Molecular Bases of Mechanisms Accounting for Drug Resistance in Gastric Adenocarcinoma

 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
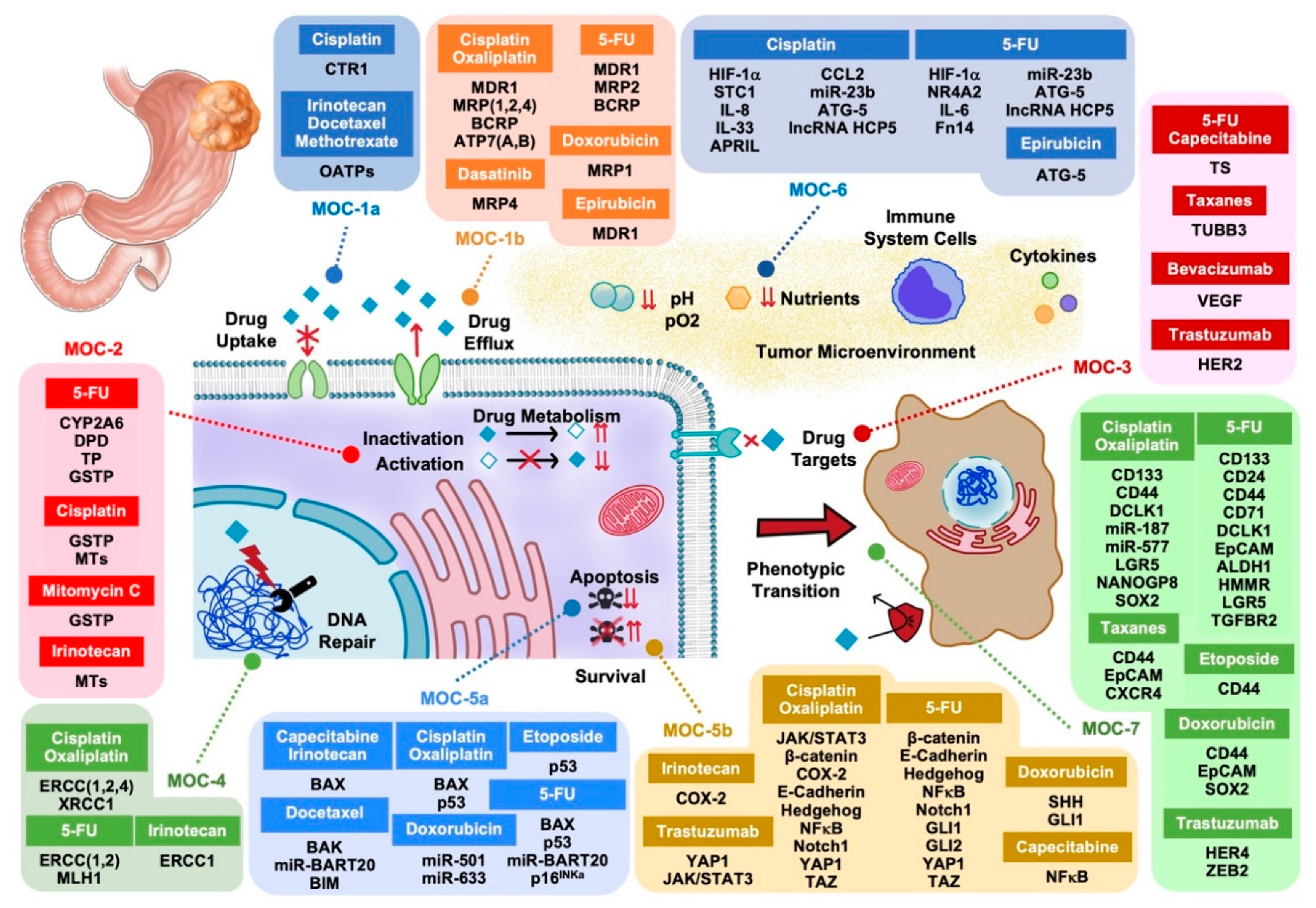
2. Mechanisms of Chemoresistance Type 1 (MOC-1)
2.1. Uptake Transporters (MOC-1a)
2.2. Export Pumps (MOC-1b)
3. Mechanisms of Chemoresistance Type 2 (MOC-2)
4. Mechanisms of Chemoresistance Type 3 (MOC-3)
5. Mechanisms of Chemoresistance Type 4 (MOC-4)
6. Mechanisms of Chemoresistance Type 5 (MOC-5)
6.1. Pro-Apoptotic Factors (MOC-5a)
6.2. Survival Pathways (MOC-5b)
7. Mechanisms of Chemoresistance Type 6 (MOC-6)
8. Mechanisms of Chemoresistance Type 7 (MOC-7)
9. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Rawla, P.; Barsouk, A. Epidemiology of gastric cancer: Global trends, risk factors and prevention. Prz. Gastroenterol. 2019, 14, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsina, M.; Miquel, J.M.; Diez, M.; Castro, S.; Tabernero, J. How I treat gastric adenocarcinoma. ESMO Open 2019, 4, e000521. [Google Scholar] [CrossRef] [Green Version]
- Marin, J.J.; Al-Abdulla, R.; Lozano, E.; Briz, O.; Bujanda, L.; Banales, J.M.; Macias, R.I. Mechanisms of Resistance to Chemotherapy in Gastric Cancer. Anticancer Agents Med. Chem. 2016, 16, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Marin, J.J.G.; Briz, O.; Herraez, E.; Lozano, E.; Asensio, M.; Di Giacomo, S.; Romero, M.R.; Osorio-Padilla, L.M.; Santos-Llamas, A.I.; Serrano, M.A.; et al. Molecular bases of the poor response of liver cancer to chemotherapy. Clin. Res. Hepatol. Gastroenterol. 2018, 42, 182–192. [Google Scholar] [CrossRef]
- Marin, J.J.; Romero, M.R.; Martinez-Becerra, P.; Herraez, E.; Briz, O. Overview of the molecular bases of resistance to chemotherapy in liver and gastrointestinal tumours. Curr. Mol. Med. 2009, 9, 1108–1129. [Google Scholar] [CrossRef]
- Marin, J.J.; Briz, O.; Monte, M.J.; Blazquez, A.G.; Macias, R.I. Genetic variants in genes involved in mechanisms of chemoresistance to anticancer drugs. Curr. Cancer Drug Targets 2012, 12, 402–438. [Google Scholar] [CrossRef]
- Obuchi, W.; Ohtsuki, S.; Uchida, Y.; Ohmine, K.; Yamori, T.; Terasaki, T. Identification of transporters associated with Etoposide sensitivity of stomach cancer cell lines and methotrexate sensitivity of breast cancer cell lines by quantitative targeted absolute proteomics. Mol. Pharmacol. 2013, 83, 490–500. [Google Scholar] [CrossRef] [Green Version]
- Al-Abdulla, R.; Perez-Silva, L.; Lozano, E.; Macias, R.I.R.; Herraez, E.; Abad, M.; Segues, N.; Bujanda, L.; Briz, O.; Marin, J.J.G. Sensitizing gastric adenocarcinoma to chemotherapy by pharmacological manipulation of drug transporters. Biochem. Pharmacol. 2020, 171, 113682. [Google Scholar] [CrossRef]
- Holzer, A.K.; Varki, N.M.; Le, Q.T.; Gibson, M.A.; Naredi, P.; Howell, S.B. Expression of the human copper influx transporter 1 in normal and malignant human tissues. J. Histochem. Cytochem. 2006, 54, 1041–1049. [Google Scholar] [CrossRef] [Green Version]
- Takechi, T.; Koizumi, K.; Tsujimoto, H.; Fukushima, M. Screening of differentially expressed genes in 5-fluorouracil-resistant human gastrointestinal tumor cells. JPN J. Cancer Res. 2001, 92, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Zhang, C.; Zhao, Y.; Bai, B.; An, J.; Zhang, H.; Wu, J.B.; Shi, C. Optical imaging of gastric cancer with near-infrared heptamethine carbocyanine fluorescence dyes. Oncotarget 2016, 7, 57277–57289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira, J.; Felipe, A.V.; Neto, R.A.; Oshima, C.T.; De Souza Silva, M.; Forones, N.M. Association between ABCB1 immunohistochemical expression and overall survival in gastric cancer patients. Asian Pac. J. Cancer Prev. 2014, 15, 6935–6938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, J.; Shen, J.; Xie, G.; Wu, J.; He, M.; Gao, L.; Zhang, Y.; Yao, X.; Shen, L. Cancer-associated fibroblasts-derived IL-8 mediates resistance to cisplatin in human gastric cancer. Cancer Lett. 2019, 454, 37–43. [Google Scholar] [CrossRef]
- Mo, D.; Fang, H.; Niu, K.; Liu, J.; Wu, M.; Li, S.; Zhu, T.; Aleskandarany, M.A.; Arora, A.; Lobo, D.N.; et al. Human Helicase RECQL4 Drives Cisplatin Resistance in Gastric Cancer by Activating an AKT-YB1-MDR1 Signaling Pathway. Cancer Res. 2016, 76, 3057–3066. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Zheng, Y.; Han, B.; Dong, X. Long noncoding RNA BLACAT1 modulates ABCB1 to promote oxaliplatin resistance of gastric cancer via sponging miR-361. Biomed. Pharmacother. 2018, 99, 832–838. [Google Scholar] [CrossRef]
- Xia, L.L.; Tang, Y.B.; Song, F.F.; Xu, L.; Ji, P.; Wang, S.J.; Zhu, J.M.; Zhang, Y.; Zhao, G.P.; Wang, Y.; et al. DCTPP1 attenuates the sensitivity of human gastric cancer cells to 5-fluorouracil by up-regulating MDR1 expression epigenetically. Oncotarget 2016, 7, 68623–68637. [Google Scholar] [CrossRef]
- Felipe, A.V.; Oliveira, J.; Moraes, A.A.; Franca, J.P.; Silva, T.D.; Forones, N.M. Reversal of Multidrug Resistance in an Epirubicin-Resistant Gastric Cancer Cell Subline. Asian Pac. J. Cancer Prev. 2018, 19, 1237–1242. [Google Scholar] [CrossRef]
- Huang, Y.S.; Xue, Z.; Zhang, H. Sorafenib reverses resistance of gastric cancer to treatment by cisplatin through down-regulating MDR1 expression. Med. Oncol. 2015, 32, 470. [Google Scholar] [CrossRef]
- Hotta, T.; Tanimura, H.; Yamaue, H.; Iwahashi, M.; Tani, M.; Tsunoda, T.; Tamai, M.; Noguchi, K.; Mizobata, S.; Arii, K.; et al. Tamoxifen circumvents the multidrug resistance in fresh human gastrointestinal cancer cells. J. Surg. Res. 1996, 66, 31–35. [Google Scholar] [CrossRef]
- Mao, Z.; Zhou, J.; Luan, J.; Sheng, W.; Shen, X.; Dong, X. Tamoxifen reduces P-gp-mediated multidrug resistance via inhibiting the PI3K/Akt signaling pathway in ER-negative human gastric cancer cells. Biomed. Pharmacother. 2014, 68, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Wongsirisin, P.; Limpakan Yamada, S.; Yodkeeree, S.; Punfa, W.; Limtrakul, P. Association of DNA Repair and Drug Transporter in Relation to Chemosensitivity in Primary Culture of Thai Gastric Cancer Patients. Biol. Pharm. Bull. 2018, 41, 360–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Dang, Y.; Liu, S.; Zhang, Y.; Zhang, G. LncRNA HOTAIR promotes cisplatin resistance in gastric cancer by targeting miR-126 to activate the PI3K/AKT/MRP1 genes. Tumour Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.W.; Liu, L.; Zhang, X.Z.; Bo, P. Kanglaite inhibits the expression of drug resistance genes through suppressing PVT1 in cisplatin-resistant gastric cancer cells. Exp. Ther. Med. 2017, 14, 1789–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Chen, L.; Xiao, Z.; Zhu, Y.; Jiang, H.; Jin, Y.; Gu, C.; Wu, Y.; Wang, L.; Zhang, W.; et al. Potentiation of the anticancer effect of doxorubicinin drug-resistant gastric cancer cells by tanshinone IIA. Phytomedicine 2018, 51, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xing, X.; Shan, F.; Li, S.; Xiao, A.; Xing, Z.; Xue, K.; Hu, Y.; Jia, Y.; Miao, R.; et al. ABCC2-24C > T polymorphism is associated with the response to platinum/5-Fu-based neoadjuvant chemotherapy and better clinical outcomes in advanced gastric cancer patients. Oncotarget 2016, 7, 55449–55457. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.H.; Wu, Q.; Xiao, X.Y.; Li, D.W.; Wang, X.P. Silencing MRP4 by small interfering RNA reverses acquired DDP resistance of gastric cancer cell. Cancer Lett. 2010, 291, 76–82. [Google Scholar] [CrossRef]
- Furmanski, B.D.; Hu, S.; Fujita, K.I.; Li, L.; Gibson, A.A.; Janke, L.J.; Williams, R.T.; Schuetz, J.D.; Sparreboom, A.; Baker, S.D. Contribution of ABCC4-mediated gastric transport to the absorption and efficacy of dasatinib. Clin. Cancer Res. 2013, 19, 4359–4370. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Guo, X.; Zhang, D.; Fan, Y.; Qin, L.; Dong, S. Upregulated miR-132 in Lgr5(+) gastric cancer stem cell-like cells contributes to cisplatin-resistance via SIRT1/CREB/ABCG2 signaling pathway. Mol. Carcinog. 2017, 56, 2022–2034. [Google Scholar] [CrossRef]
- Yu, B.; Gu, D.; Zhang, X.; Li, J.; Liu, B.; Xie, J. GLI1-mediated regulation of side population is responsible for drug resistance in gastric cancer. Oncotarget 2017, 8, 27412–27427. [Google Scholar] [CrossRef] [Green Version]
