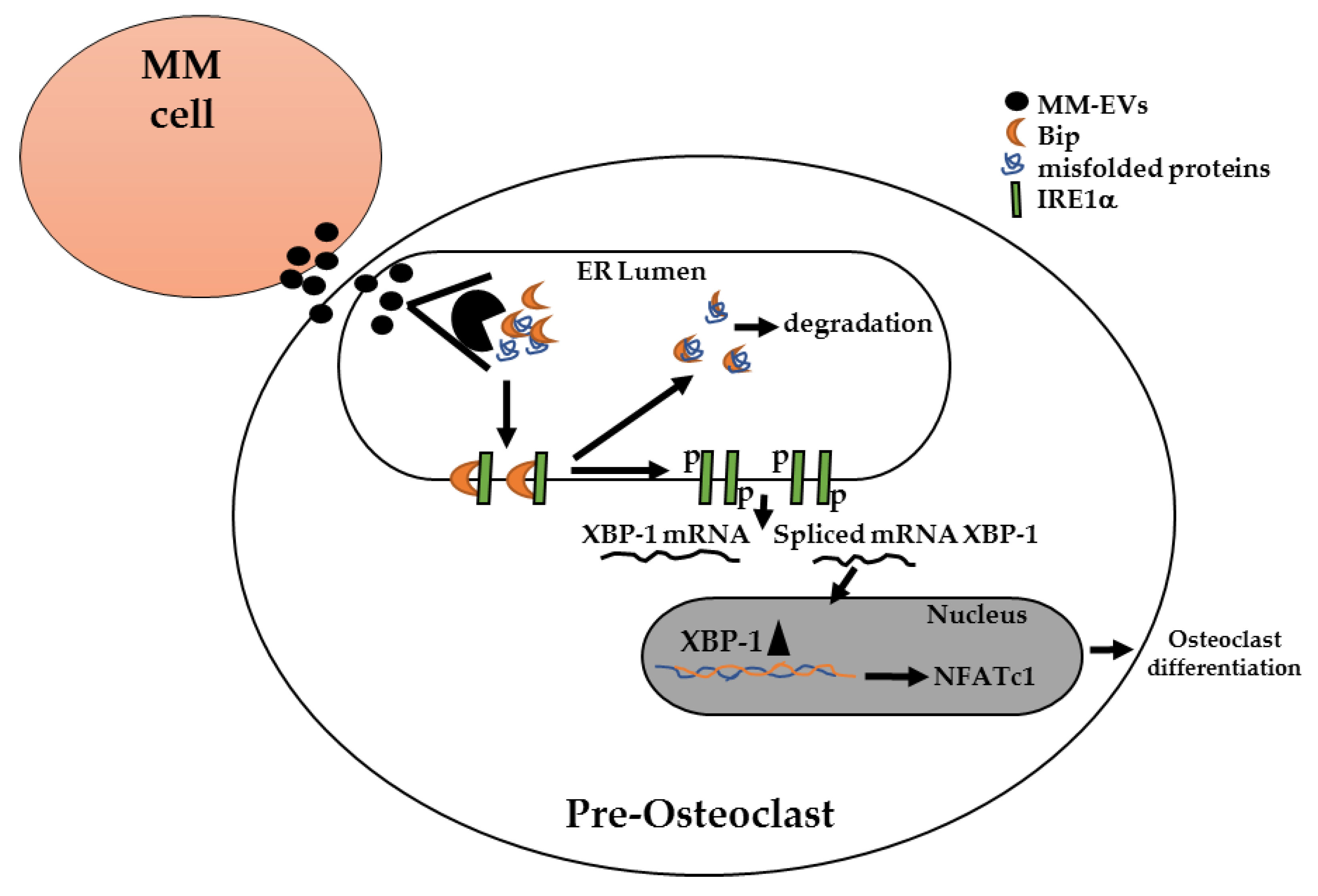
2.2. MM-EVs Affect IRE1α-XBP1 Pathway in Raw264.7 Cells
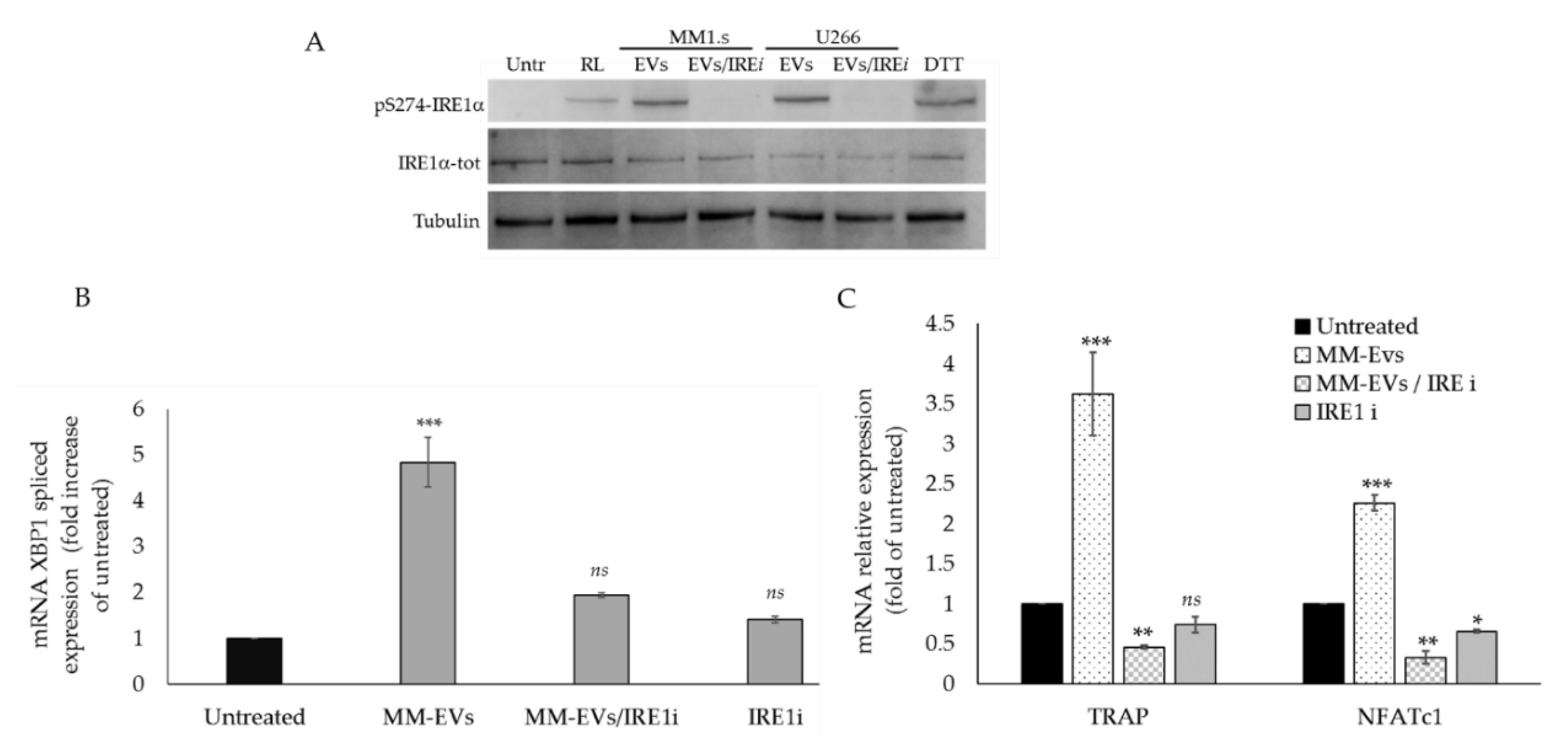
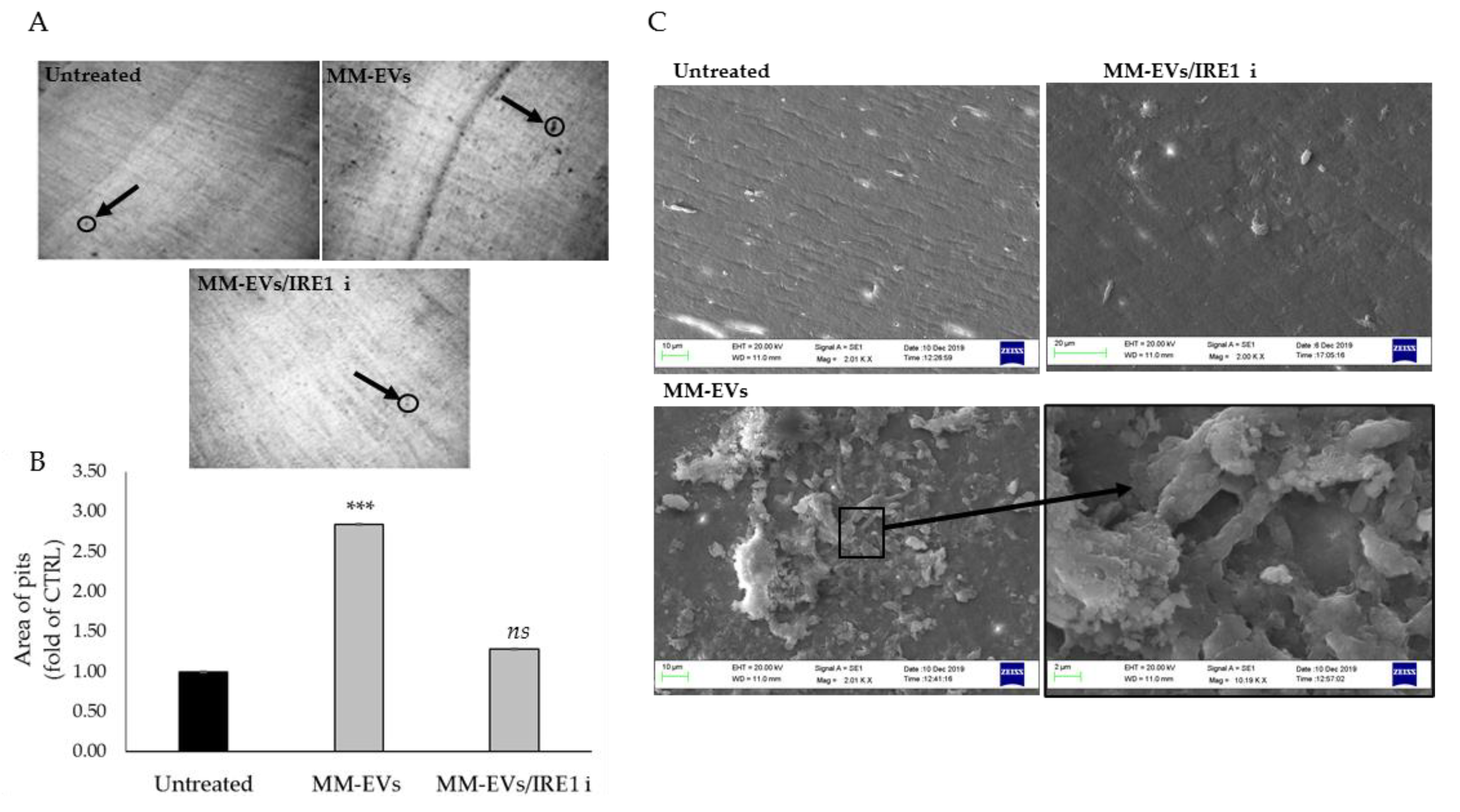
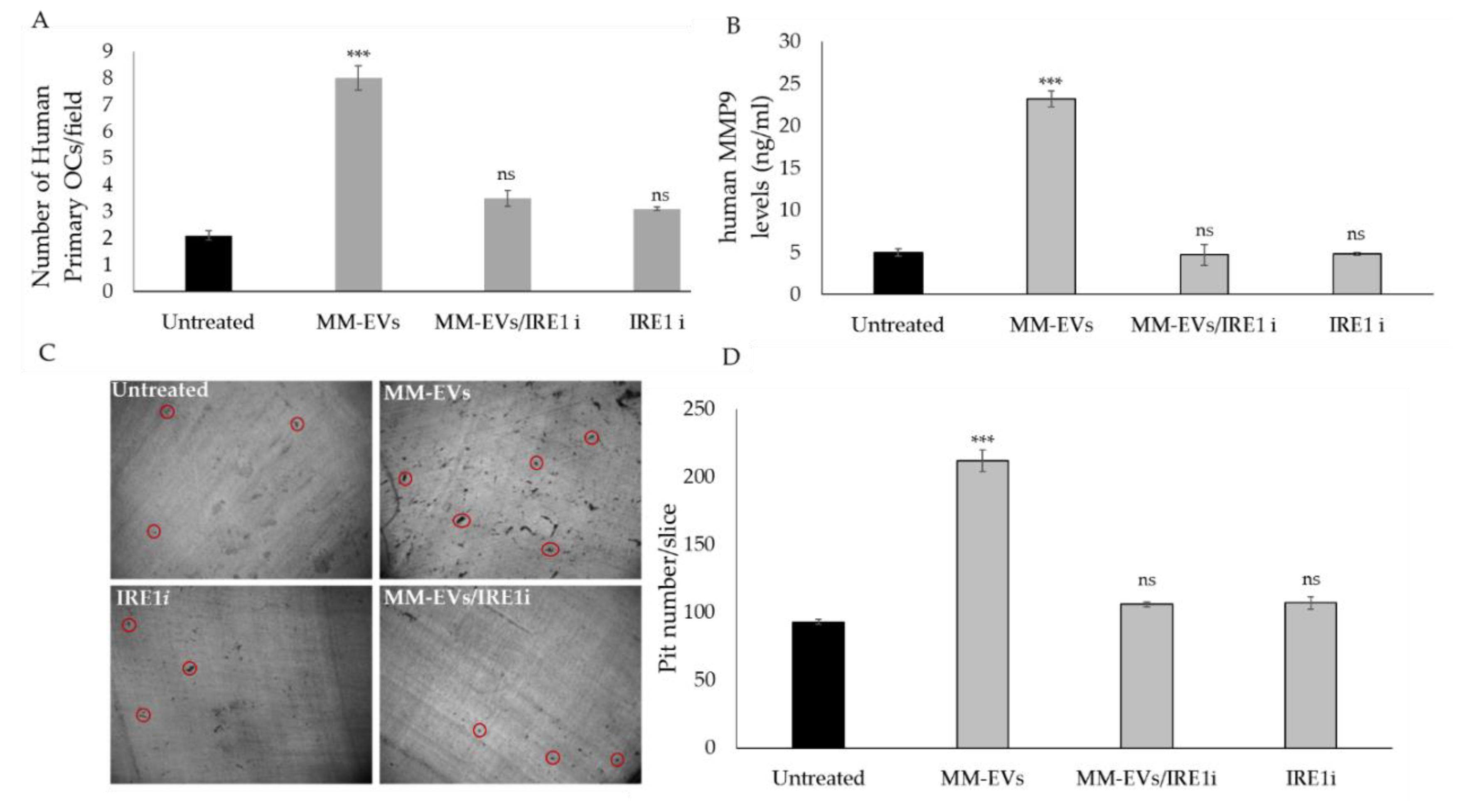
2.3. The Chemical Block of the IRE1α-XBP1 Pathway Perturbs MM-EV -Induced OC Differentiation
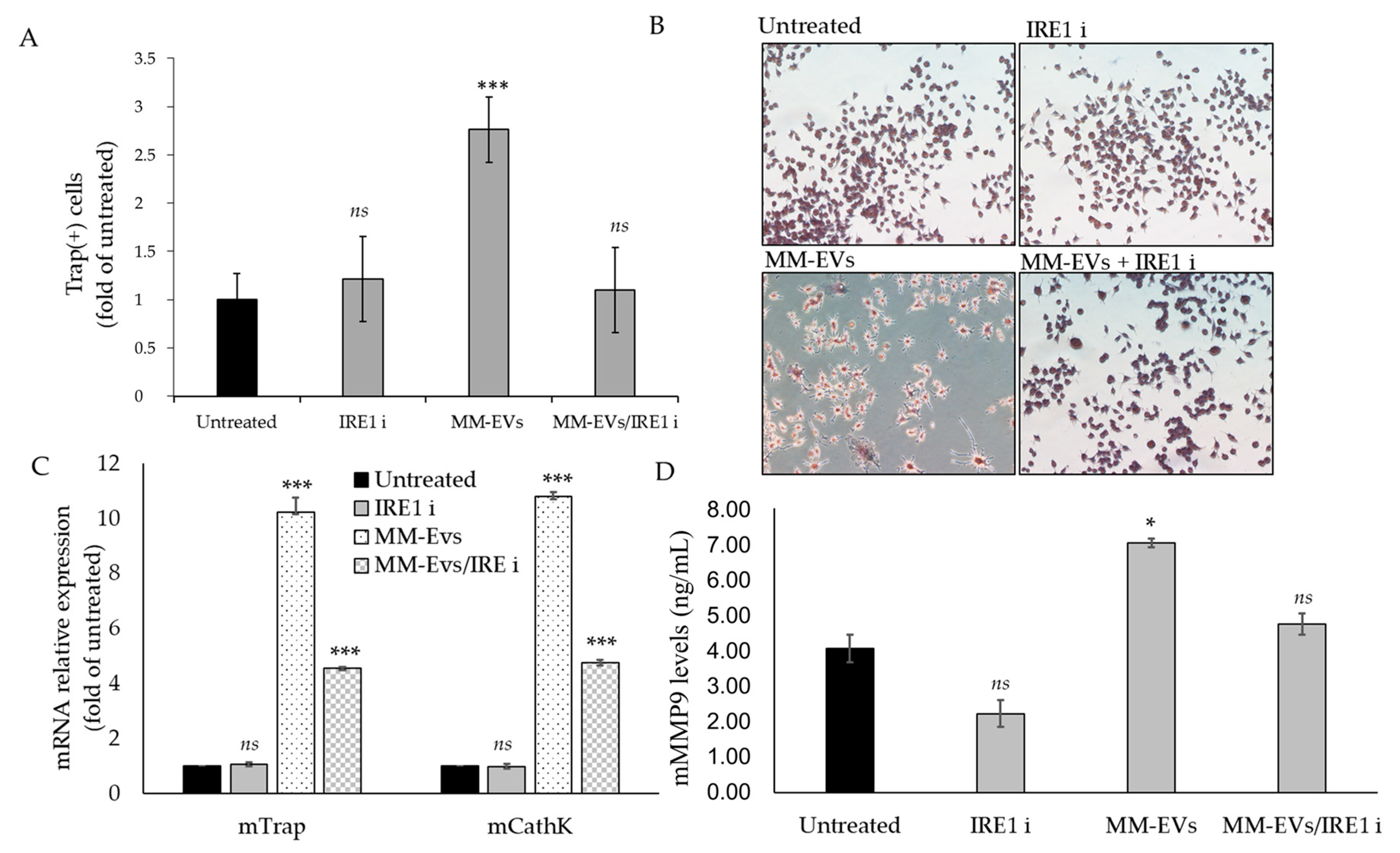
2.4. Raw264.7 Cells Co-Treated with MM1.s-EVs and IRE1α-Inhibitor Impair Terminal OC Differentiation and Reduce Bone Resorption Activity
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Small Extracellular-Vesicles Purification
4.3. Isolation of Human Peripheral Blood Mononuclear Cells
4.4. Preparation of Human Primary pOC and OCs
4.5. Uptake of Multiple Myeloma Exosomes by Raw264.7 Cells
4.6. Proteomic Analyses of MM1.s EVs: Sample Preparation, IDA and Data Analysis
4.7. Protein Identification
4.8. Bionformatic Analysis
4.9. Viability Assay (WST-1 Test)
4.10. TRAP Staining Assay
4.11. Bone Resorption Assay
4.12. ELISA Assay
4.13. Western Blot Analysis
4.14. RNA Extraction and Real-Time PCR
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Raje, N.; Roodman, G.D. Advances in the biology and treatment of bone disease in multiple myeloma. Clin. Cancer Res. 2011, 17, 1278–1286. [Google Scholar] [CrossRef] [Green Version]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; Dimopoulos, M.A. Pathogenesis of bone disease in multiple myeloma: From bench to bedside. Blood Cancer J. 2018, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Lemaire, M.; Deleu, S.; De Bruyne, E.; Van Valckenborgh, E.; Menu, E.; Vanderkerken, K. The microenvironment and molecular biology of the multiple myeloma tumor. Adv. Cancer Res. 2011, 110, 19–42. [Google Scholar] [CrossRef]
- Terpos, E.; Morgan, G.; Dimopoulos, M.A.; Drake, M.T.; Lentzsch, S.; Raje, N.; Sezer, O.; García-Sanz, R.; Shimizu, K.; Turesson, I.; et al. International Myeloma Working Group recommendations for the treatment of multiple myeloma-related bone disease. J. Clin. Oncol. 2013, 31, 2347–2357. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.; Di Martino, M.T.; Morelli, E.; Leotta, M.; Rizzo, A.; Grimaldi, A.; Misso, G.; Tassone, P.; Caraglia, M. Molecular targets for the treatment of multiple myeloma. Curr. Cancer Drug Targets 2012, 12, 757–767. [Google Scholar] [CrossRef]
- Ring, E.S.; Lawson, M.A.; Snowden, J.A.; Jolley, I.; Chantry, A.D. New agents in the Treatment of Myeloma Bone Disease. Calcif. Tissue Int. 2018, 102, 196–209. [Google Scholar] [CrossRef] [Green Version]
- Faict, S.; Muller, J.; De Veirman, K.; De Bruyne, E.; Maes, K.; Vrancken, L.; Heusschen, R.; De Raeve, H.; Schots, R.; Vanderkerken, K.; et al. Exosomes play a role in multiple myeloma bone disease and tumor development by targeting osteoclasts and osteoblasts. Blood Cancer J. 2018, 8, 105. [Google Scholar] [CrossRef] [Green Version]
- Bai, H.; Lei, K.; Huang, F.; Jiang, Z.; Zhou, X. Exo-circRNAs: A new paradigm for anticancer therapy. Mol. Cancer 2019, 18, 56. [Google Scholar] [CrossRef]
- Raimondi, L.; De Luca, A.; Amodio, N.; Manno, M.; Raccosta, S.; Taverna, S.; Bellavia, D.; Naselli, F.; Fontana, S.; Schillaci, O.; et al. Involvement of multiple myeloma cell-derived exosomes in osteoclast differentiation. Oncotarget 2015, 6, 13772–13789. [Google Scholar] [CrossRef] [Green Version]
- Raimondo, S.; Saieva, L.; Vicario, E.; Pucci, M.; Toscani, D.; Manno, M.; Raccosta, S.; Giuliani, N.; Alessandro, R. Multiple myeloma-derived exosomes are enriched of amphiregulin (AREG) and activate the epidermal growth factor pathway in the bone microenvironment leading to osteoclastogenesis. J. Hematol. Oncol. 2019, 12, 2. [Google Scholar] [CrossRef]
- Li, B.; Xu, H.; Han, H.; Song, S.; Zhang, X.; Ouyang, L.; Qian, C.; Hong, Y.; Qiu, Y.; Zhou, W.; et al. Exosome-mediated transfer of lncRUNX2-AS1 from multiple myeloma cells to MSCs contributes to osteogenesis. Oncogene 2018, 37, 5508–5519. [Google Scholar] [CrossRef]