- Yu, B.; Gu, D.; Zhang, X.; Liu, B.; Xie, J. The role of GLI2-ABCG2 signaling axis for 5Fu resistance in gastric cancer. J. Genet. Genom. 2017, 44, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, P.; Stein, U.; Scheffer, G.L.; Lage, H. Modulation of the atypical multidrug-resistant phenotype by a hammerhead ribozyme directed against the ABC transporter BCRP/MXR/ABCG2. Cancer Gene Ther. 2002, 9, 579–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.C.; Chen, L.T.; Tsou, T.C.; Pan, W.Y.; Kuo, C.C.; Liu, J.F.; Yeh, S.C.; Tsai, F.Y.; Hsieh, H.P.; Chang, J.Y. Combined modalities of resistance in an oxaliplatin-resistant human gastric cancer cell line with enhanced sensitivity to 5-fluorouracil. Br. J. Cancer 2007, 97, 334–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonhardt, K.; Gebhardt, R.; Mossner, J.; Lutsenko, S.; Huster, D. Functional interactions of Cu-ATPase ATP7B with cisplatin and the role of ATP7B in the resistance of cells to the drug. J. Biol. Chem. 2009, 284, 7793–7802. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Tanimoto, K.; Otani, K.; Satoh, K.; Ohtaki, M.; Yoshida, K.; Toge, T.; Yahata, H.; Tanaka, S.; Chayama, K.; et al. Concise prediction models of anticancer efficacy of 8 drugs using expression data from 12 selected genes. Int. J. Cancer 2004, 111, 617–626. [Google Scholar] [CrossRef]
- Ikeda, K.; Yoshisue, K.; Matsushima, E.; Nagayama, S.; Kobayashi, K.; Tyson, C.A.; Chiba, K.; Kawaguchi, Y. Bioactivation of tegafur to 5-fluorouracil is catalyzed by cytochrome P-450 2A6 in human liver microsomes in vitro. Clin. Cancer Res. 2000, 6, 4409–4415. [Google Scholar]
- Daigo, S.; Takahashi, Y.; Fujieda, M.; Ariyoshi, N.; Yamazaki, H.; Koizumi, W.; Tanabe, S.; Saigenji, K.; Nagayama, S.; Ikeda, K.; et al. A novel mutant allele of the CYP2A6 gene (CYP2A6*11) found in a cancer patient who showed poor metabolic phenotype towards tegafur. Pharmacogenetics 2002, 12, 299–306. [Google Scholar] [CrossRef]
- Jeong, J.H.; Park, S.R.; Ahn, Y.; Ryu, M.H.; Ryoo, B.Y.; Kong, S.Y.; Yook, J.H.; Yoo, M.W.; Kim, B.S.; Kang, Y.K. Associations between CYP2A6 polymorphisms and outcomes of adjuvant S-1 chemotherapy in patients with curatively resected gastric cancer. Gastric Cancer 2017, 20, 146–155. [Google Scholar] [CrossRef]
- Yang, L.; Zou, S.; Shu, C.; Song, Y.; Sun, Y.K.; Zhang, W.; Zhou, A.; Yuan, X.; Yang, Y.; Hu, S. CYP2A6 Polymorphisms Associate with Outcomes of S-1 Plus Oxaliplatin Chemotherapy in Chinese Gastric Cancer Patients. Genom. Proteom. Bioinf. 2017, 15, 255–262. [Google Scholar] [CrossRef]
- Kim, Y.W.; Kim, M.J.; Ryu, K.W.; Lim, H.S.; Lee, J.H.; Kong, S.Y.; Lee, J.S.; Choi, I.J.; Kim, C.G.; Lee, J.Y.; et al. A phase II study of perioperative S-1 combined with weekly docetaxel in patients with locally advanced gastric carcinoma: Clinical outcomes and clinicopathological and pharmacogenetic predictors for survival. Gastric Cancer 2016, 19, 586–596. [Google Scholar] [CrossRef] [Green Version]
- Kikuyama, S.; Inada, T.; Shimizu, K.; Miyakita, M.; Ogata, Y. p53, bcl-2 and thymidine phosphorylase as predictive markers of chemotherapy in patients with advanced and recurrent gastric cancer. Anticancer Res. 2001, 21, 2149–2153. [Google Scholar] [PubMed]
- Noguchi, T.; Fujiwara, S.; Takeno, S.; Kai, S.; Mizuta, A.; Nagao, Y.; Uchida, Y. Clinical impact of thymidine phosphorylase expression in gastric cancer. Oncol. Rep. 2003, 10, 561–566. [Google Scholar] [PubMed]
- Sasako, M.; Terashima, M.; Ichikawa, W.; Ochiai, A.; Kitada, K.; Kurahashi, I.; Sakuramoto, S.; Katai, H.; Sano, T.; Imamura, H. Impact of the expression of thymidylate synthase and dihydropyrimidine dehydrogenase genes on survival in stage II/III gastric cancer. Gastric Cancer 2015, 18, 538–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terashima, M.; Fujiwara, H.; Takagane, A.; Abe, K.; Irinoda, T.; Nakaya, T.; Yonezawa, H.; Oyama, K.; Saito, K.; Kanzaki, N.; et al. Prediction of sensitivity to fluoropyrimidines by metabolic and target enzyme activities in gastric cancer. Gastric Cancer 2003, 6, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Napieralski, R.; Ott, K.; Kremer, M.; Specht, K.; Vogelsang, H.; Becker, K.; Muller, M.; Lordick, F.; Fink, U.; Rudiger Siewert, J.; et al. Combined GADD45A and thymidine phosphorylase expression levels predict response and survival of neoadjuvant-treated gastric cancer patients. Clin. Cancer Res. 2005, 11, 3025–3031. [Google Scholar] [CrossRef] [Green Version]
- Hu, F.K.; Shen, J.; Gao, S.W. [The influence of CYP2A6 polymorphism on adjuvant S-1 chemotherapy outcomes in patients with curatively resected gastric cancer]. Zhonghua Yi Xue Za Zhi 2018, 98, 1915–1918. [Google Scholar] [CrossRef]
- Shen, X.M.; Zhou, C.; Lian, L.; Li, L.Q.; Li, W.; Tao, M. Relationship Between the DPD and TS mRNA Expression and the Response to S-1-Based Chemotherapy and Prognosis in Patients with Advanced Gastric Cancer. Cell Biochem. Biophys. 2015, 71, 1653–1661. [Google Scholar] [CrossRef]
- Wang, D.; Yu, X.; Wang, X. High/positive expression of 5-fluorouracil metabolic enzymes predicts better response to S-1 in patients with gastric cancer: A meta-analysis. Int. J. Biol. Markers 2016, 31, e101–e109. [Google Scholar] [CrossRef]
- Okuyama, T.; Maehara, Y.; Endo, K.; Baba, H.; Adachi, Y.; Kuwano, M.; Sugimachi, K. Expression of glutathione S-transferase-pi and sensitivity of human gastric cancer cells to cisplatin. Cancer 1994, 74, 1230–1236. [Google Scholar] [CrossRef]
- Geng, M.; Wang, L.; Chen, X.; Cao, R.; Li, P. The association between chemosensitivity and Pgp, GST-pi and Topo II expression in gastric cancer. Diagn. Pathol. 2013, 8, 198. [Google Scholar] [CrossRef] [Green Version]
- Chun, J.H.; Kim, H.K.; Kim, E.; Kim, I.H.; Kim, J.H.; Chang, H.J.; Choi, I.J.; Lim, H.S.; Kim, I.J.; Kang, H.C.; et al. Increased expression of metallothionein is associated with irinotecan resistance in gastric cancer. Cancer Res. 2004, 64, 4703–4706. [Google Scholar] [CrossRef] [Green Version]
- Suganuma, K.; Kubota, T.; Saikawa, Y.; Abe, S.; Otani, Y.; Furukawa, T.; Kumai, K.; Hasegawa, H.; Watanabe, M.; Kitajima, M.; et al. Possible chemoresistance-related genes for gastric cancer detected by cDNA microarray. Cancer Sci. 2003, 94, 355–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinney, S.K.; Sanghani, S.P.; Davis, W.I.; Hurley, T.D.; Sun, Z.; Murry, D.J.; Bosron, W.F. Hydrolysis of capecitabine to 5’-deoxy-5-fluorocytidine by human carboxylesterases and inhibition by loperamide. J. Pharmacol. Exp. Ther. 2005, 313, 1011–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, G.; Zhang, W.; Ma, M.K.; McLeod, H.L. Human carboxylesterase 2 is commonly expressed in tumor tissue and is correlated with activation of irinotecan. Clin. Cancer Res. 2002, 8, 2605–2611. [Google Scholar] [PubMed]
- Fox, C.A.; Sapinoso, L.M.; Zhang, H.; Zhang, W.; McLeod, H.L.; Petroni, G.R.; Mullick, T.; Moskaluk, C.A.; Frierson, H.F.; Hampton, G.M.; et al. Altered expression of TFF-1 and CES-2 in Barrett’s Esophagus and associated adenocarcinomas. Neoplasia 2005, 7, 407–416. [Google Scholar] [CrossRef] [Green Version]
- Galizia, G.; Ferraraccio, F.; Lieto, E.; Orditura, M.; Castellano, P.; Imperatore, V.; La Manna, G.; Pinto, M.; Ciardiello, F.; La Mura, A.; et al. p27 downregulation and metallothionein overexpression in gastric cancer patients are associated with a poor survival rate. J. Surg. Oncol. 2006, 93, 241–252. [Google Scholar] [CrossRef]
- Lin, S.; Wang, X.; Pan, Y.; Tian, R.; Lin, B.; Jiang, G.; Chen, K.; He, Y.; Zhang, L.; Zhai, W.; et al. Transcription Factor Myeloid Zinc-Finger 1 Suppresses Human Gastric Carcinogenesis by Interacting with Metallothionein 2A. Clin. Cancer Res. 2019, 25, 1050–1062. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Lin, S.; Xing, R.; Zhu, M.; Lin, B.; Cui, J.; Li, W.; Gao, J.; Shen, L.; Zhao, Y.; et al. Epigenetic Upregulation of Metallothionein 2A by Diallyl Trisulfide Enhances Chemosensitivity of Human Gastric Cancer Cells to Docetaxel Through Attenuating NF-kappaB Activation. Antioxid. Redox. Signal. 2016, 24, 839–854. [Google Scholar] [CrossRef] [Green Version]
- Tuccari, G.; Giuffre, G.; Arena, F.; Barresi, G. Immunohistochemical detection of metallothionein in carcinomatous and normal human gastric mucosa. Histol. Histopathol. 2000, 15, 1035–1041. [Google Scholar] [CrossRef]
- De Bruin, W.C.; Wagenmans, M.J.; Peters, W.H. Expression of glutathione S-transferase alpha, P1-1 and T1-1 in the human gastrointestinal tract. JPN J. Cancer Res. 2000, 91, 310–316. [Google Scholar] [CrossRef]
- Goto, S.; Iida, T.; Cho, S.; Oka, M.; Kohno, S.; Kondo, T. Overexpression of glutathione S-transferase pi enhances the adduct formation of cisplatin with glutathione in human cancer cells. Free Radic. Res. 1999, 31, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.F.; Yao, R.Y.; Liu, K.W.; Lv, H.Y.; Jiang, T.; Liang, J. Genetic polymorphism of GSTP1: Prediction of clinical outcome to oxaliplatin/5-FU-based chemotherapy in advanced gastric cancer. J. Korean Med. Sci. 2010, 25, 846–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cengiz, B.; Yumrutas, O.; Bozgeyik, E.; Borazan, E.; Igci, Y.Z.; Bozgeyik, I.; Oztuzcu, S. Differential expression of the UGT1A family of genes in stomach cancer tissues. Tumour Biol. 2015, 36, 5831–5837. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Iwasa, S.; Shoji, H.; Honma, Y.; Takashima, A.; Kato, K.; Hamaguchi, T.; Higuchi, K.; Boku, N. Association between UGT1A1 gene polymorphism and safety and efficacy of irinotecan monotherapy as the third-line treatment for advanced gastric cancer. Gastric Cancer 2019, 22, 778–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, H.C.; Roh, M.S.; Oh, S.Y.; Kim, S.H.; Kim, M.C.; Kim, J.S.; Kim, H.J. Prognostic value of expression of ERCC1, thymidylate synthase, and glutathione S-transferase P1 for 5-fluorouracil/oxaliplatin chemotherapy in advanced gastric cancer. Ann. Oncol. 2007, 18, 504–509. [Google Scholar] [CrossRef]
- Huang, J.; Hu, H.; Xie, Y.; Tang, Y.; Liu, W.; Zhong, M. Effect of TUBB3, TS and ERCC1 mRNA expression on chemoresponse and clinical outcome of advanced gastric cancer by multiplex branched-DNA liquid chip technology. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2013, 38, 582–589. [Google Scholar] [CrossRef]
- Kim, M.H.; Zhang, X.; Jung, M.; Jung, I.; Park, H.S.; Beom, S.H.; Kim, H.S.; Rha, S.Y.; Kim, H.; Choi, Y.Y.; et al. Immunohistochemistry Biomarkers Predict Survival in Stage II/III Gastric Cancer Patients: From a Prospective Clinical Trial. Cancer Res. Treat. 2019, 51, 819–831. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, J.Q.; Liu, J.L.; Qin, X.G.; Huang, Y. Polymorphisms in ERCC1, GSTs, TS and MTHFR predict clinical outcomes of gastric cancer patients treated with platinum/5-Fu-based chemotherapy: A systematic review. BMC Gastroenterol. 2012, 12, 137. [Google Scholar] [CrossRef] [Green Version]
- Skarlos, D.V.; Bai, M.; Goussia, A.; Samantas, E.; Galani, E.; Tsavdaridis, D.; Karina, M.; Papakostas, P.; Konstantara, A.; Fountzilas, G. Expression of a molecular marker panel as a prognostic tool in gastric cancer patients treated postoperatively with docetaxel and irinotecan. A study of the Hellenic Cooperative Oncology Group. Anticancer Res. 2007, 27, 2973–2983. [Google Scholar]
- Zheng, W.E.; Chen, H.; Yuan, S.F.; Wu, L.L.; Zhang, W.; Sun, H.Y.; Chen, W.J. Overexpression of betaIII-tubulin and survivin associated with drug resistance to docetaxel-based chemotherapy in advanced gastric cancer. J. BUON 2012, 17, 284–290. [Google Scholar]
- Urano, N.; Fujiwara, Y.; Doki, Y.; Kim, S.J.; Miyoshi, Y.; Noguchi, S.; Miyata, H.; Takiguchi, S.; Yasuda, T.; Yano, M.; et al. Clinical significance of class III beta-tubulin expression and its predictive value for resistance to docetaxel-based chemotherapy in gastric cancer. Int. J. Oncol. 2006, 28, 375–381. [Google Scholar] [PubMed]
- Hwang, J.E.; Hong, J.Y.; Kim, K.; Kim, S.H.; Choi, W.Y.; Kim, M.J.; Jung, S.H.; Shim, H.J.; Bae, W.K.; Hwang, E.C.; et al. Class III beta-tubulin is a predictive marker for taxane-based chemotherapy in recurrent and metastatic gastric cancer. BMC Cancer 2013, 13, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Zhang, D.; Jiang, J.; Liu, P.; Wu, C. The relationships between the chemosensitivity of human gastric cancer to paclitaxel and the expressions of class III beta-tubulin, MAPT, and survivin. Med. Oncol. 2014, 31, 950. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.A.; Lee, H.S.; Lee, H.E.; Jeon, Y.K.; Yang, H.K.; Kim, W.H. EGFR in gastric carcinomas: Prognostic significance of protein overexpression and high gene copy number. Histopathology 2008, 52, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Lordick, F.; Kang, Y.K.; Chung, H.C.; Salman, P.; Oh, S.C.; Bodoky, G.; Kurteva, G.; Volovat, C.; Moiseyenko, V.M.; Gorbunova, V.; et al. Capecitabine and cisplatin with or without cetuximab for patients with previously untreated advanced gastric cancer (EXPAND): A randomised, open-label phase 3 trial. Lancet Oncol. 2013, 14, 490–499. [Google Scholar] [CrossRef]
- Waddell, T.; Chau, I.; Cunningham, D.; Gonzalez, D.; Okines, A.F.; Okines, C.; Wotherspoon, A.; Saffery, C.; Middleton, G.; Wadsley, J.; et al. Epirubicin, oxaliplatin, and capecitabine with or without panitumumab for patients with previously untreated advanced oesophagogastric cancer (REAL3): A randomised, open-label phase 3 trial. Lancet Oncol. 2013, 14, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Stahl, M.; Maderer, A.; Lordick, F.; Mihaljevic, A.L.; Kanzler, S.; Hoehler, T.; Thuss-Patience, P.; Monig, S.; Kunzmann, V.; Schroll, S.; et al. Perioperative chemotherapy with or without epidermal growth factor receptor blockade in unselected patients with locally advanced oesophagogastric adenocarcinoma: Randomized phase II study with advanced biomarker program of the German Cancer Society (AIO/CAO STO-0801). Eur. J. Cancer 2018, 93, 119–126. [Google Scholar] [CrossRef]
- Bang, Y.J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Bang, Y.J.; Feng-Yi, F.; Xu, J.M.; Lee, K.W.; Jiao, S.C.; Chong, J.L.; Lopez-Sanchez, R.I.; Price, T.; Gladkov, O.; et al. HER2 screening data from ToGA: Targeting HER2 in gastric and gastroesophageal junction cancer. Gastric Cancer 2015, 18, 476–484. [Google Scholar] [CrossRef]
- Wang, D.S.; Liu, Z.X.; Lu, Y.X.; Bao, H.; Wu, X.; Zeng, Z.L.; Liu, Z.; Zhao, Q.; He, C.Y.; Lu, J.H.; et al. Liquid biopsies to track trastuzumab resistance in metastatic HER2-positive gastric cancer. Gut 2019, 68, 1152–1161. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Tabernero, J.; Tomasek, J.; Chau, I.; Melichar, B.; Safran, H.; Tehfe, M.A.; Filip, D.; Topuzov, E.; Schlittler, L.; et al. Biomarker analyses in REGARD gastric/GEJ carcinoma patients treated with VEGFR2-targeted antibody ramucirumab. Br. J. Cancer 2016, 115, 974–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, D.; Hou, H.; Zhang, X. Progress in the treatment of solid tumors with apatinib: A systematic review. Onco. Targets Ther. 2018, 11, 4137–4147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, T.T.; Xu, H.; Xu, H.M.; Wang, Z.N.; Xu, Y.Y.; Song, Y.X.; Yin, S.C.; Liu, X.Y.; Miao, Z.F. The efficacy and safety of targeted therapy with or without chemotherapy in advanced gastric cancer treatment: A network meta-analysis of well-designed randomized controlled trials. Gastric Cancer 2018, 21, 361–371. [Google Scholar] [CrossRef] [Green Version]
- Mi, Y.J.; Liang, Y.J.; Huang, H.B.; Zhao, H.Y.; Wu, C.P.; Wang, F.; Tao, L.Y.; Zhang, C.Z.; Dai, C.L.; Tiwari, A.K.; et al. Apatinib (YN968D1) reverses multidrug resistance by inhibiting the efflux function of multiple ATP-binding cassette transporters. Cancer Res. 2010, 70, 7981–7991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cutsem, E.; De Haas, S.; Kang, Y.K.; Ohtsu, A.; Tebbutt, N.C.; Ming Xu, J.; Peng Yong, W.; Langer, B.; Delmar, P.; Scherer, S.J.; et al. Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: A biomarker evaluation from the AVAGAST randomized phase III trial. J. Clin. Oncol. 2012, 30, 2119–2127. [Google Scholar] [CrossRef] [PubMed]
- De Haas, S.; Delmar, P.; Bansal, A.T.; Moisse, M.; Miles, D.W.; Leighl, N.; Escudier, B.; Van Cutsem, E.; Carmeliet, P.; Scherer, S.J.; et al. Genetic variability of VEGF pathway genes in six randomized phase III trials assessing the addition of bevacizumab to standard therapy. Angiogenesis 2014, 17, 909–920. [Google Scholar] [CrossRef]
- Liu, J.; He, C.; Xing, C.; Yuan, Y. Nucleotide excision repair related gene polymorphisms and genetic susceptibility, chemotherapeutic sensitivity and prognosis of gastric cancer. Mutat. Res. 2014, 765, 11–21. [Google Scholar] [CrossRef]
- Wei, K.K.; Jiang, L.; Wei, Y.Y.; Wang, Y.F.; Qian, X.K.; Dai, Q.; Guan, Q.L. The prognostic value of ERCC1 expression in gastric cancer patients treated with platinum-based chemotherapy: A meta-analysis. Tumour Biol. 2014, 35, 8721–8731. [Google Scholar] [CrossRef]