- Gupta, A.; Read, D.E.; Gupta, S. Assays for induction of the unfolded protein response and selective activation of the three major pathways. Methods Mol. Biol. 2015, 1292, 19–38. [Google Scholar] [CrossRef]
- Liu, C.Y.; Kaufman, R.J. The unfolded protein response. J. Cell Sci. 2003, 116, 1861–1862. [Google Scholar] [CrossRef] [Green Version]
- Carrasco, D.R.; Sukhdeo, K.; Protopopova, M.; Sinha, R.; Enos, M.; Carrasco, D.E.; Zheng, M.; Mani, M.; Henderson, J.; Pinkus, G.S.; et al. The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer Cell 2007, 11, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Vincenz, L.; Jäger, R.; O’Dwyer, M.; Samali, A. Endoplasmic reticulum stress and the unfolded protein response: Targeting the Achilles heel of multiple myeloma. Mol. Cancer Ther. 2013, 12, 831–843. [Google Scholar] [CrossRef] [Green Version]
- Aronson, L.I.; Davies, F.E. DangER: Protein ovERload. Targeting protein degradation to treat myeloma. Haematologica 2012, 97, 1119–1130. [Google Scholar] [CrossRef]
- Tohmonda, T.; Yoda, M.; Iwawaki, T.; Matsumoto, M.; Nakamura, M.; Mikoshiba, K.; Toyama, Y.; Horiuchi, K. IRE1α/XBP1-mediated branch of the unfolded protein response regulates osteoclastogenesis. J. Clin. Investig. 2015, 125, 3269–3279. [Google Scholar] [CrossRef] [Green Version]
- Song, I.; Kim, J.H.; Kim, K.; Jin, H.M.; Youn, B.U.; Kim, N. Regulatory mechanism of NFATc1 in RANKL-induced osteoclast activation. FEBS Lett. 2009, 583, 2435–2440. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Kim, N. Regulation of NFATc1 in Osteoclast Differentiation. J. Bone Metab. 2014, 21, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Hino, S.; Kondo, S.; Yoshinaga, K.; Saito, A.; Murakami, T.; Kanemoto, S.; Sekiya, H.; Chihara, K.; Aikawa, Y.; Hara, H.; et al. Regulation of ER molecular chaperone prevents bone loss in a murine model for osteoporosis. J. Bone Miner. Metab. 2010, 28, 131–138. [Google Scholar] [CrossRef]