- Yamada, Y.; Boku, N.; Nishina, T.; Yamaguchi, K.; Denda, T.; Tsuji, A.; Hamamoto, Y.; Konishi, K.; Tsuji, Y.; Amagai, K.; et al. Impact of excision repair cross-complementing gene 1 (ERCC1) on the outcomes of patients with advanced gastric cancer: Correlative study in Japan Clinical Oncology Group Trial JCOG9912. Ann. Oncol. 2013, 24, 2560–2565. [Google Scholar] [CrossRef]
- Song, A.L.; Zhao, L.; Wang, Y.W.; He, D.Q.; Li, Y.M. Chemoresistance in gastric cancer is attributed to the overexpression of excision repair cross-complementing 1 (ERCC1) caused by microRNA-122 dysregulation. J. Cell. Physiol. 2019, 234, 22485–22492. [Google Scholar] [CrossRef]
- Ning, J.; Jiao, Y.; Xie, X.; Deng, X.; Zhang, Y.; Yang, Y.; Zhao, C.; Wang, H.; Gu, K. miR1385p modulates the expression of excision repair crosscomplementing proteins ERCC1 and ERCC4, and regulates the sensitivity of gastric cancer cells to cisplatin. Oncol. Rep. 2019, 41, 1131–1139. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Li, C.H.; Jin, T.F.; Xu, D.Y. Study on the ERCC1 gene polymorphism response to chemotherapy and prognosis of gastric cancer. Genet. Mol. Res. 2014, 13, 8722–8728. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.C.; Zhao, Y.; Zhang, T.; Ling, X.L.; Zhao, D. Association between the ERCC1 rs11615 polymorphism and clinical outcomes of oxaliplatin-based chemotherapies in gastrointestinal cancer: A meta-analysis. Onco Targets Ther. 2015, 8, 641–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, M.H.; Li, G.Y.; Wu, X.J.; Zhang, C.X.; Zhang, C.F.; Zhu, K.X. Genetic variability of genes in NER pathway influences the treatment outcome of gastric cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 5563–5569. [Google Scholar] [PubMed]
- Bai, Y.; Wang, L.; Li, G.; Fang, X.; Li, Y.; Yang, S. Genetic variability of ERCC1 genes in NER pathway influences the treatment outcome of gastric cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 13367–13373. [Google Scholar]
- Park, S.R.; Kong, S.Y.; Nam, B.H.; Choi, I.J.; Kim, C.G.; Lee, J.Y.; Cho, S.J.; Kim, Y.W.; Ryu, K.W.; Lee, J.H.; et al. CYP2A6 and ERCC1 polymorphisms correlate with efficacy of S-1 plus cisplatin in metastatic gastric cancer patients. Br. J. Cancer 2011, 104, 1126–1134. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Wang, H.; Wang, Y.; Wang, X. ERCC1 rs3212986 A/C polymorphism is not associated with chemotherapy treatment outcomes in gastric cancer patients: Evidence from 11 publications in Chinese populations. Oncol. Targets Ther. 2018, 11, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Yeh, Y.S.; Chen, Y.T.; Tsai, H.L.; Huang, C.W.; Ma, C.J.; Su, W.C.; Huang, C.M.; Huang, M.Y.; Hu, H.M.; Lu, C.Y.; et al. Predictive Value of ERCC1, ERCC2, and XRCC Expression for Patients with Locally Advanced or Metastatic Gastric Cancer Treated with Neoadjuvant mFOLFOX-4 Chemotherapy. Pathol. Oncol. Res. 2019, 26, 1105–1116. [Google Scholar] [CrossRef]
- Li, M.; Gao, M.; Xie, X.; Zhang, Y.; Ning, J.; Liu, P.; Gu, K. MicroRNA-200c reverses drug resistance of human gastric cancer cells by targeting regulation of the NER-ERCC3/4 pathway. Oncol. Lett. 2019, 18, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Wang, S.; Chen, Q.; Zhang, Y.; Ni, P.; Wu, X.; Zhang, J.; Qiang, F.; Li, A.; Roe, O.D.; et al. TXNL1-XRCC1 pathway regulates cisplatin-induced cell death and contributes to resistance in human gastric cancer. Cell Death Dis. 2014, 5, e1055. [Google Scholar] [CrossRef]
- Zhang, X.; Jiang, L.P.; Yin, Y.; Wang, Y.D. XRCC1 and XPD genetic polymorphisms and clinical outcomes of gastric cancer patients treated with oxaliplatin-based chemotherapy: A meta-analysis. Tumour Biol. 2014, 35, 5637–5645. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Kurokawa, Y.; Takahashi, T.; Miyazaki, Y.; Tanaka, K.; Makino, T.; Yamasaki, M.; Nakajima, K.; Ikeda, J.I.; Mori, M.; et al. Predictive value of MLH1 and PD-L1 expression for prognosis and response to preoperative chemotherapy in gastric cancer. Gastric Cancer 2019, 22, 785–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, J.Y.; Kim, H.; Cheong, J.H.; Hyung, W.J.; Noh, S.H. Microsatellite instability in sporadic gastric cancer: Its prognostic role and guidance for 5-FU based chemotherapy after R0 resection. Int. J. Cancer 2012, 131, 505–511. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.M.; Luo, T.H.; Nie, M.M.; Fang, G.E.; Ma, L.Y.; Xue, X.C.; Wei, G.; Ke, C.W.; Bi, J.W. Influence of ERCC1 and ERCC4 polymorphisms on response to prognosis in gastric cancer treated with FOLFOX-based chemotherapy. Tumour Biol. 2014, 35, 2941–2948. [Google Scholar] [CrossRef] [PubMed]
- Young, K.; Starling, N.; Cunningham, D. Targeting deficient DNA damage repair in gastric cancer. Expert Opin. Pharmacother. 2016, 17, 1757–1766. [Google Scholar] [CrossRef]
- Yao, Y.; Tao, H.; Kim, J.J.; Burkhead, B.; Carloni, E.; Gasbarrini, A.; Sepulveda, A.R. Alterations of DNA mismatch repair proteins and microsatellite instability levels in gastric cancer cell lines. Lab. Investig. 2004, 84, 915–922. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Hwang, I.G.; Min, H.Y.; Bang, Y.J.; Kim, W.H. Clinical significance of BRCA1 and BRCA2 mRNA and protein expression in patients with sporadic gastric cancer. Oncol. Lett. 2019, 17, 4383–4392. [Google Scholar] [CrossRef]
- Shim, H.J.; Yun, J.Y.; Hwang, J.E.; Bae, W.K.; Cho, S.H.; Lee, J.H.; Kim, H.N.; Shin, M.H.; Kweon, S.S.; Kim, H.J.; et al. BRCA1 and XRCC1 polymorphisms associated with survival in advanced gastric cancer treated with taxane and cisplatin. Cancer Sci. 2010, 101, 1247–1254. [Google Scholar] [CrossRef]
- Fenoglio-Preiser, C.M.; Wang, J.; Stemmermann, G.N.; Noffsinger, A. TP53 and gastric carcinoma: A review. Hum. Mutat. 2003, 21, 258–270. [Google Scholar] [CrossRef]
- Liu, K.J.; Qi, H.Z.; Yao, H.L.; Lei, S.L.; Lei, Z.D.; Li, T.G.; Zhao, H. An updated meta-analysis of the p53 codon 72 polymorphism and gastric cancer risk. Mol. Biol. Rep. 2012, 39, 8265–8275. [Google Scholar] [CrossRef]
- Huang, Z.H.; Hua, D.; Li, L.H.; Zhu, J.D. Prognostic role of p53 codon 72 polymorphism in gastric cancer patients treated with fluorouracil-based adjuvant chemotherapy. J. Cancer Res. Clin. Oncol. 2008, 134, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.G.; Sohn, S.K.; Chae, Y.S.; Song, H.S.; Kwon, K.Y.; Do, Y.R.; Kim, M.K.; Lee, K.H.; Hyun, M.S.; Lee, W.S.; et al. TP53 codon 72 polymorphism associated with prognosis in patients with advanced gastric cancer treated with paclitaxel and cisplatin. Cancer Chemother. Pharmacol. 2009, 64, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Cascinu, S.; Graziano, F.; Del Ferro, E.; Staccioli, M.P.; Ligi, M.; Carnevali, A.; Muretto, P.; Catalano, G. Expression of p53 protein and resistance to preoperative chemotherapy in locally advanced gastric carcinoma. Cancer 1998, 83, 1917–1922. [Google Scholar] [CrossRef]
- Xu, H.Y.; Xu, W.L.; Wang, L.Q.; Chen, M.B.; Shen, H.L. Relationship between p53 status and response to chemotherapy in patients with gastric cancer: A meta-analysis. PLoS ONE 2014, 9, e95371. [Google Scholar] [CrossRef] [PubMed]
- Roman-Rosales, A.A.; Garcia-Villa, E.; Herrera, L.A.; Gariglio, P.; Diaz-Chavez, J. Mutant p53 gain of function induces HER2 over-expression in cancer cells. BMC Cancer 2018, 18, 709. [Google Scholar] [CrossRef] [Green Version]
- Tahara, T.; Tahara, S.; Horiguchi, N.; Okubo, M.; Terada, T.; Yamada, H.; Yoshida, D.; Omori, T.; Osaki, H.; Maeda, K.; et al. Molecular subtyping of gastric cancer combining genetic and epigenetic anomalies provides distinct clinicopathological features and prognostic impacts. Hum. Mutat. 2019, 40, 347–354. [Google Scholar] [CrossRef]
- Tahara, T.; Shibata, T.; Okamoto, Y.; Yamazaki, J.; Kawamura, T.; Horiguchi, N.; Okubo, M.; Nakano, N.; Ishizuka, T.; Nagasaka, M.; et al. Mutation spectrum of TP53 gene predicts clinicopathological features and survival of gastric cancer. Oncotarget 2016, 7, 42252–42260. [Google Scholar] [CrossRef] [Green Version]
- Shim, Y.H.; Kang, G.H.; Ro, J.Y. Correlation of p16 hypermethylation with p16 protein loss in sporadic gastric carcinomas. Lab. Investig. 2000, 80, 689–695. [Google Scholar] [CrossRef] [Green Version]
- He, D.; Zhang, Y.W.; Zhang, N.N.; Zhou, L.; Chen, J.N.; Jiang, Y.; Shao, C.K. Aberrant gene promoter methylation of p16, FHIT, CRBP1, WWOX, and DLC-1 in Epstein-Barr virus-associated gastric carcinomas. Med. Oncol. 2015, 32, 92. [Google Scholar] [CrossRef]
- Wang, M.; Li, Y.; Gao, J.; Zhou, J.; Gu, L.; Shen, L.; Deng, D. p16 Methylation is associated with chemosensitivity to fluorouracil in patients with advanced gastric cancer. Med. Oncol. 2014, 31, 988. [Google Scholar] [CrossRef]
- Mitsuno, M.; Kitajima, Y.; Ide, T.; Ohtaka, K.; Tanaka, M.; Satoh, S.; Miyazaki, K. Aberrant methylation of p16 predicts candidates for 5-fluorouracil-based adjuvant therapy in gastric cancer patients. J. Gastroenterol. 2007, 42, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Bartchewsky, W., Jr.; Martini, M.R.; Squassoni, A.C.; Alvarez, M.C.; Ladeira, M.S.; Salvatore, D.M.; Trevisan, M.A.; Pedrazzoli, J., Jr.; Ribeiro, M.L. Effects of Helicobacter pylori infection on the expressions of Bax and Bcl-2 in patients with chronic gastritis and gastric cancer. Dig. Dis. Sci. 2010, 55, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lin, Y.; Lan, F.; Yu, Y.; Ouyang, X.; Liu, W.; Xie, F.; Huang, Q. BAX and CDKN1A polymorphisms correlated with clinical outcomes of gastric cancer patients treated with postoperative chemotherapy. Med. Oncol. 2014, 31, 249. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.H.; Han, J.H.; Kim, J.H.; Ahn, M.S.; Hwang, Y.H.; Lee, H.W.; Kang, S.Y.; Park, J.S.; Choi, J.H.; Lee, K.J.; et al. Bax predicts outcome in gastric cancer patients treated with 5-fluorouracil, leucovorin, and oxaliplatin palliative chemotherapy. Dig. Dis. Sci. 2011, 56, 131–138. [Google Scholar] [CrossRef]
- Pietrantonio, F.; Biondani, P.; De Braud, F.; Pellegrinelli, A.; Bianchini, G.; Perrone, F.; Formisano, B.; Di Bartolomeo, M. Bax expression is predictive of favorable clinical outcome in chemonaive advanced gastric cancer patients treated with capecitabine, oxaliplatin, and irinotecan regimen. Transl. Oncol. 2012, 5, 155–159. [Google Scholar] [CrossRef] [Green Version]
- Kubo, T.; Kawano, Y.; Himuro, N.; Sugita, S.; Sato, Y.; Ishikawa, K.; Takada, K.; Murase, K.; Miyanishi, K.; Sato, T.; et al. BAK is a predictive and prognostic biomarker for the therapeutic effect of docetaxel treatment in patients with advanced gastric cancer. Gastric Cancer 2016, 19, 827–838. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.; Huang, Y.; Zou, Z.; Gimenez-Capitan, A.; Yu, L.; Hu, W.; Zhu, L.; Sun, X.; Sanchez, J.J.; Guan, W.; et al. High BIM mRNA levels are associated with longer survival in advanced gastric cancer. Oncol. Lett. 2017, 13, 1826–1834. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Choi, H.; Lee, S.K. Epstein-Barr virus miR-BART20-5p regulates cell proliferation and apoptosis by targeting BAD. Cancer Lett. 2015, 356, 733–742. [Google Scholar] [CrossRef]
- Xu, Y.C.; Liu, X.; Li, M.; Li, Y.; Li, C.Y.; Lu, Y.; Sanches, J.; Wang, L.; Du, Y.; Mao, L.M.; et al. A Novel Mechanism of Doxorubicin Resistance and Tumorigenesis Mediated by MicroRNA-501-5p-Suppressed BLID. Mol. Ther. Nucleic Acids 2018, 12, 578–590. [Google Scholar] [CrossRef] [Green Version]
- Qiao, L.; Wong, B.C. Targeting apoptosis as an approach for gastrointestinal cancer therapy. Drug Resist. Updat. 2009, 12, 55–64. [Google Scholar] [CrossRef]
- Yoo, N.J.; Lee, S.H.; Jeong, E.G.; Lee, J.W.; Soung, Y.H.; Nam, S.W.; Kim, S.H.; Lee, J.Y. Expression of nuclear and cytoplasmic phosphorylated FADD in gastric cancers. Pathol. Res. Pract. 2007, 203, 73–78. [Google Scholar] [CrossRef]
- Pang, X.; Zhou, Z.; Yu, Z.; Han, L.; Lin, Z.; Ao, X.; Liu, C.; He, Y.; Ponnusamy, M.; Li, P.; et al. Foxo3a-dependent miR-633 regulates chemotherapeutic sensitivity in gastric cancer by targeting Fas-associated death domain. RNA Biol. 2019, 16, 233–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.H.; Liu, S.Q.; Qin, M.B.; Huang, J.A.; Xu, C.Y.; Wu, W.H.; Zhu, L.Y.; Qin, N.; Lai, M.Y. NIK and IKKbetabinding protein contributes to gastric cancer chemoresistance by promoting epithelialmesenchymal transition through the NFkappaB signaling pathway. Oncol. Rep. 2018, 39, 2721–2730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, F.; Nishizuka, S.S.; Kume, K.; Ishida, K.; Katagiri, H.; Sato, K.; Iwaya, T.; Koeda, K.; Wakabayashi, G. A compensatory role of NF-kappaB to p53 in response to 5-FU-based chemotherapy for gastric cancer cell lines. PLoS ONE 2014, 9, e90155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manu, K.A.; Shanmugam, M.K.; Li, F.; Chen, L.; Siveen, K.S.; Ahn, K.S.; Kumar, A.P.; Sethi, G. Simvastatin sensitizes human gastric cancer xenograft in nude mice to capecitabine by suppressing nuclear factor-kappa B-regulated gene products. J. Mol. Med. 2014, 92, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Bozkaya, Y.; Ozdemir, N.Y.; Sezer, S.; Kostek, O.; Demirci, N.S.; Yazici, O.; Erdem, G.U.; Eren, T.; Zengin, N. Is serum survivin expression a predictive biomarker in locally advanced gastric cancer patients treated with neoadjuvant chemotherapy? Cancer Biomark 2018, 22, 143–149. [Google Scholar] [CrossRef]
- Bataille, F.; Rummele, P.; Dietmaier, W.; Gaag, D.; Klebl, F.; Reichle, A.; Wild, P.; Hofstadter, F.; Hartmann, A. Alterations in p53 predict response to preoperative high dose chemotherapy in patients with gastric cancer. Mol. Pathol. 2003, 56, 286–292. [Google Scholar] [CrossRef] [Green Version]
- Yashiro, M.; Inoue, T.; Nishioka, N.; Matsuoka, T.; Boland, C.R.; Hirakawa, K. Allelic imbalance at p53 and microsatellite instability are predictive markers for resistance to chemotherapy in gastric carcinoma. Ann. Surg. Oncol. 2009, 16, 2926–2935. [Google Scholar] [CrossRef] [Green Version]
- Melucci, E.; Casini, B.; Ronchetti, L.; Pizzuti, L.; Sperati, F.; Pallocca, M.; De Nicola, F.; Goeman, F.; Gallo, E.; Amoreo, C.A.; et al. Expression of the Hippo transducer TAZ in association with WNT pathway mutations impacts survival outcomes in advanced gastric cancer patients treated with first-line chemotherapy. J. Transl. Med. 2018, 16, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Tan, B.B.; Fan, L.Q.; Zhao, Q.; Liu, Y.; Wang, D. Heterogeneity of COX-2 and multidrug resistance between primary tumors and regional lymph node metastases of gastric cancer. Tumori 2012, 98, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Lian, G.; Li, L.; Ye, F.; Wang, D.; Liu, J.; Shi, Y.; Jing, C.; Suo, J.; Zhang, D.Y.; Chen, M. The screening and analysis of protein signatures and signaling associated with chemoresistance based on Protein Pathway Array technology in gastric cancer. Oncol. Rep. 2018, 39, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.; Park, D.J.; Schmidt, B.; Thomas, N.J.; Lee, H.J.; Kim, T.S.; Janjigian, Y.Y.; Cohen, D.J.; Yoon, S.S. CD44 expression denotes a subpopulation of gastric cancer cells in which Hedgehog signaling promotes chemotherapy resistance. Clin. Cancer Res. 2014, 20, 3974–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Q.; Zhang, T.; Li, C. LncRNA MALAT1 Regulates the Cell Proliferation and Cisplatin Resistance in Gastric Cancer via PI3K/AKT Pathway. Cancer Manag. Res. 2020, 12, 1929–1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, L.; Takacs, A.; Slotta-Huspenina, J.; Langer, R.; Becker, K.; Novotny, A.; Ott, K.; Walch, A.; Hapfelmeier, A.; Keller, G. Clinical Significance of NOTCH1 and NOTCH2 Expression in Gastric Carcinomas: An Immunohistochemical Study. Front. Oncol. 2015, 5, 94. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Serrano, A.; Angulo, B.; Dominguez, C.; Pazo-Cid, R.; Salud, A.; Jimenez-Fonseca, P.; Leon, A.; Galan, M.C.; Alsina, M.; Rivera, F.; et al. Genomic Profiling of HER2-Positive Gastric Cancer: PI3K/Akt/mTOR Pathway as Predictor of Outcomes in HER2-Positive Advanced Gastric Cancer Treated with Trastuzumab. Oncologist 2018, 23, 1092–1102. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Gong, A.; Yang, H.; George, S.K.; Jiao, Z.; Huang, H.; Jiang, X.; Zhang, Y. Sonic hedgehog-glioma associated oncogene homolog 1 signaling enhances drug resistance in CD44(+)/Musashi-1(+) gastric cancer stem cells. Cancer Lett. 2015, 369, 124–133. [Google Scholar] [CrossRef]
- Santos, J.C.; Carrasco-Garcia, E.; Garcia-Puga, M.; Aldaz, P.; Montes, M.; Fernandez-Reyes, M.; De Oliveira, C.C.; Lawrie, C.H.; Arauzo-Bravo, M.J.; Ribeiro, M.L.; et al. SOX9 Elevation Acts with Canonical WNT Signaling to Drive Gastric Cancer Progression. Cancer Res. 2016, 76, 6735–6746. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Li, F.; Yao, X.; Mou, T.; Xu, Z.; Han, Z.; Chen, S.; Li, W.; Yu, J.; Qi, X.; et al. The HER4-YAP1 axis promotes trastuzumab resistance in HER2-positive gastric cancer by inducing epithelial and mesenchymal transition. Oncogene 2018, 37, 3022–3038. [Google Scholar] [CrossRef] [Green Version]