- Shi, M.; Song, W.; Han, T.; Chang, B.; Li, G.; Jin, J.; Zhang, Y. Role of the unfolded protein response in topography-induced osteogenic differentiation in rat bone marrow mesenchymal stem cells. Acta Biomat. 2017, 54, 175–185. [Google Scholar] [CrossRef]
- Wu, C.H.; Silvers, C.R.; Messing, E.M.; Lee, Y.F. Bladder cancer extracellular vesicles drive tumorigenesis by inducing the unfolded protein response in endoplasmic reticulum of nonmalignant cells. J. Biol. Chem. 2019, 294, 3207–3218. [Google Scholar] [CrossRef] [Green Version]
- Raimondo, S.; Saieva, L.; Corrado, C.; Fontana, S.; Flugy, A.; Rizzo, A.; De Leo, G.; Alessandro, R. Chronic myeloid leukemia-derived exosomes promote tumor growth through an autocrine mechanism. Cell Commun. Signal. 2015, 13, 8. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell. Biol. 2006. [Google Scholar] [CrossRef]
- Collin-Osdoby, P.; Osdoby, P. RANKL-mediated osteoclast formation from murine RAW 264.7 cells. Methods Mol. Biol. 2012, 816, 187–202. [Google Scholar] [CrossRef]
- Cuetara, B.L.; Crotti, T.N.; O’Donoghue, A.J.; McHugh, K.P. Cloning and characterization of osteoclast precursors from the RAW264.7 cell line. In Vitro Cell Dev. Biol. Anim. 2006, 42, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Tohmonda, T.; Miyauchi, Y.; Ghosh, R.; Yoda, M.; Uchikawa, S.; Takito, J.; Morioka, H.; Nakamura, M.; Iwawaki, T.; Chiba, K.; et al. The IRE1α-XBP1 pathway is essential for osteoblast differentiation through promoting transcription of Osterix. EMBO Rep. 2011, 12, 451–457. [Google Scholar] [CrossRef] [Green Version]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, M.; Kogawa, M.; Wada, S.; Takayanagi, H.; Tsujimoto, M.; Katayama, S.; Hisatake, K.; Nogi, Y. Essential role of p38 mitogen-activated protein kinase in cathepsin K gene expression during osteoclastogenesis through association of NFATc1 and PU.1. J. Biol. Chem. 2004, 279, 45969–45979. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Sato, K.; Asagiri, M.; Morita, I.; Soma, K.; Takayanagi, H. Contribution of nuclear factor of activated T cells c1 to the transcriptional control of immunoreceptor osteoclast-associated receptor but not triggering receptor expressed by myeloid cells-2 during osteoclastogenesis. J. Biol. Chem. 2005, 280, 32905–32913. [Google Scholar] [CrossRef] [Green Version]
- Yaccoby, S.; Wezeman, M.J.; Henderson, A.; Cottler-Fox, M.; Yi, Q.; Barlogie, B.; Epstein, J. Cancer and the microenvironment: Myeloma-osteoclast interactions as a model. Cancer Res. 2004, 64, 2016–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Concha, N.O.; Smallwood, A.; Bonnette, W.; Totoritis, R.; Zhang, G.; Federowicz, K.; Yang, J.; Qi, H.; Chen, S.; Campobasso, N.; et al. Long-Range Inhibitor-Induced Conformational Regulation of Human IRE1α Endoribonuclease Activity. Mol. Pharmacol. 2015, 88, 1011–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Børset, M.; Sundan, A.; Waage, A.; Standal, T. Why do myeloma patients have bone disease? A historical perspective. Blood Rev. 2019, 100646. [Google Scholar] [CrossRef] [PubMed]
- Lomas, O.C.; Tahri, S.; Ghobrial, I.M. The microenvironment in myeloma. Curr. Opin. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.; Petrusca, D.N.; Roodman, G.D. Therapeutic targets in myeloma bone disease. Br. J. Pharmacol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Giannandrea, D.; Lesma, E.; Basile, A.; Chiaramonte, R. Extracellular Vesicles Enhance Multiple Myeloma Metastatic Dissemination. Int. J. Mol. Sci. 2019, 20, 3236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.E. Extracellular Vesicles and Metastasis. Cold Spring Harb. Perspect. Med. 2019. [Google Scholar] [CrossRef]
- Feng, W.; Dean, D.C.; Hornicek, F.J.; Shi, H.; Duan, Z. Exosomes promote pre-metastatic niche formation in ovarian cancer. Mol. Cancer 2019, 18, 124. [Google Scholar] [CrossRef] [Green Version]