- Ooi, C.H.; Ivanova, T.; Wu, J.; Lee, M.; Tan, I.B.; Tao, J.; Ward, L.; Koo, J.H.; Gopalakrishnan, V.; Zhu, Y.; et al. Oncogenic pathway combinations predict clinical prognosis in gastric cancer. PLoS Genet. 2009, 5, e1000676. [Google Scholar] [CrossRef] [Green Version]
- Akyala, A.I.; Peppelenbosch, M.P. Gastric cancer and Hedgehog signaling pathway: Emerging new paradigms. Genes Cancer 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Yeh, T.S.; Wu, C.W.; Hsu, K.W.; Liao, W.J.; Yang, M.C.; Li, A.F.; Wang, A.M.; Kuo, M.L.; Chi, C.W. The activated Notch1 signal pathway is associated with gastric cancer progression through cyclooxygenase-2. Cancer Res. 2009, 69, 5039–5048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hang, Q.; Sun, R.; Jiang, C.; Li, Y. Notch 1 promotes cisplatin-resistant gastric cancer formation by upregulating lncRNA AK022798 expression. Anticancer Drugs 2015, 26, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Yuan, W.; Lai, C.; Zhong, S.; Yang, C.; Wang, R.; Mao, L.; Chen, Z. EphA2-to-YAP pathway drives gastric cancer growth and therapy resistance. Int. J. Cancer 2020, 146, 1937–1949. [Google Scholar] [CrossRef] [PubMed]
- Russi, S.; Verma, H.K.; Laurino, S.; Mazzone, P.; Storto, G.; Nardelli, A.; Zoppoli, P.; Calice, G.; La Rocca, F.; Sgambato, A.; et al. Adapting and Surviving: Intra and Extra-Cellular Remodeling in Drug-Resistant Gastric Cancer Cells. IJMS 2019, 20, 3736. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Ning, X.; Sun, L.; Zhang, H.; Shi, Y.; Guo, C.; Han, S.; Liu, J.; Sun, S.; Han, Z.; et al. Hypoxia-inducible factor-1 alpha contributes to hypoxia-induced chemoresistance in gastric cancer. Cancer Sci. 2008, 99, 121–128. [Google Scholar]
- Danza, K.; Silvestris, N.; Simone, G.; Signorile, M.; Saragoni, L.; Brunetti, O.; Monti, M.; Mazzotta, A.; De Summa, S.; Mangia, A.; et al. Role of miR-27a, miR-181a and miR-20b in gastric cancer hypoxia-induced chemoresistance. Cancer Biol. Ther. 2016, 17, 400–406. [Google Scholar] [CrossRef] [Green Version]
- Ge, X.; Liu, X.; Lin, F.; Li, P.; Liu, K.; Geng, R.; Dai, C.; Lin, Y.; Tang, W.; Wu, Z.; et al. MicroRNA-421 regulated by HIF-1alpha promotes metastasis, inhibits apoptosis, and induces cisplatin resistance by targeting E-cadherin and caspase-3 in gastric cancer. Oncotarget 2016, 7, 24466–24482. [Google Scholar] [CrossRef]
- Zhang, X.W.; Bu, P.; Liu, L.; Zhang, X.Z.; Li, J. Overexpression of long non-coding RNA PVT1 in gastric cancer cells promotes the development of multidrug resistance. Biochem. Biophys. Res. Commun. 2015, 462, 227–232. [Google Scholar] [CrossRef]
- Rohwer, N.; Dame, C.; Haugstetter, A.; Wiedenmann, B.; Detjen, K.; Schmitt, C.A.; Cramer, T. Hypoxia-inducible factor 1alpha determines gastric cancer chemosensitivity via modulation of p53 and NF-kappaB. PLoS ONE 2010, 5, e12038. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Tan, B.B.; Li, Y.; Fan, L.Q.; Yang, P.G.; Tian, Y. Enhancement of Drug Sensitivity by Knockdown of HIF-1alpha in Gastric Carcinoma Cells. Oncol. Res. 2016, 23, 129–136. [Google Scholar] [CrossRef]
- Nakamura, J.; Kitajima, Y.; Kai, K.; Hashiguchi, K.; Hiraki, M.; Noshiro, H.; Miyazaki, K. HIF-1alpha is an unfavorable determinant of relapse in gastric cancer patients who underwent curative surgery followed by adjuvant 5-FU chemotherapy. Int. J. Cancer 2010, 127, 1158–1171. [Google Scholar] [CrossRef]
- Wang, Y.; Qi, Z.; Zhou, M.; Yang, W.; Hu, R.; Li, G.; Ma, X.; Zhang, Z. Stanniocalcin1 promotes cell proliferation, chemoresistance and metastasis in hypoxic gastric cancer cells via Bcl2. Oncol. Rep. 2019, 41, 1998–2008. [Google Scholar] [CrossRef]
- Han, Y.; Cai, H.; Ma, L.; Ding, Y.; Tan, X.; Chang, W.; Guan, W.; Liu, Y.; Shen, Q.; Yu, Y.; et al. Expression of orphan nuclear receptor NR4A2 in gastric cancer cells confers chemoresistance and predicts an unfavorable postoperative survival of gastric cancer patients with chemotherapy. Cancer 2013, 119, 3436–3445. [Google Scholar] [CrossRef] [Green Version]
- Zhi, X.; Tao, J.; Xiang, G.; Cao, H.; Liu, Z.; Yang, K.; Lv, C.; Ni, S. APRIL induces cisplatin resistance in gastric cancer cells via activation of the NF-kappaB pathway. Cell Physiol. Biochem. 2015, 35, 571–585. [Google Scholar] [CrossRef]
- Xu, W.; Wei, Q.; Han, M.; Zhou, B.; Wang, H.; Zhang, J.; Wang, Q.; Sun, J.; Feng, L.; Wang, S.; et al. CCL2-SQSTM1 positive feedback loop suppresses autophagy to promote chemoresistance in gastric cancer. Int. J. Biol. Sci. 2018, 14, 1054–1066. [Google Scholar] [CrossRef]
- Kwon, O.H.; Kim, J.H.; Kim, S.Y.; Kim, Y.S. TWEAK/Fn14 signaling mediates gastric cancer cell resistance to 5-fluorouracil via NF-kappaB activation. Int. J. Oncol. 2014, 44, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Ham, I.H.; Oh, H.J.; Jin, H.; Bae, C.A.; Jeon, S.M.; Choi, K.S.; Son, S.Y.; Han, S.U.; Brekken, R.A.; Lee, D.; et al. Targeting interleukin-6 as a strategy to overcome stroma-induced resistance to chemotherapy in gastric cancer. Mol. Cancer 2019, 18, 68. [Google Scholar] [CrossRef]
- Kuai, W.X.; Wang, Q.; Yang, X.Z.; Zhao, Y.; Yu, R.; Tang, X.J. Interleukin-8 associates with adhesion, migration, invasion and chemosensitivity of human gastric cancer cells. World J. Gastroenterol. 2012, 18, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Song, X.; Xu, X.; Mou, Y. Cancer-Associated Fibroblasts Promote the Chemo-resistance in Gastric Cancer through Secreting IL-11 Targeting JAK/STAT3/Bcl2 Pathway. Cancer Res. Treat. 2019, 51, 194–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.L.; Zhao, Y.R.; Weng, G.B.; Chen, Y.C.; Wei, X.N.; Shao, J.P.; Ji, H. IL-33-induced JNK pathway activation confers gastric cancer chemotherapy resistance. Oncol. Rep. 2015, 33, 2746–2752. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Chen, Z.; Huang, J.; Chen, J.; Yuan, W.; Deng, Z. Upregulation of autophagy-related gene-5 (ATG-5) is associated with chemoresistance in human gastric cancer. PLoS ONE 2014, 9, e110293. [Google Scholar] [CrossRef]
- Bhattacharya, B.; Low, S.H.; Soh, C.; Kamal Mustapa, N.; Beloueche-Babari, M.; Koh, K.X.; Loh, J.; Soong, R. Increased drug resistance is associated with reduced glucose levels and an enhanced glycolysis phenotype. Br. J. Pharmacol. 2014, 171, 3255–3267. [Google Scholar] [CrossRef]
- Chen, F.; Zhuang, M.; Zhong, C.; Peng, J.; Wang, X.; Li, J.; Chen, Z.; Huang, Y. Baicalein reverses hypoxia-induced 5-FU resistance in gastric cancer AGS cells through suppression of glycolysis and the PTEN/Akt/HIF-1alpha signaling pathway. Oncol. Rep. 2015, 33, 457–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Liu, B.; Chen, Z.; Li, G.; Zhang, Z. MSC-induced lncRNA HCP5 drove fatty acid oxidation through miR-3619-5p/AMPK/PGC1alpha/CEBPB axis to promote stemness and chemo-resistance of gastric cancer. Cell Death Dis. 2020, 11, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, R.; Zhang, B.; Zhang, X.; Xue, J.; Yuan, X.; Yan, Y.; Wang, M.; Zhu, W.; Qian, H.; Xu, W. Exosomes derived from human mesenchymal stem cells confer drug resistance in gastric cancer. Cell Cycle 2015, 14, 2473–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, P.; Chen, L.; Yuan, X.; Luo, Q.; Liu, Y.; Xie, G.; Ma, Y.; Shen, L. Exosomal transfer of tumor-associated macrophage-derived miR-21 confers cisplatin resistance in gastric cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, Y.; Zhang, Z.; Shang, Y.; Jiang, X.; Dong, J.; Yu, P.; Nie, Y.; Zhao, Q. miR-23b-3p regulates the chemoresistance of gastric cancer cells by targeting ATG12 and HMGB2. Cell Death Dis. 2015, 6, e1766. [Google Scholar] [CrossRef] [Green Version]
- Xia, P.; Xu, X.Y. Epithelial-mesenchymal transition and gastric cancer stem cell. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Ren, L.; Ding, Y.; Li, F.; Chen, X.; Ouyang, Y.; Zhang, Y.; Zhang, D. Hyaluronan-mediated motility receptor confers resistance to chemotherapy via TGFbeta/Smad2-induced epithelial-mesenchymal transition in gastric cancer. FASEB J. 2019, 33, 6365–6377. [Google Scholar] [CrossRef]
- Li, L.Q.; Pan, D.; Chen, Q.; Zhang, S.W.; Xie, D.Y.; Zheng, X.L.; Chen, H. Sensitization of Gastric Cancer Cells to 5-FU by MicroRNA-204 Through Targeting the TGFBR2-Mediated Epithelial to Mesenchymal Transition. Cell Physiol. Biochem. 2018, 47, 1533–1545. [Google Scholar] [CrossRef]
- Luo, Y.; Wu, J.; Wu, Q.; Li, X.; Zhang, J.; Rong, X.; Rao, J.; Liao, Y.; Bin, J.; Huang, N.; et al. miR-577 Regulates TGF-beta Induced Cancer Progression through a SDPR-Modulated Positive-Feedback Loop with ERK-NF-kappaB in Gastric Cancer. Mol. Ther. 2019, 27, 1166–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.L.; Li, Z.; Lv, C.M.; Wang, W. MiR-187 influences cisplatin-resistance of gastric cancer cells through regulating the TGF-beta/Smad signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 9907–9914. [Google Scholar] [CrossRef] [PubMed]
- Takaishi, S.; Okumura, T.; Tu, S.; Wang, S.S.; Shibata, W.; Vigneshwaran, R.; Gordon, S.A.; Shimada, Y.; Wang, T.C. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 2009, 27, 1006–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; Bu, L.; Yasuda, T.; Koiwa, M.; Akiyama, T.; Uchihara, T.; Baba, H.; Ishimoto, T. Gastric Cancer Stem Cells: Current Insights into the Immune Microenvironment and Therapeutic Targets. Biomedicines 2020, 8, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, H.S.; Park, D.J.; Kim, H.H.; Kim, W.H.; Lee, H.S. Combination of epithelial-mesenchymal transition and cancer stem cell-like phenotypes has independent prognostic value in gastric cancer. Hum. Pathol. 2012, 43, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Zavros, Y. Initiation and Maintenance of Gastric Cancer: A Focus on CD44 Variant Isoforms and Cancer Stem Cells. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, M.E.; Jeon, T.Y.; Hwang, S.H.; Lee, Y.S.; Kim, H.J.; Shim, H.E.; Yoon, S.; Baek, S.Y.; Kim, B.S.; Kang, C.D.; et al. Cancer spheres from gastric cancer patients provide an ideal model system for cancer stem cell research. Cell. Mol. Life Sci. 2011, 68, 3589–3605. [Google Scholar] [CrossRef]
- Ma, Y.; Zhu, J.; Chen, S.; Ma, J.; Zhang, X.; Huang, S.; Hu, J.; Yue, T.; Zhang, J.; Wang, P.; et al. Low expression of SPARC in gastric cancer-associated fibroblasts leads to stemness transformation and 5-fluorouracil resistance in gastric cancer. Cancer Cell Int. 2019, 19, 137. [Google Scholar] [CrossRef] [Green Version]
- Fujikuni, N.; Yamamoto, H.; Tanabe, K.; Naito, Y.; Sakamoto, N.; Tanaka, Y.; Yanagihara, K.; Oue, N.; Yasui, W.; Ohdan, H. Hypoxia-mediated CD24 expression is correlated with gastric cancer aggressiveness by promoting cell migration and invasion. Cancer Sci. 2014, 105, 1411–1420. [Google Scholar] [CrossRef]
- Lee, H.H.; Seo, K.J.; An, C.H.; Kim, J.S.; Jeon, H.M. CD133 expression is correlated with chemoresistance and early recurrence of gastric cancer. J. Surg. Oncol. 2012, 106, 999–1004. [Google Scholar] [CrossRef]
- Song, S.; Pei, G.; Du, Y.; Wu, J.; Ni, X.; Wang, S.; Jiang, B.; Luo, M.; Yu, J. Interaction between CD133 and PI3K-p85 promotes chemoresistance in gastric cancer cells. Am. J. Transl. Res. 2018, 10, 304–314. [Google Scholar] [PubMed]
- Jiang, J.; Zhang, Y.; Chuai, S.; Wang, Z.; Zheng, D.; Xu, F.; Li, C.; Liang, Y.; Chen, Z. Trastuzumab (herceptin) targets gastric cancer stem cells characterized by CD90 phenotype. Oncogene 2012, 31, 671–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohkuma, M.; Haraguchi, N.; Ishii, H.; Mimori, K.; Tanaka, F.; Kim, H.M.; Shimomura, M.; Hirose, H.; Yanaga, K.; Mori, M. Absence of CD71 transferrin receptor characterizes human gastric adenosquamous carcinoma stem cells. Ann. Surg. Oncol. 2012, 19, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Chiwaki, F.; Takahashi, R.U.; Aoyagi, K.; Yanagihara, K.; Nishimura, T.; Tamaoki, M.; Komatsu, M.; Komatsuzaki, R.; Matsusaki, K.; et al. Identification and Characterization of CXCR4-Positive Gastric Cancer Stem Cells. PLoS ONE 2015, 10, e0130808. [Google Scholar] [CrossRef]
- Li, K.; Guo, X.; Wang, Z.; Li, X.; Bu, Y.; Bai, X.; Zheng, L.; Huang, Y. The prognostic roles of ALDH1 isoenzymes in gastric cancer. Onco. Targets Ther. 2016, 9, 3405–3414. [Google Scholar] [CrossRef] [Green Version]
- Xi, H.Q.; Cui, J.X.; Shen, W.S.; Wu, X.S.; Bian, S.B.; Li, J.Y.; Song, Z.; Wei, B.; Chen, L. Increased expression of Lgr5 is associated with chemotherapy resistance in human gastric cancer. Oncol. Rep. 2014, 32, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Men, X.; Zhao, R.; Han, J.; Fan, Z.; Wang, Y.; Lv, Y.; Zuo, J.; Zhao, L.; Sang, M.; et al. miR-200c inhibits TGF-beta-induced-EMT to restore trastuzumab sensitivity by targeting ZEB1 and ZEB2 in gastric cancer. Cancer Gene Ther. 2018, 25, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Wang, B.; Wang, X.; Luo, Y.; Fan, W. NANOGP8 is the key regulator of stemness, EMT, Wnt pathway, chemoresistance, and other malignant phenotypes in gastric cancer cells. PLoS ONE 2018, 13, e0192436. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Zhang, Y.; Wang, S.; Zhou, J.; Xu, S. Sox2 enhances the tumorigenicity and chemoresistance of cancer stem-like cells derived from gastric cancer. J. Biomed. Res. 2012, 26, 336–345. [Google Scholar] [CrossRef]
- Wang, B.; Chen, Q.; Cao, Y.; Ma, X.; Yin, C.; Jia, Y.; Zang, A.; Fan, W. LGR5 Is a Gastric Cancer Stem Cell Marker Associated with Stemness and the EMT Signature Genes NANOG, NANOGP8, PRRX1, TWIST1, and BMI1. PLoS ONE 2016, 11, e0168904. [Google Scholar] [CrossRef] [Green Version]
- Chandrakesan, P.; Panneerselvam, J.; Qu, D.; Weygant, N.; May, R.; Bronze, M.S.; Houchen, C.W. Regulatory Roles of Dclk1 in Epithelial Mesenchymal Transition and Cancer Stem Cells. J. Carcinog. Mutagen. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, Y.; Seno, H.; Fukuoka, A.; Ueo, T.; Yamaga, Y.; Maruno, T.; Nakanishi, N.; Kanda, K.; Komekado, H.; Kawada, M.; et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat. Genet. 2013, 45, 98–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.Q.; He, W.F.; Wu, Y.J.; Zhao, S.L.; Wang, L.; Ouyang, Y.Y.; Tang, S.Y. LncRNA SNHG1 promotes EMT process in gastric cancer cells through regulation of the miR-15b/DCLK1/Notch1 axis. BMC Gastroenterol. 2020, 20, 156. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Qu, D.; Weygant, N.; Peng, J.; Houchen, C.W. Cancer Stem Cell Marker DCLK1 Correlates with Tumorigenic Immune Infiltrates in the Colon and Gastric Adenocarcinoma Microenvironments. Cancers 2020, 12, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Jones, K.; Mei, H. Doublecotin-Like Kinase 1 Increases Chemoresistance of Colorectal Cancer Cells through the Anti-Apoptosis Pathway. J. Stem Cell Res. Ther. 2019, 9. [Google Scholar] [CrossRef]
- Panneerselvam, J.; Mohandoss, P.; Patel, R.; Gillan, H.; Li, M.; Kumar, K.; Nguyen, D.; Weygant, N.; Qu, D.; Pitts, K.; et al. DCLK1 Regulates Tumor Stemness and Cisplatin Resistance in Non-small Cell Lung Cancer via ABCD-Member-4. Mol. Ther. Oncol. 2020, 18, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.X.; Cai, Y.D.; Wang, Y.D.; Cui, X.B.; Xie, T.T.; Li, W.J.; Peng, L.; Zhang, Y.; Wang, Z.Q.; Wang, J.; et al. Fas signaling promotes motility and metastasis through epithelial-mesenchymal transition in gastrointestinal cancer. Oncogene 2013, 32, 1183–1192. [Google Scholar] [CrossRef] [Green Version]
- Han, R.; Xiong, J.; Xiao, R.; Altaf, E.; Wang, J.; Liu, Y.; Xu, H.; Ding, Q.; Zhang, Q. Activation of beta-catenin signaling is critical for doxorubicin-induced epithelial-mesenchymal transition in BGC-823 gastric cancer cell line. Tumour Biol. 2013, 34, 277–284. [Google Scholar] [CrossRef]
- Briz, O.; Macias, R.I.; Vallejo, M.; Silva, A.; Serrano, M.A.; Marin, J.J. Usefulness of liposomes loaded with cytostatic bile acid derivatives to circumvent chemotherapy resistance of enterohepatic tumors. Mol. Pharmacol. 2003, 63, 742–750. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Protein | Feature | Drugs Affected | Consequences | Ref. |
|---|---|---|---|---|
| Uptake carriers (MOC-1a) | ||||
| CTR1 | Down-regulation | Cisplatin | Decreased sensitivity | [9,10] |
| OATP1B3 | Alternative TSS | Irinotecan, Docetaxel, Methotrexate | Unknown | [9] |
| Export pumps (MOC-1b) | ||||
| MDR1 | Up-regulation * | Platinum derivatives, | Reduced clinical response | [14] |
| 5-FU, Epirubicin | Decreased cell sensitivity in vitro | [17,18] | ||
| MRP1 | Up-regulation | Cisplatin, Doxorubicin | Decreased cell sensitivity in vitro | [22,23,24,25] |
| MRP2 | GV (rs717620; CC) | 5-FU, Oxaliplatin | Worse response | [26] |
| MRP4 | Up-regulation | Cisplatin, Dasatinib, | Decreased cell sensitivity in vitro | [27,28] |
| 5-FU | Increased risk of cancer relapse | [31] | ||
| BCRP | Up-regulation | Cisplatin | Reduced OS | [30] |
| ATP7A | Up-regulation | Oxaliplatin | Decreased cell sensitivity in vitro | [33] |