- Raimondi, L.; De Luca, A.; Gallo, A.; Costa, V.; Russelli, G.; Cuscino, N.; Manno, M.; Raccosta, S.; Carina, V.; Bellavia, D.; et al. Osteosarcoma cell-derived exosomes affect tumor microenvironment by specific packaging of microRNAs. Carcinogenesis 2019. [Google Scholar] [CrossRef]
- Li, F.X.; Liu, J.J.; Xu, F.; Lin, X.; Zhong, J.Y.; Wu, F.; Yuan, L.Q. Role of tumor-derived exosomes in bone metastasis. Oncol. Lett. 2019, 18, 3935–3945. [Google Scholar] [CrossRef]
- Siwecka, N.; Rozpędek, W.; Pytel, D.; Wawrzynkiewicz, A.; Dziki, A.; Dziki, Ł.; Diehl, J.A.; Majsterek, I. Dual role of Endoplasmic Reticulum Stress-Mediated Unfolded Protein Response Signaling Pathway in Carcinogenesis. Int. J. Mol. Sci. 2019, 20, 4354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, S.K.; Chiu, C.C.; Dahms, H.U.; Chou, C.K.; Cheng, C.M.; Chang, W.T.; Cheng, K.C.; Wang, H.D.; Lin, I.L. Unfolded Protein Response (UPR) in Survival, Dormancy, Immunosuppression, Metastasis, and Treatments of Cancer Cells. Int. J. Mol. Sci. 2019, 20, 2518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doron, B.; Abdelhamed, S.; Butler, J.T.; Hashmi, S.K.; Horton, T.M.; Kurre, P. Transmissible ER stress reconfigures the AML bone marrow compartment. Leukemia 2018. [Google Scholar] [CrossRef]
- Hosoi, T.; Nakashima, M.; Ozawa, K. Incorporation of the Endoplasmic Reticulum Stress-Induced Spliced Form of XBP1 mRNA in the Exosomes. Front. Physiol. 2018, 9, 1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahadevan, N.R.; Rodvold, J.; Sepulveda, H.; Rossi, S.; Drew, A.F.; Zanetti, M. Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc. Natl. Acad. Sci. USA 2011, 108, 6561–6566. [Google Scholar] [CrossRef] [Green Version]
- Butler, J.T.; Kurre, P. Transmissible ER stress shapes the leukemic microenvironment. Oncotarget 2019, 10, 4080–4082. [Google Scholar] [CrossRef]
- Heusermann, W.; Hean, J.; Trojer, D.; Steib, E.; von Bueren, S.; Graff-Meyer, A.; Genoud, C.; Martin, K.; Pizzato, N.; Voshol, J.; et al. Exosomes surf on filopodia to enter cells at endocytic hot spots, traffic within endosomes, and are targeted to the ER. J. Cell. Biol. 2016, 213, 173–184. [Google Scholar] [CrossRef]