| ATP7B | Up-regulation | Cisplatin | Decreased cell sensitivity in vitro | [34] |
| Protein | Feature | Drugs affected | Consequences | Ref. |
|---|---|---|---|---|
| CYP2A6 | Inactivating GVs | Tegafur (5-FU) | Reduced DFS and OS | [37,38,46] |
| DPD | Up-regulation | 5-FU | Reduced OS | [47,48] |
| GST-pi | Up-regulation | 5-FU, Cisplatin, Mitomycin C | Increased resistance in vitro | [49,50] |
| MTs | Up-regulation * | Cisplatin, Irinotecan (SN-38) | Reduced clinical response * | [51,52] |
| TP | Down-regulation * | 5-FU | Reduced OS * | [41,42] |
| Protein | Feature | Drug Affected | Consequences | Ref. |
|---|---|---|---|---|
| HER2 | Low expression | Trastuzumab | Reduced OS | [78,79] |
| TS | High expression | Capecitabine, Oxaliplatin | Worse outcome * | [67] |
| TS | GV: 2R/2R or 2R/3R | Cisplatin, Oxaliplatin, 5-FU | Reduced OS | [68] |
| TUBB3 | High expression | Taxanes, Cisplatin, 5-FU | Worse clinical outcome | [66,70,71,72] |
| VEGF | Low expression | Bevacizumab | Worse clinical outcome | [85] |
| VEGFR-2 | High expression | Ramucirumab | Shorter PFS | [81] |
| Factor | Feature | Drugs Affected | Consequences | Ref. |
|---|---|---|---|---|
| Nucleotide-excision DNA repair (NER) | ||||
| ERCC1 | High expression | FOLFOX | Reduced OS and TTP | [65] |
| ERCC1 | High expression | Platinum derivatives | Reduced OS and response | [88] |
| ERCC1 | GV:rs11615 | 5-FU, Oxaliplatin | Reduced OS and response * | [92,93] |
| ERCC1 | GV:rs3212986 | Cisplatin, FOLFOX | Worse clinical outcome * | [94,96] |
| ERCC2 | Up-regulation | FOLFOX | Reduced OS, PFS and response | [98] |
| ERCC4 | High expression | Cisplatin | Decreased sensitivity in vitro | [91,99] |
| Base-excision DNA repair (BER) | ||||
| XRCC1 | Up-regulation | Cisplatin | Increased drug resistance in vitro | [100] |
| XRCC1 | GV:rs25487 | Oxaliplatin | Worse clinical outcome | [101] |
| Mismatch repair (MMR) | ||||
| MLH1 | Lack of expression | 5-FU | Reduced response | [102] |
| MSI | Appearance | 5-FU | Reduced DFS | [103] |
| Factor | Feature | Drugs Affected | Consequences | Ref. |
|---|---|---|---|---|
| Pro-apoptotic factors (MOC-5a) | ||||
| BAK | Down-regulation | Docetaxel | Decreased sensitivity in vitro | [126] |
| BAX | Down-regulation | 5-FU, Capecitabine, Cisplatin, Irinotecan, Oxaliplatin | Reduced OS and PFS | [123,124,125] |
| BIM | Down-regulation | Docetaxel | Reduced OS | [127] |
| miR-501 | Up-regulation | Doxorubicin | Decreased sensitivity in vitro | [129] |
| miR-633 | Up-regulation | Doxorubicin | Decreased sensitivity in vitro and in vivo | [132] |
| miR-BART20-5p | Up-regulation | 5-FU, Docetaxel | Decreased sensitivity in vitro | [128] |
| p16INKa | Down-regulation | 5-FU | Reduced response * | [120,121] |
| p53 | Down-regulation | 5-FU, Cisplatin, Etoposide, Mitomycin C | Reduced response | [137,138] |
| p53 | Loss of activity | 5-FU, Cisplatin, Paclitaxel | Reduced OS | [111,112] |
| p53 | Gain-of-function GV | First-line chemotherapy | Reduced OS and PFS | [116,117] |
| Survival pathways (MOC-5b) | ||||
| β-catenin, APC, FBXW7 | Mutations | First-line chemotherapy | Reduced OS and PFS | [139] |
| COX-2 | Up-regulation | Oxaliplatin, Irinotecan | Decreased sensitivity in vitro | [140] |
| E-cadherin | Down-regulation | 5-FU, Cisplatin | Reduced DFS | [141] |
| GLI1, GLI2 | Up-regulation | 5-FU | Reduced clinical response | [31] |
| Hedgehog | Increased activity | 5-FU, Cisplatin | Reduced OS | [142] |
| JAK/STAT3 | Increased activity | Cisplatin | Reduced OS | [143] |
| NFκB | Increased activity | 5-FU, Capecitabine, Cisplatin | Decreased sensitivity in vitro | [14,134,135] |
| Notch 1 | Up-regulation | 5-FU, Cisplatin | Reduced OS | [144] |
| PI3K/AKT | Increased activity | Trastuzumab | Reduced OS and PFS | [145] |
| SHH, GLI1 | Up-regulation | Doxorubicin | Decreased sensitivity in vitro | [146] |
| Survivin | Up-regulation | 5-FU, Cisplatin, Docetaxel | Increased DPR | [136] |
| WNT/β-catenin | Increased activity | Cisplatin | Reduced OS and DFS | [147] |
| YAP1 | Up-regulation | Trastuzumab | Decreased sensitivity in vitro | [148] |
| YAP1, TAZ | Up-regulation | 5-FU, Cisplatin | Reduced OS and PFS | [139] |
| Factor | Feature | Drugs Affected | Consequences | Ref. |
|---|---|---|---|---|
| Hypoxia | ||||
| HIF-1α | Up-regulation | 5-FU, Platinum derivatives | Apoptosis inhibition | [156,157,158,159] |
| Up-regulation | 5-FU | Relapse after treatment | [161] | |
| STC1 | Up-regulation | Cisplatin | Apoptosis inhibition | [162] |
| Immune system and inflammation | ||||
| APRIL | Increased production | Cisplatin | Apoptosis inhibition | [164] |
| CCL2 | Increased production | Cisplatin | Apoptosis inhibition | [165] |
| Fn14 | Increased production | 5-FU | Apoptosis inhibition | [166] |
| IL-6 | Increased production | 5-FU | Poor response | [167] |
| IL-8 | Increased production | Platinum derivatives | ABCB1 overexpression and Apoptosis inhibition | [14,168] |
| IL-11 | Increased production | Several drugs | Apoptosis inhibition | [169] |
| IL-33 | Increased production | Platinum derivatives | Apoptosis inhibition | [170] |
| NR4A2 | High expression | 5-FU | Apoptosis inhibition and worse survival rates | [163] |
| Others | ||||
| ATG-5 | High expression | 5-FU, Cisplatin, Epirubicin | Poor survival | [171] |
| Glycemia | Low levels | 5-FU | Metabolic reprogramming and activation of survival | [172] |
| Glycolysis enzymes | Up-regulation | 5-FU | Metabolic reprogramming | [173] |
| lncRNA HCP5 | Production | 5-FU, Oxaliplatin | Metabolic reprogramming | [174] |
| MSC-Exosomes | Production | 5-FU | Activation of other MOCs | [175] |
| TAM-Exosomes | miR-21a-5p transfer | Cisplatin | Apoptosis inhibition | [176] |
| Factor | Feature | Drugs Affected | Consequences | Ref. |
|---|---|---|---|---|
| Cell adhesion proteins | ||||
| CD133 | Up-regulation | 5-FU, Cisplatin | Reduced OS and DFS | [190] |
| CD44 | Up-regulation | 5-FU, Etoposide | Decreased sensitivity in vitro | [183] |
| CD44 | Up-regulation | 5-FU, Oxaliplatin | Decreased clinical response | [142] |
| CD44/CD24 | Up-/Down-regulation | 5-FU | Reduced OS | [188] |
| CD44/EpCAM | Up-regulation | 5-FU, Doxorubicin, Paclitaxel | Decreased sensitivity in vitro | [187] |
| CD71 | Down-regulation | 5-FU | Decreased sensitivity in vitro and in vivo | [193] |
| CXCR4 | Up-regulation | Docetaxel | Decreased sensitivity in vitro | [194] |
| Enzymes | ||||
| ALDH1 | Up-regulation | 5-FU | Reduced OS | [195] |
| Survival pathways | ||||
| Hedgehog | Increased activity | 5-FU | Decreased sensitivity in vitro | [31] |
| HER4 | Up-regulation | Trastuzumab | Decreased sensitivity in vitro and in vivo | [148] |
| HMMR | Up-regulation | 5-FU | Reduced OS | [179] |
| LGR5 | Up-regulation | 5-FU, Oxaliplatin | Reduced OS | [196] |
| miR-187 | Down-regulation | Cisplatin | Decreased sensitivity in vitro | [182] |
| miR-577 | Up-regulation | Oxaliplatin | Decreased sensitivity in vitro | [181] |
| TGF-β/ZEB2 | Increased activity | Trastuzumab | Decreased sensitivity in vitro | [197] |
| TGFBR2 | Up-regulation | 5-FU | Decreased sensitivity in vitro | [180] |
| Transcription factors | ||||
| NANOGP8 | Up-regulation | Oxaliplatin | Decreased sensitivity in vitro | [198] |
| SOX2 | Up-regulation | Cisplatin, Doxorubicin | Decreased sensitivity in vitro and in vivo | [199] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marin, J.J.G.; Perez-Silva, L.; Macias, R.I.R.; Asensio, M.; Peleteiro-Vigil, A.; Sanchez-Martin, A.; Cives-Losada, C.; Sanchon-Sanchez, P.; Sanchez De Blas, B.; Herraez, E.; et al. Molecular Bases of Mechanisms Accounting for Drug Resistance in Gastric Adenocarcinoma. Cancers 2020, 12, 2116. https://doi.org/10.3390/cancers12082116
Marin JJG, Perez-Silva L, Macias RIR, Asensio M, Peleteiro-Vigil A, Sanchez-Martin A, Cives-Losada C, Sanchon-Sanchez P, Sanchez De Blas B, Herraez E, et al. Molecular Bases of Mechanisms Accounting for Drug Resistance in Gastric Adenocarcinoma. Cancers. 2020; 12(8):2116. https://doi.org/10.3390/cancers12082116
Chicago/Turabian StyleMarin, Jose J. G., Laura Perez-Silva, Rocio I. R. Macias, Maitane Asensio, Ana Peleteiro-Vigil, Anabel Sanchez-Martin, Candela Cives-Losada, Paula Sanchon-Sanchez, Beatriz Sanchez De Blas, Elisa Herraez, and et al. 2020. "Molecular Bases of Mechanisms Accounting for Drug Resistance in Gastric Adenocarcinoma" Cancers 12, no. 8: 2116. https://doi.org/10.3390/cancers12082116