- Jagannathan, S.; Hsu, J.C.; Reid, D.W.; Chen, Q.; Thompson, W.J.; Moseley, A.M.; Nicchitta, C.V. Multifunctional roles for the protein translocation machinery in RNA anchoring to the endoplasmic reticulum. J. Biol. Chem. 2014, 289, 25907–25924. [Google Scholar] [CrossRef] [Green Version]
- Reid, D.W.; Nicchitta, C.V. Diversity and selectivity in mRNA translation on the endoplasmic reticulum. Nat. Rev. Mol. Cell. Biol. 2015, 16, 221–231. [Google Scholar] [CrossRef]
- Tokutake, Y.; Yamada, K.; Hayash, S.; Arai, W.; Watanabe, T.; Yonekura, S. IRE1-XBP1 Pathway of the Unfolded Protein Response Is Required during Early Differentiation of C2C12 Myoblasts. Int. J. Mol. Sci. 2020, 21, 182. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.; Smith, W.; Hao, D. Overview of RAW264.7 for osteoclastogensis study: Phenotype and stimuli. J. Cell. Mol. Med. 2019, 23, 3077–3087. [Google Scholar] [CrossRef] [Green Version]
- Tomasetti, M.; Lee, W.; Santarelli, L.; Neuzil, J. Exosome-derived microRNAs in cancer metabolism: Possible implications in cancer diagnostics and therapy. Exp. Mol. Med. 2017, 49, e285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, I.; Nabet, B.Y. Exosomes in the tumor microenvironment as mediators of cancer therapy resistance. Mol. Cancer 2019, 18, 32. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Chen, M.; Jiang, R.; Guo, Y.; Wu, M.; Zhang, X. Exosome-related tumor microenvironment. J. Cancer 2018, 9, 3084–3092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taverna, S.; Flugy, A.; Saieva, L.; Kohn, E.C.; Santoro, A.; Meraviglia, S.; De Leo, G.; Alessandro, R. Role of exosomes released by chronic myelogenous leukemia cells in angiogenesis. Int. J. Cancer 2012, 130, 2033–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schillaci, O.; Fontana, S.; Monteleone, F.; Taverna, S.; Di Bella, M.A.; Di Vizio, D.; Alessandro, R. Exosomes from metastatic cancer cells transfer amoeboid phenotype to non-metastatic cells and increase endothelial permeability: Their emerging role in tumor heterogeneity. Sci. Rep. 2017, 7, 4711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Pan, I.; Roitenberg, N.; Cohen, E. Vesicle-mediated secretion of misfolded prion protein molecules from cyclosporin A-treated cells. FASEB J. 2018, 32, 1479–1492. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| NFATc1 | GGGTCAGTGTGACCGAAGAT | GGAAGTCAGAAGTGGGTGGA |
| Trap | GCGACCATTGTTAGCCACATACG | CGTTGATGTCGCACAGAGGGAT |
| uXBP1 | CCGCAGCACTCAGACTATG | GGGTCCAACTTGTCCAGAAT |
| sXBP1 | CTGAGTCCGCAGCAGGT | AAACATGACAGGGTCCAACTT |
| GAPDH | CCCAGAAGACTGTGGATGG | CAGATTGGGGGTAGGAACAC |
| Hu GAPDH | ATGGGGAAGGTGAAGGTCG | GGGTCATTGATGGCAACAATAT |
| Hu sXBP1 | AGACAGCGCTTGGGGATGGAT | CCTGCACCTGCTGCGGACTC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raimondi, L.; De Luca, A.; Fontana, S.; Amodio, N.; Costa, V.; Carina, V.; Bellavia, D.; Raimondo, S.; Siragusa, S.; Monteleone, F.; et al. Multiple Myeloma-Derived Extracellular Vesicles Induce Osteoclastogenesis through the Activation of the XBP1/IRE1α Axis. Cancers 2020, 12, 2167. https://doi.org/10.3390/cancers12082167
Raimondi L, De Luca A, Fontana S, Amodio N, Costa V, Carina V, Bellavia D, Raimondo S, Siragusa S, Monteleone F, et al. Multiple Myeloma-Derived Extracellular Vesicles Induce Osteoclastogenesis through the Activation of the XBP1/IRE1α Axis. Cancers. 2020; 12(8):2167. https://doi.org/10.3390/cancers12082167
Chicago/Turabian StyleRaimondi, Lavinia, Angela De Luca, Simona Fontana, Nicola Amodio, Viviana Costa, Valeria Carina, Daniele Bellavia, Stefania Raimondo, Sergio Siragusa, Francesca Monteleone, and et al. 2020. "Multiple Myeloma-Derived Extracellular Vesicles Induce Osteoclastogenesis through the Activation of the XBP1/IRE1α Axis" Cancers 12, no. 8: 2167. https://doi.org/10.3390/cancers12082167